
Abstract
Mitochondrial dysfunction has been reported to be involved in the pathophysiology of autism spectrum disorder (ASD). Studies investigating the possible association between ASD and polymorphism in SLC25A12, which encodes the mitochondrial aspartate/glutamate carrier, have yielded inconsistent results. We conducted a systematic review and meta-analysis of such studies to elucidate if and which SLC25A12 single nucleotide polymorphisms (SNPs) are associated with ASD. We searched PubMed, Ovid, Web of Science, and ERIC databases through September 20th, 2014. Odds ratios (ORs) were aggregated using random effect models. Sensitivity analyses were conducted based on study design (family-based or case-control). Fifteen out of 79 non-duplicate records were retained for qualitative synthesis. We pooled 10 datasets from 9 studies with 2001 families, 735 individuals with ASD and 632 typically developing (TD) individuals for the meta-analysis of rs2292813, as well as 11 datasets from 10 studies with 2016 families, 852 individuals with ASD and 1058 TD individuals for the meta-analysis of rs2056202. We found a statistically significant association between ASD and variant in rs2292813 (OR = 1.190, 95 % CI 1.052–1.346, P = 0.006) as well as in rs2056202 (OR = 1.206, 95 % CI 1.035–1.405, P = 0.016). Sensitivity analyses including only studies with family-based design demonstrated significant association between ASD and polymorphism in rs2292813 (OR = 1.216, 95 % CI 1.075–1.376, P = 0.002) and rs2056202 (OR = 1.267, 95 % CI 1.041–1.542, P = 0.018). In contrast, sensitivity analyses including case-control design studies only failed to find a significant association. Further research on the role of SLC25A12 and ASD may pave the way for potential innovative therapeutic interventions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental condition characterized by impairments in social communication and interaction, as well as repetitive and restricted behavior [1, 2]. Currently, the disorder is estimated to affect about 1 % of the general population [3]. ASD is recognized as a heritable condition [4–6], and genetics is reported to play an important role in its etiology [7].
Research on the genetics of ASD has taken several approaches, including genome-wide association studies (GWAS). Although the GWAS approach is suited to comprehensively investigate the genetic background of human diseases, it faces the problem that common disease-common variant model does not yield large effect size to reach statistical threshold in GWAS study [8]. In this context, an alternative approach that investigates a priori a specific gene, based on its biological function, may also be fruitful. To date, a number of genes have been investigated for their possible role in the etiology of ASD in the light of pathophysiological hypotheses on this disorder [9].
Mitochondrial dysfunction has been demonstrated to be associated with neuropsychiatric conditions [10, 11]. In the case of ASD, the hypothesis of an involvement of mitochondrial dysfunction has been supported by findings of significantly higher prevalence of mitochondrial diseases in individuals with ASD compared to typically developing (TD) individuals [12]. In addition, it has been reported that individuals with ASD showed atypical serum metabolites associated with mitochondrial function, such as lactate and pyruvate [12–15]. Furthermore, neuroimaging studies have consistently shown atypical levels of metabolites associated with mitochondrial function, including N-actylaspartate (NAA) and lactate [16–19]. Finally, an increasing number of genetic studies in ASD have focused on mitochondrial DNA [14, 20] and on genes that are associated with mitochondrial function [21], such as SLC25A12, which encodes the brain mitochondrial aspartate/glutamate carrier (AGC) [22–24]. However, so far, studies investigating a possible association between single nucleotide polymorphisms (SNPs) in SLC25A12 and ASD have yielded inconsistent results (e.g., [25–27]).
To clarify a possible role of SLC25A12 in ASD, we conducted a systematic review and meta-analysis of studies that investigated the association between SNPs in SLC25A12 and ASD.
Methods and Materials
Methods for this meta-analysis have been developed according to recommendations from the Preferred Reporting Items for Systematic Reviews and Meta-Analyses Statement [28].
Selection Criteria
Study Type
Studies were included if they:
1) were peer-reviewed in order to ensure high levels of methodological adequacy and to avoid the inevitable bias caused by dependence on investigators agreeing to provide data from unpublished studies, as suggested by the Cochrane Group [29].
2) adopted either a “family-based association” or “case-control” design.
Population
Individuals, of any age and with no restriction of gender, diagnosed with ASD, including Asperger’s syndrome, or pervasive developmental disorders according to standardized tools.
Outcome
Association between SNPs in SLC25A12 and ASD. We did not select a priori any specific SNPs. However, in order to provide a robust estimate of effects, consistently with other recent meta-analyses (e.g. [9]), we performed meta-analyses only if four or more datasets were available for each single SNP.
Search Methods for Identification of Studies
Electronic searches were conducted by the two authors independently in the following databases, available via the University of Tokyo Medical Library and the New York University (NYU) Medical Library: PubMed, Ovid databases (Ovid MEDLINE®, EMBASE Classic + EMBASE, PsycINFO), Web of knowledge databases (including Web of Science, Biological Abstracts, BIOSIS, Current Contents Connect, Data Citation Index, Derwet Innovations Index, FSTA, INspec, MEDLINE, and SciELO), and ERIC. The last search was performed on September 20th, 2014. Supplement 1 reports the search terms and syntax for each database.
Identification and Selection of Studies
In stage 1, the two authors screened title and abstracts of all non-duplicated papers and agreed on a final list of references to assess. The full-text version of the articles passing stage 1 was assessed for eligibility by the two authors, independently. Discrepancies were resolved with consensus. Reference lists of the retained papers were also scanned to determine if any relevant studies had been missed during the database searches. Data from multiple reports of the same study were linked together. Where required, the corresponding author was contacted to inquire on study eligibility.
Data Extraction
The two authors independently extracted the following data: 1) names of the first authors, 2) year of publication, 3) study design (i.e., family-based association or case-control), 4) number of participants, 5) ethnicity of participants, 6) diagnostic criteria, and 7) transmitted or non-transmitted risk allele counts from heterozygous parents to individuals with ASD for family-based studies and risk and non-risk allele count or risk allele frequency in case-control studies. When the study did not report sufficient data to calculate effect size, we contacted corresponding authors via e-mail to request additional information. If the contact with the author was not successful, we could not include the study in the meta-analysis.
Statistical Analysis
We adopted a previously reported method to integrate results from family-based design and case-control design [30]. Odds ratio (OR) was used as an effect size [9, 31]. Specifically, we calculated logarithm of ORs and standard error from each retained study using number of transmitted and non-transmitted risk alleles in family-based studies and count or frequency of risk allele in case-control studies. For one family-based study, where number of transmitted and non-transmitted risk alleles were not available, we calculated OR using chi-square and number of families [32]. In one study reporting result of transmission disequilibrium test (TDT) and family-based association tests (FBAT) [33], we chose TDT with the aim to decrease between-study heterogeneity of included studies, because the large majority of included studies reported results based on TDT design [25, 26, 34]. We calculated standardized error of logarithm of OR using p value for the study by Carayol et al. [35] in order to include it into the meta-analysis.
We aggregated effect sizes using Comprehensive Meta-Analysis software (Biostat, Inc., Englewood, New Jersey, USA), following the procedure described in a recent meta-analysis of SNPs of oxytocin receptor gene in individuals with ASD [9].
We used a random effect model to account for heterogeneity in results of included studies. Statistical threshold for significance was set at P < 0.025 (=0.05/2, corrected for multiple comparisons).
Publication Bias Assessment
We examined publication bias quantitatively according to the Egger’s liner regression method [36]. Significance for publication bias was set at P < 0.10 [36].
Between-Study Heterogeneity Assessment
Given the considerable between-study heterogeneity in participants, such as ethnicity, and study design (i.e., family-based design or case-control design), we employed I 2 score to evaluate between-study heterogeneity [37, 38]. Although there is no definite score for cutoff, the following categorization has been suggested for interpretation: “0 to 40 %: might not be important”, “30 to 60 %: may represent moderate heterogeneity”, “50 to 90 %: may represent substantial heterogeneity”, and “75 to 100 %: considerable heterogeneity” [29].
Sensitivity Analysis
In order to test if findings were influenced by study design, we conducted a sensitivity analysis including only family-based design and another one focusing only on case-control design. Statistical threshold for significance was set for P = 0.0125 (=0.05/4, case-control and family-based designs for two SNPs).
One-Study Removed Sensitivity Analysis
To challenge the robustness of the results, we conducted “one-study removed” sensitivity analysis. Using this method, we evaluate whether results were influenced by only one single study [39, 40]. According to this approach, the more the sensitivity analysis preserves significance, the more the result is replicable. We applied the “one-study removed” procedure to the sensitivity analysis including only studies with family-based design. We did not apply this procedure to the sensitivity analysis including only case-control studies, since this did not yield significant results (see “Results” section). We set a statistical threshold at P < 0.025.
Results
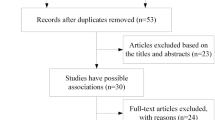
Figure 1 shows the PRISMA studies selection flowchart. Supplemental Table 1 reports studies excluded from the qualitative synthesis, with reasons.

The process of systematic screening of studies. A comprehensive literature search firstly identified 154 studies from databases. After removing duplicates, there were 79 independent studies for further screening. Among them, 52 studies were excluded after reading abstract, which left 27 studies for full-text screening. Twelve studies were excluded from the qualitative synthesis as a result of full-text screening. Among the remaining 15 studies, three studies were not included in the meta-analysis because they did not provide sufficient data to calculate effect sizes. Therefore, 12 studies were identified to be eligible for meta-analysis
Our search identified a total of 15 studies for qualitative synthesis and potentially relevant for the meta-analysis (Table 1) [25–27, 32–35, 41–48]. Twenty-three types of SNP (rs11757, rs12692976, rs17499593, rs17581284, rs1878583, rs2056202, rs2271758, rs2292813, rs35678, rs3749004, rs3765166, rs3770445, rs3821095, rs4307059, rs6433317, rs6716901, rs6724337, rs6758704, rs7573003, rs7586207, rs925881, rs908670, and rs970948) were investigated once or more in the identified 15 studies. Among these 23 SNPs, only rs2292813 and rs2056202 were investigated in four or more datasets. Eleven studies, with a total of 12 datasets (either family-based or case-control datasets; the study by Blasi et al. [25] provided both types of datasets) focused on rs2292813 [25–27, 32–35, 41, 44, 46, 47]. Two datasets from two studies [34, 46] were excluded given lack of complete data necessary to carry out the analysis (We sent an e-mail to corresponding authors to gather such data on 22nd Sep 2014 and waited for their response for one month. If the authors did not respond to our e-mail, we did not include the study). Therefore, 9 studies were used for the meta-analysis of rs2292813.
Twelve studies with a total of 14 datasets investigated rs2056202 [25–27, 32–34, 41–44, 46, 47] (the studies by Blasi et al. [25] and Correia et al. [42] both provided family-based as well as case-control datasets). Two datasets from two studies [46, 47] could not be retained for the meta-analysis since it was not possible to obtain additional necessary information from the authors. As for the study by Correia et al. that reported results of both family-based design and case-control design, we extracted the results from case-control dataset only, given lack of necessary data from the family-based design dataset [42]. Therefore, ten studies were used for the meta-analysis of rs2056202.
In total, 12 studies were used for the meta-analyses.
Meta-Analysis of Datasets on rs2292813
Nine studies, including seven family-based design datasets with a total of 2001 families [25–27, 32, 33, 35, 47] and three case-control design datasets recruiting 735 individuals with ASD and 632 individuals with TD [25, 41, 44], were eligible to the meta-analysis (Table 2). A random effect model meta-analysis demonstrated a significant association between rs2292813 and ASD (“G” allele increasing the risk for ASD, OR = 1.190, 95 % CI 1.052–1.346, P = 0.006). Between-study heterogeneity was small (I 2 = 13.9), and there was no significant publication bias (P = 0.427) (Fig. 2a).

Results of meta-analyses. a A random effect model demonstrated a significant association between polymorphism of rs2292813 and autism spectrum disorder. b A sensitivity analysis with studies with family-based design also demonstrated a significant association between polymorphism of rs2292813 and autism spectrum disorder. In contrast, c a sensitivity analysis with studies with case-control design revealed no significant association between polymorphism of rs2292813 and autism spectrum disorder. d A random effect model showed a statistically significant association between polymorphism in rs2056202 and autism spectrum disorder. e A sensitivity analysis with studies with family-based design preserved a statistically significant association between polymorphism in rs2056202 and autism spectrum disorder. On the other hand, no significant association between polymorphism in rs2056202 and autism spectrum disorder was detected in a sensitivity analysis with studies with case-control design (f)
A sensitivity analysis focusing only on datasets with family-based design confirmed the significant association between rs2292813 and ASD (“G” allele increasing the risk for ASD, OR = 1.216, 95 % CI 1.075–1.376, P = 0.002), with negligible between-study heterogeneity (I 2 = 12.6) and no significant publication bias (P = 0.178) (Table 2 and Fig. 2b). On the other hand, a sensitivity analysis focusing only on case-control design datasets showed no significant association between “G” allele in rs2292813 and ASD (OR = 0.899, 95 % CI 0.594–1.360, P = 0.615) (Table 2 and Fig. 2c).
Meta-Analysis of Datasets on rs2056202
Ten studies, including six family-based design datasets with 2016 families [25–27, 32–34] and five case-control design datasets recruiting 852 individuals with ASD and 1058 individuals with TD [25, 41–44], were amenable to meta-analysis (Table 2). A random effect model meta-analysis showed a significant association between rs2056202 and ASD (“G” allele increasing the risk for ASD, OR = 1.206, 95 % CI 1.035–1.405, P = 0.016). There was moderate between-study heterogeneity (I 2 = 44.6), and no publication bias was observed (P = 0.373) (Fig. 2d).
A sensitivity analysis focusing only on datasets with family-based design confirmed a significant association between polymorphism in rs2056202 and ASD (“G” allele increasing the risk for ASD, OR = 1.267, 95 % CI 1.041–1.542, P = 0.018), with substantial between-study heterogeneity (I 2 = 61.7) but no significant publication bias (P = 0.269) (Table 2 and Fig. 2e). On the other hand, a sensitivity analysis including only case-control design datasets revealed no significant association between polymorphism in rs2056202 and ASD (OR = 1.071, 95 % CI 0.854–1.343, P = 0.552) (Table 2 and Fig. 2f).
One-Study Removed Sensitivity Analysis
rs2292813
Using, as mentioned, a strict threshold at P < 0.025, all “one-study removed” sensitivity analyses, except one, preserved the significant association between “G” allele in rs2292813 and development of ASD. In addition, all the “one-study removed” analyses applied to the sensitivity analysis of rs2292813 with family-based design preserved the significance of the association between polymorphism in rs2292813 and ASD (Fig. 3b).

Results of “one-study removed” sensitivity analysis. a “One-study removed” sensitivity analysis of rs2292813 showed that all the sensitivity analyses, except one, preserved statistically significant association between polymorphism in rs2292813 and autism spectrum disorder. b “One-study removed” procedure applied to sensitivity analysis of family-based design studies of rs229813 has demonstrated that all the sensitivity analyses preserved the significance of the association. c “One-study removed” sensitivity analysis of rs2056202 demonstrated that six out of 11 preserved the significant association between polymorphism in rs2056202 and autism spectrum disorder. d “One-study removed” procedure applied to sensitivity analysis of family-based design studies of rs2056202 showed that two out of the six sensitivity analyses have replicated the significant association
rs2056202
Six out of the 11 “one-study removed” sensitivity analyses showed a statistically significant association (P < 0.025) between polymorphism in rs2056202 and ASD (Fig. 3c). In contrast, only two out of the six “one-study removed” analyses preserved the significant association between “G” allele in rs2056202 and ASD (Fig. 3d).
Discussion
The present meta-analysis showed a significant association between polymorphism of both rs2292813 and rs2056202 in SLC25A12 and ASD, with small between-study heterogeneity and without significant publication bias. Sensitivity analyses focusing only on family-based design showed that both polymorphism of rs2292813 and rs2056202 in SLC25A12 were associated with ASD, with low between-study heterogeneity and without significant publication bias.
As we expected, the analyses demonstrated significance albeit small pooled OR for the association between SNPs variants in SLC25A12 and ASD. The small effect size is a possible reason why available studies have yielded inconsistent results. In addition, the small effect size might contribute to explain why SLC25A12 did not reach the threshold in existing GWAS studies of ASD [49–51]. Although effect size was small, the present result supports the possibility that SLC25A12 is involved in the etiology of ASD.
Our results are consistent with the findings from several lines of research. Postmortem studies demonstrated atypical brain expression of AGC1 in individuals with ASD compared with TD individuals [22, 33, 44]. Although it is not possible to explore activation of AGC1 in vivo, results from several neuroimaging studies are in line with these postmortem studies. For instance, NAA is a metabolite that contributes to regulate osmotic pressure of neurons and becomes a precursor of N-acetylaspartylglutamate (NAAG) [52]. Magnetic resonance spectroscopy studies have recognized these as reflection of neuronal density and function [52–54]. As NAA is synthesized in neuronal mitochondria from aspartate and acetyl-coenzyme A and is coupled with glutamate to become NAAG [52, 54], NAA is reported to have a link with mitochondrial function [55]. Basic animal experiments suggested that AGC1 is associated with NAA synthesis in the brain; furthermore, evidence in human showed that individuals with AGC deficiency had significantly lower NAA levels than those with normal levels of AGC [56–59]. In line with the notion that AGC1 is atypically expressed or activated in individuals with ASD, a number of studies have reported atypical NAA and NAAG levels in individuals with ASD [16–18, 60, 61]. It is assumed that abnormality in AGC1 activation may result in abnormal aspartate/glutamate exchange rate, which induces abnormal respiratory chain activity and enhances oxidative stress, which eventually results in neuronal neurofilamentous accumulations and myelination deficits (reviewed in [12, 33]).
Although the general meta-analysis and sensitivity analysis focusing only on family-based design studies demonstrated significant association between polymorphism in SLC25A12 and ASD, sensitivity analysis focusing only on case-control design studies did not show any significant association. This may have been accounted for by the fact that family-based design studies compare individuals with ASD to their relatives (mainly their parents), who may share common genetic characteristics. In contrast, in case-control design studies, it is required to conduct population stratification analysis to ensure that cases and controls share common genetic characteristics other than the SNPs of interest [62]. Not all case-control design studies included in the current meta-analysis have implemented population stratification analysis. Thus, in comparison to case-control design studies, it might be possible that family-based design studies had more statistical power to detect the potential effect of SLC25A12.
Our results should be considered in the light of some limitations. First, although I 2 scores of the two general meta-analyses (the one on rs2292813 and the other on rs2056202, including all available studies) were small, the meta-analyses might have been hampered by considerable inherent between-study heterogeneity, since they integrated studies with two different study designs, namely family-based design and case-control design. Second, in addition to differences in design, included studies were also heterogeneous in terms of type of tissue collecting. Particularly, one study with case-control design by Lepagnol-Bestel et al. collected brain tissue, rather than blood or buccal swabs, to investigate variants in SNPs [44]. In addition, from the forest plot, this study may be considered as an outlier. However, it is worth noting that in the “one-study removed” sensitivity analysis that excluded the study by Lepagnol-Bestel et al. [44], the association between ASD and polymorphisms of rs2292813 (OR = 1.186, 95 % CI 1.051–1.337, P = 0.006, Fig. 3a) or rs2056202 (OR = 1.199, 95 % CI 1.035–1.389, P = 0.016, Fig. 3b) remained significant. Furthermore, we note that our approach of pooling studies based on different tissues is consistent with a recent meta-analysis of SNPs of oxytocin receptor in ASD [9]. Third, with regard to one study by Blasi et al. [25], we used datasets of both family-based design and case-control design. However, as shown in one-study removed sensitivity analysis, it should be emphasized that both the sensitivity analysis discarding family-based design dataset from Blasi et al. [25] and the sensitivity analysis excluding case-control design dataset from the study preserved the statistical conclusion that polymorphism in rs2292813 (OR = 1.184, 95 % CI 1.028–1.363, P = 0.019 and OR = 1.209, 95 % CI 1.070–1.366, P = 0.002, respectively, Fig. 3a) and rs2056202 (OR = 1.219, 95 % CI 1.024–1.452, P = 0.026 and OR = 1.235, 95 % CI 1.061–1.438, P = 0.007, Fig. 3b) in SLC25A12 is associated with ASD. Fourth, although we have demonstrated statistically robust association between variants in SNPs in SLC25A12 and ASD, it was not possible to conduct sensitivity analysis based on ethnicity due to insufficient information from the retained studies.
Conclusion
The present meta-analysis suggests that polymorphism in SLC25A12 deserves further attention as possible mechanism involved in the etiopathophysiology of ASD.
References
American Pychiatric Association (2013) Diagnostic and statistical manual of mental disorders, 5th edn. American Psychiatric Publishing, Arlington, VA, USA
Aoki Y, Yahata N, Watanabe T, Takano Y, Kawakubo Y, Kuwabara H, Iwashiro N, Natsubori T et al (2014) Oxytocin improves behavioural and neural deficits in inferring others' social emotions in autism. Brain 137(Pt 11):3073–3086
Centers for Disease Control and Prevention (2012) Prevalence of autism spectrum disorders—autism and developmental disabilities monitoring network, 14 sites, United States, 2008. MMWR Surveill Summ 61(3):1–19
Frazier TW, Thompson L, Youngstrom EA, Law P, Hardan AY, Eng C, Morris N (2014) A twin study of heritable and shared environmental contributions to autism. J Autism Dev Disord 44(8):2013–2025
Robinson EB, Koenen KC, McCormick MC, Munir K, Hallett V, Happé F, Plomin R, Ronald A (2011) Evidence that autistic traits show the same etiology in the general population and at the quantitative extremes (5 %, 2.5 %, and 1 %). Arch Gen Psychiatry 68(11):1113–1121
Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A et al (2011) Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 68(11):1095–1102
Lai M-C, Lombardo MV, Baron-Cohen S (2014) Autism. Lancet 383(9920):896–910
Visscher PM, Brown MA, McCarthy MI, Yang J (2012) Five years of GWAS discovery. Am J Hum Genet 90(1):7–24
LoParo D, Waldman ID (2014) The oxytocin receptor gene (OXTR) is associated with autism spectrum disorder: a meta-analysis. Mol Psychiatry. doi:10.1038/mp.2014.77
Quiroz JA, Gray NA, Kato T, Manji HK (2008) Mitochondrially mediated plasticity in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology 33(11):2551–2565
Munakata K, Iwamoto K, Bundo M, Kato T (2005) Mitochondrial DNA 3243A > G mutation and increased expression of LARS2 gene in the brains of patients with bipolar disorder and schizophrenia. Biol Psychiatry 57(5):525–532
Rossignol DA, Frye RE (2012) Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry 17(3):290–314
Manji H, Kato T, Di Prospero NA, Ness S, Beal MF, Krams M, Chen G (2012) Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci 13(5):293–307
Giulivi C, Zhang YF, Omanska-Klusek A, Ross-Inta C, Wong S, Hertz-Picciotto I, Tassone F, Pessah IN (2010) Mitochondrial dysfunction in autism. JAMA 304(21):2389–2396
Kuwabara H, Yamasue H, Koike S, Inoue H, Kawakubo Y, Kuroda M, Takano Y, Iwashiro N et al (2013) Altered metabolites in the plasma of autism spectrum disorder: a capillary electrophoresis time-of-flight mass spectroscopy study. PLoS One 8(9):e73814
Aoki Y, Kasai K, Yamasue H (2012) Age-related change in brain metabolite abnormalities in autism: a meta-analysis of proton magnetic resonance spectroscopy studies. Transl Psychiatry 2:e69
Aoki Y, Abe O, Yahata N, Kuwabara H, Natsubori T, Iwashiro N, Takano Y, Inoue H et al (2012) Absence of age-related prefrontal NAA change in adults with autism spectrum disorders. Transl Psychiatry 2:e178
Aoki Y, Watanabe T, Abe O, Kuwabara H, Yahata N, Takano Y, Iwashiro N, Natsubori T et al (2014) Oxytocin's neurochemical effects in the medial prefrontal cortex underlie recovery of task-specific brain activity in autism: a randomized controlled trial. Mol Psychiatry. doi:10.1038/mp.2014.74
Goh S, Dong Z, Zhang Y, DiMauro S, Peterson BS (2014) Mitochondrial dysfunction as a neurobiological subtype of autism spectrum disorder: evidence from brain imaging. JAMA Psychiatry 71(6):665–671
Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, O'Hearn S, Levy S et al (2014) Progressive increase in mtDNA 3243A > G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci U S A 111(38):E4033–4042
Napolioni V, Persico AM, Porcelli V, Palmieri L (2011) The mitochondrial aspartate/glutamate carrier AGC1 and calcium homeostasis: physiological links and abnormalities in autism. Mol Neurobiol 44(1):83–92
Silverman JM, Buxbaum JD, Ramoz N, Schmeidler J, Reichenberg A, Hollander E, Angelo G, Smith CJ et al (2008) Autism-related routines and rituals associated with a mitochondrial aspartate/glutamate carrier SLC25A12 polymorphism. Am J Med Genet B Neuropsychiatr Genet 147(3):408–410
Anitha A, Nakamura K, Thanseem I, Yamada K, Iwayama Y, Toyota T, Matsuzaki H, Miyachi T et al (2012) Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol Autism 3(1):12
del Arco A, Satrustegui J (1998) Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J Biol Chem 273(36):23327–23334
Blasi F, Bacchelli E, Carone S, Toma C, Monaco AP, Bailey AJ, Maestrini E, International Molecular Genetic Study of Autism Consortium (IMGSAC) (2006) SLC25A12 and CMYA3 gene variants are not associated with autism in the IMGSAC multiplex family sample. Eur J Hum Genet 14(1):123–126
Ramoz N, Reichert JG, Smith CJ, Silverman JM, Bespalova IN, Davis KL, Buxbaum JD (2004) Linkage and association of the mitochondrial aspartate/glutamate carrier SLC25A12 gene with autism. Am J Psychiatry 161(4):662–669
Segurado R, Conroy J, Meally E, Fitzgerald M, Gill M, Gallagher L (2005) Confirmation of association between autism and the mitochondrial aspartate/glutamate carrier SLC25A12 gene on chromosome 2q31. Am J Psychiatry 162(11):2182–2184
Moher D, Cook DJ, Eastwood S, Olkin I, Rennie D, Stroup DF (1999) Improving the quality of reports of meta-analyses of randomised controlled trials: the QUOROM statement. Quality of reporting of meta-analyses. Lancet 354(9193):1896–1900
Higgins J, Green S (2008) Cochrane handbook for systematic reviews of interventions. Wiley, Chichester, UK
Kazeem GR, Farrall M (2005) Integrating case-control and TDT studies. Ann Hum Genet 69(Pt3):329–335
Brandys MK, Kas MJ, van Elburg AA, Ophoff R, Slof-Op't Landt MC, Middeldorp CM, Boomsma DI, van Furth EF et al (2013) The Val66Met polymorphism of the BDNF gene in anorexia nervosa: new data and a meta-analysis. World J Biol Psychiatry 14(6):441–451
Kim SJ, Silva RM, Flores CG, Jacob S, Guter S, Valcante G, Zaytoun AM, Cook EH et al (2011) A quantitative association study of SLC25A12 and restricted repetitive behavior traits in autism spectrum disorders. Mol Autism 2(1):8
Palmieri L, Papaleo V, Porcelli V, Scarcia P, Gaita L, Sacco R, Hager J, Rousseau F et al (2010) Altered calcium homeostasis in autism-spectrum disorders: evidence from biochemical and genetic studies of the mitochondrial aspartate/glutamate carrier AGC1. Mol Psychiatry 15(1):38–52
Ramoz N, Cai G, Reichert JG, Silverman JM, Buxbaum JD (2008) An analysis of candidate autism loci on chromosome 2q24-q33: evidence for association to the STK39 gene. Am J Med Genet B Neuropsychiatr Genet 147B(7):1152–1158
Carayol J, Schellenberg GD, Tores F, Hager J, Ziegler A, Dawson G (2010) Assessing the impact of a combined analysis of four common low-risk genetic variants on autism risk. Mol Autism 1:4
Egger M, Davey Smith G, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ 315(7109):629–634
Aoki Y, Aoki A, Suwa H (2012) Reduction of N-acetylaspartate in the medial prefrontal cortex correlated with symptom severity in obsessive-compulsive disorder: meta-analyses of 1H-MRS studies. Transl Psychiatry 2:1–10
Aoki Y, Inokuchi R, Suwa H, Aoki A (2013) Age-related change of neurochemical abnormality in attention-deficit hyperactivity disorder: a meta-analysis. Neurosci Biobehav Rev 37(8):1692–1701
Aoki Y, Abe O, Nippashi Y, Yamasue H (2013) Comparison of white matter integrity between autism spectrum disorder subjects and typically developing individuals: a meta-analysis of diffusion tensor imaging tractography studies. Mol Autism 4(1):25
Aoki Y, Inokuchi R, Gunshin M, Yahagi N, Suwa H (2012) Diffusion tensor imaging studies of mild traumatic brain injury: a meta-analysis. J Neurol Neurosurg Psychiatry 83(9):870–876
Chien WH, Wu YY, Gau SS, Huang YS, Soong WT, Chiu YN, Chen CH (2010) Association study of the SLC25A12 gene and autism in Han Chinese in Taiwan. Prog Neuropsychopharmacol Biol Psychiatry 34(1):189–192
Correia C, Coutinho AM, Diogo L, Grazina M, Marques C, Miguel T, Ataíde A, Almeida J et al (2006) Brief report: High frequency of biochemical markers for mitochondrial dysfunction in autism: no association with the mitochondrial aspartate/glutamate carrier SLC25A12 gene. J Autism Dev Disord 36(8):1137–1140
Durdiakova J, Warrier V, Baron-Cohen S, Chakrabarti B (2014) Single nucleotide polymorphism rs6716901 in SLC25A12 gene is associated with Asperger syndrome. Mol Autism 5(1):25
Lepagnol-Bestel AM, Maussion G, Boda B, Cardona A, Iwayama Y, Delezoide AL, Moalic JM, Muller D et al (2008) SLC25A12 expression is associated with neurite outgrowth and is upregulated in the prefrontal cortex of autistic subjects. Mol Psychiatry 13(4):385–397
Prandini P, Pasquali A, Malerba G, Marostica A, Zusi C, Xumerle L, Muglia P, Da Ros L et al (2012) The association of rs4307059 and rs35678 markers with autism spectrum disorders is replicated in Italian families. Psychiatr Genet 22(4):177–181
Rabionet R, McCauley JL, Jaworski JM, Ashley-Koch AE, Martin ER, Sutcliffe JS, Haines JL, DeLong GR et al (2006) Lack of association between autism and SLC25A12. Am J Psychiatry 163(5):929–931
Turunen JA, Rehnstrom K, Kilpinen H, Kuokkanen M, Kempas E, Ylisaukko-Oja T (2008) Mitochondrial aspartate/glutamate carrier SLC25A12 gene is associated with autism. Autism Res 1(3):189–192
Yan Z-H, Xing J, Luo H-Y, Yang T-S, Sakamoto Y, Nanba E (2006) Association between SLC25A12 and SCN2A2 gene polymorphisms and autism. J Jilin Univ Med Ed 32:313–315
Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M et al (2009) Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459(7246):528–533
Weiss LA, Arking DE, Gene Discovery Project of Johns H, the Autism C, Daly MJ, Chakravarti A (2009) A genome-wide linkage and association scan reveals novel loci for autism. Nature 461(7265):802–808
Wittkowski KM, Sonakya V, Bigio B, Tonn MK, Shic F, Ascano M, Nasca C, Gold-Von SG (2014) A novel computational biostatistics approach implies impaired dephosphorylation of growth factor receptors as associated with severity of autism. Transl Psychiatry 4:e354
Baslow MH (2011) The vertebrate brain, evidence of its modular organization and operating system: insights into the brain’s basic units of structure, function, and operation and how they influence neuronal signaling and behavior. Front Behav Neurosci 5:5
Sager T, Hansen A, Laursen H (2000) Correlation between N-acetylaspartate levels and histopathologic changes in cortical infarcts of mice after middle cerebral artery occlusion. J Cereb Blood Flow Metab 20(5):780–788
Moffett JR, ROSS B, Arun P, Madhavarao CN, Namboodiri AMA (2007) N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol 81(2):89–131
Kamada K, Takeuchi F, Houkin K, Kitagawa M, Kuriki S, Ogata A, Tashiro K, Koyanagi I et al (2001) Reversible brain dysfunction in MELAS: MEG, and (1)H MRS analysis. J Neurol Neurosurg Psychiatry 70(5):675–678
Satrústegui J, Contreras L, Ramos M, Marmol P, del Arco A, Saheki T, Pardo B (2007) Role of aralar, the mitochondrial transporter of aspartate-glutamate, in brain N-acetylaspartate formation and Ca(2+) signaling in neuronal mitochondria. J Neurosci Res 85(15):3359–3366
Jalil MA, Begum L, Contreras L, Pardo B, Iijima M, Li MX, Ramos M, Marmol P et al (2005) Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J Biol Chem 280(35):31333–31339
Ramos M, Pardo B, Llorente-Folch I, Saheki T, Del Arco A, Satrustegui J (2011) Deficiency of the mitochondrial transporter of aspartate/glutamate aralar/AGC1 causes hypomyelination and neuronal defects unrelated to myelin deficits in mouse brain. J Neurosci Res 89(12):2008–2017
Falk MJ, Li D, Gai X, McCormick E, Place E, Lasorsa FM, Otieno FG, Hou C et al (2014) AGC1 deficiency causes infantile epilepsy, abnormal myelination, and reduced N-acetylaspartate. JIMD Rep 14:77–85
Tebartz van Elst L, Maier S, Fangmeier T, Endres D, Mueller GT, Nickel K, Ebert D, Lange T et al (2014) Disturbed cingulate glutamate metabolism in adults with high-functioning autism spectrum disorder: evidence in support of the excitatory/inhibitory imbalance hypothesis. Mol Psychiatry. doi:10.1038/mp.2014.62
Horder J, Lavender T, Mendez MA, O'Gorman R, Daly E, Craig MC, Lythgoe DJ, Barker GJ et al (2013) Reduced subcortical glutamate/glutamine in adults with autism spectrum disorders: a [(1)H]MRS study. Transl Psychiatry 3:e279
McGinnis R, Shifman S, Darvasi A (2002) Power and efficiency of the TDT and case-control design for association scans. Behav Genet 32(2):135–144
Author contributions
YA conceived the study design. YA and SC have independently screened and extracted the data. YA and SC wrote the paper.
Conflict of Interest
Dr. Samuele Cortese has received royalties from Aargon Healthcare Italy.
Dr. Yuta Aoki declares no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 33 kb)
Rights and permissions
About this article

Cite this article
Aoki, Y., Cortese, S. Mitochondrial Aspartate/Glutamate Carrier SLC25A12 and Autism Spectrum Disorder: a Meta-Analysis. Mol Neurobiol 53, 1579–1588 (2016). https://doi.org/10.1007/s12035-015-9116-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9116-3