
Abstract
Studies have reported typically biophysical lysosomal cation channels including TPCs. Their plausible biological roles are being elucidated by pharmacological, genetic and conventional patch clamp procedures. The best characterized so far among these channels is the ML1 isoform of TRP. The reported TRPs and TPCs are bypass for cation fluxes and are strategic for homeostasis of ionic milieu of the acidic organelles they confine to. Ca2+ homeostasis and adequate acidic pHL are critically influential for the regulation of a plethora of biological functions these intracellular cation channels perform. In lysosomal ion channel biology, we review: ML1 and TPC2 in Ca2+ signaling, ML1 and TPC2 in pHL regulation. Using Aβ42 and tau proteins found along clathrin endolysosomal internalization pathway (Fig. 3), we proffer a mechanism of abnormal pHL and ML1/TPC2-dependent cation homeostasis in AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
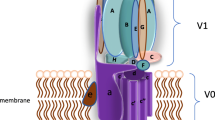
Lysosomes are membrane-enclosed acidic organelles and contain acid hydrolases (Fig. 1). The acid hydrolases lend lysosomes the ability to degrade and process biological materials including polysaccharides, proteins and lipids. The degradative ability is a function of 4.5–5.0 acidic pH in the lumen of the lysosome (Fig. 1). In this acidic optima, lysosomal acid hydrolases are optimally active [1]. Apart from the acid hydrolase contents, lysosomes house Ca2+ [2, 3], and cation channels [4–6]. Some of the cation channels among others are transient receptor potential mucolipin 1 (ML1) and two-pore channel 2 (TPC2). These channels are well known to perform multifarious functions and a single distinctive physiological function could not yet be completely ascribed to either of these channels despite huge intense research work.

Structure and magnitude of a mechanized pHL. A stringent requirement for a functional luminal acid hydrolases is low pHL, of 4.5 to 5.0, maintained by ATP-mediated proton pump and leak hypothesis. Either pump in or leak out of H+ is electrogenic requiring a counter mechanism (ML1/TPC2-mediated cation efflux or anion influx that may not be the rheogenic Cl_ antiporter) to banish a positive net charge inside the lumen generated by ATP hydrolysis to regulate V-ATPase, the principle regulator of pHL
Accordingly, it is been reported that the intracellular lysosomal cation channels play roles in autophagy [7], migration and cell polarity [8], morphogenesis [9], cancer, neurodegeneration [10], stroke/ischemia and even in aging through a coordinated action of Ca2+ homeostasis, membrane lipids and proteins. Other roles of the channels may include: neuronal communication [2, 6], membrane trafficking [11, 12], regulation of gene expression and nociception, temperature [13], and sensation controls, muscle contraction, and vaso-motor actions.
The regulatory mechanisms underlying the roles are being hugely investigated and are not clearly understood. Phosphoinositides [4, 5, 14, 15] and GTPases [16, 17] can potentiate MLl/TPC2 specific activation in the functions. The phosphoinositides are membrane-bound lipid that prescribes signature identify for cytoplasmic proteins and potentiates the activity of the cation channels with specificity and potency [14]. The GTPases are well known regulators of membrane trafficking that switch between active and inactive forms in a GTP/GDP-dependent manner respectively to regulate membrane trafficking. The classical switches underline a process that manifests discrete signaling to regulate the ML1/TPC2 activity in the functions. Furthermore, by working hand-in-hand in a combined coordinated fashion rather than just single entities in signaling processes, intracellular dependent activations by the GTPases, the Ca2+-mediated and the phosphoinositides can fine-tune the channels mediated activities. Juxta-organellar levels of Ca2+ that fixes the spatio and temporary signal required for membrane trafficking is also a way. Another indispensable crucial means is mechanization of the structure and magnitude of lysosomal pH (pHL). The maintenance of pHL is largely done by vacuolar-type ATPase (V-ATPase), albeit, either influx of cations or efflux of anions to truncate electrical potential that can be generated by proton accumulation through V-ATPase across lysosomal membrane.
Abnormalities in pHL regulation, in Ca2+ second messenger signaling, in specific and potent activations by phosphoinositides and in on and off classical switches of GTPase, together can defect lysosomes function in the degradation of biological materials. Defects in the signaling networks associated with lysosomes potentially underlie how macromolecules accumulate in AD (Fig. 3). Accumulation once initiated can beget accumulation through consequent disruption of biochemical signaling. This is supported in part by a hypothesis which states that accumulation of materials that cannot be degraded by lysosomal hydrolytic enzymes causes a kind of lysosomal stress that can disrupt if not abolish completely biochemical signaling in the lysosome [18, 19].
Since luminal acidifications regulated by cation sources are necessary for the distribution and degradation of macromolecules in the endolysosomal pathway, we review ML1 and TPC2 in Ca2+ signaling, ML1 and TPC2 in pHL acidification (Fig. 1). With the identification of Ca2+ permeable channels in the lysosomes, we opine a mechanism of pHL and ML1/TPC2 -mediated aberrant cation signaling in AD for the first time that potentially provides the basis for the famous AD amyloid hypothesis.
The Channels in pHL Regulation
Because pHL regulation is important in endolysosomal biogenesis and repair, fusion and fission, trafficking, cell growth, immunity, lipid storage disease, AD, Parkinson’s disease and cystic fibrosis, we discussed how ML1 and TPC2 mediated abnormal Ca2+ release could dilute the structure and magnitude of pHL. This is believed to be the initial bases upon which acid hydrolases that degrade macromolecules including Aβ become defective. In turn, for instance, in lipid storage disease (LSD), defective acid hydrolases stimulates substrate accumulation of macromolecules. In the LSD, the role of ML1/TPC2 in such accumulation has been studied and in such accumulation there is near to complete compromised Ca2+ signaling. Ca2+ signaling per say is desperate and ubiquitous occurring in both LSD and AD. Aberrant Ca2+ homeostasis in luminal acidic organelle as found in NP are a key strategy leading to LSD [20]. In such homeostatic process, PI(3, 5)P2 could potentiate ML1/TPC2 dependent Ca2+ release but NAADP could not potentiate TPC2-dependent Ca2+ release (Wang et al. [15]). Also, in such homeostasis, and as direct evidence, NAADP and PI(3,5)P2 could potentiate TPC2 in cellular functions [21]. This resolves an existing controversy that NAADP does not activate TPC2 [7, 15] and that NAADP does activate TPC2 [6] for Ca2+ release. For ML1, much is not known about NAADP-dependent activation. Apparently, it appears that NAADP dependent endolysosomal ML1/TPC2 activation for Ca2+ release is restricted to TPC2.
Notwithstanding, it is noteworthy that both ML1 and TPC2 are strategic machineries for modulating amounts of Ca2+ necessary to elicit cellular functions. Abnormal Ca2+ effluxes by these channels can increase luminal alkalization (Fig. 1). Increase in intraluminal alkalization reduces luminal pHL acidification [22]. This negates a stringent requirement for a functional pHL system within an acidic pH optima ranging between 4.5 and 5.0. Accordingly, if the NAADP-dependent endolysosomal activation is restricted to the TPC2 or to both TPC2 and ML1 even, a common effect would be to elicit lysosomal Ca2+ release. If this adds to Ca2+ -induced release by other second messengers like cyclic nucleotides, this would further increase lysosomal Ca2+ release. Hence, to the luminal side of the acidic organelles, NAADP-induced Ca2+ release do not only deplete luminal Ca2+ but causes intraluminal alkalization as well [23].
Indeed, ML1 and TPC2 are potential sources for regulating pHL (Fig. 1). TPC2 knock-out macrophage showed pHL alkalization [7] and mucolipin 1 V, a genetic disorder caused by mutation of ML1 caused either increase [24] or decrease [25] in pHL. Another strong potential candidate of pHL regulation is P2X4. Besides, acidification of lysosomal lumen is done basically by a vacuolar-type ATPase (V-ATPase). V-ATPase is an electrogenic rotary proton transport motor that uses the energy of ATP hydrolysis to move protons across membranes [26] (Fig. 1). Mass movement of proton across the membrane generates a positive net charge — ‘a positive feedback mechanism’ in the lumen. This mechanism, if left unchecked, can destroy the V-ATPase activity and attenuate the extent of the pHL acidification (Fig. 1). To have a physiological condition, in other words to eliminate the positive feedback process, a counter-ion conductive process must exist together with the V-ATPase to dispel the deleterious positive feedback process that can exhaust cellular energy contents. It is been thought that either the efflux of cations or the influx of anion in opposite direction could salvage the positive feedback process (Fig. 1).
Studies in human and mice osteoclast (Osmt1) revealed chloride as the principal counter-ion that dispels the lysosomal lumen of the deleterious positive feedback electrical potential difference [27]. However, pHL is not abnormal in cells that do not have either Cl-7 or Osmt1 [28, 29]. It became unclear what specific biological roles the Cl-7 channels can perform. Surprisingly, Cl-7 is emerging as a likely Cl−/H+ antiporter in the lysosomal membrane instead [30]. To address this controversy, [29] constructed Cl-7 mutant named “Cl-7 uncouple” with glutamate-to-alanine substitution to switch this ClC from an ion antiporter to a Cl-7 channel. The uncouple breaks up Cl−/H+ antiporter and separates Cl− influx from H+ exchange and allows for the flow of Cl− anions, not supporting the translocation of protons across lysosomal membrane. Their elegant data showed not much differences in lysosome acidification, membrane potential, and steady-state pH between the uncouple Cl-7 mice and the wild-type. Similarly, Grinstein’s group in 2010 made similar documentation and implicated rather lysosomal cations (Na+ and K+) much more to regulate pHL. The source of these cations in the acidic organelle awaits identification.
ML1/TPC2-Dependent Cation Homeostasis and pHL Regulation as an Emerging Aspect of Neurodegenerative Diseases
We discussed how the structure and magnitude of pHL could be diluted or regulated. Since the discovery of lysosome, the role of lysosome in disease processes has been of a great interest and importance. The nature of gradual (persistent) to full disease states (permanent) or clinical syndromes, emanating from lysosomal and cell dysfunctions has remained poorly understood. We hypothesize the role of ML1/TPC2-mediated Ca2+ signaling and pHL regulation in AD (Fig. 2). AD is a neurodegenerative disorder characterized by cognitive decline. Amyloid-beta (Aβ) and Tau proteins are well known proteins of AD pathology. An unpublished data shifts our knowledge of the independent roles of Tau and Aβ in AD pathology towards them being interacting partners in AD formation. Also, in this study, heavy accumulation of amyloid-beta and Tau was observed along the clathrin-endolysosomal degradation pathway (Fig. 3). The clathrin endolysosomal pathway represents a means of internalization of macromolecules for delivery to the lysosomes for degradation and recycling. By this way, defective signaling networks within the lysosomes can potentiate accumulation of internalized macromolecules in the lysosomes (Fig. 3). Accumulated materials particularly Aβ42 and Tau in AD —the product of defective acidic hydrolases may be in part due to alteration in the pHL. Abnormal ML1/TPC2-induced Ca2+ release (Fig. 2) do not only delete luminal Ca2+ but also causes intraluminal alkalization that deranges the pHL. Conversely, accumulation when initiated will beget accumulation through the dysfunctional ML1/TPC2-mediated Ca2+ release. This illustrates the substrate (escalated) accumulation within various parts of autophagosomal-endosomal-lysosomal- system (Fig. 3).

A model for pathological features of ML1/TPC2 -mediated aberrant Ca2+ signaling in AD: each arrow starting from defective acid hydrolases by abnormal pHL forms a step in the pathogenesis of AD representing the mechanisms of AB/Tau-ML1/TPC2-mediated aberrant Ca2+ signaling and their interactive roles. The basis of this model is predicated on ML1/TPC2 as a global road network for modulating ionic meliue in AD. This is since ML1 are non-selective permeable to Ca2+, Mg2+, K+, Na+ [31] and Fe2+[32]. TPC2 are also selective permeable cation channel to Na+ [7, 15]. ML1 is particularly permeable to Fe2+ and the role of Fe2+ in the formation ROS is indispensable. Targeting ML1 selective permeability to Fe2+ pharmacologically could provide means of ameliorating AD

An ultra-structure of intracellular 24 h 10um beta Nu1-Tau-5000x in the clathrin endolysosomal internalization pathway illustrating high accumulation of Aβ (bigger in size) and Tau (smaller in size) likely mediated by abnormal ML1/TPC2-Ca2+ dependent signaling
In this line, membrane-inserted portion of Aβ42 accumulated in lysosomes may destabilize the lysosomal membrane and induce neurotoxicity [33]. A model for pathological features of AD by ML1/TPC2-mediated Ca2+ signaling via deranged pHL and Aβ/Tau accumulation shows how this membrane-inserted (deposited) portion of Aβ42 could cause cellular toxicity and final mental loss in AD (Fig. 2). Membrane-inserted portion of Aβ42 could induce altered membrane trafficking of juxtaorganellar Ca2+ in cells. Defects in membrane trafficking, accumulation of macromolecules and altered Ca2+ homeostasis are common typical features in NP form of LSD [4, 5, 20, 34]. For the AD and as observed in Fig. 3, and illustrated in Fig. 2, Aβ42 [33] and tau accumulate in the endolytic pathway. This sort of accumulations as already tested in NP form of LSD [4, 5, 20] but not in AD, are partly seriously evident in the eclectic hypothesis which states that primary accumulated materials cause a sort of lysosomal stress which can impair common biological and/or specific signaling pathways in the lysosomes [18]. Although in AD, Aβ and Tau proteins are well known to accumulate, sphingomylin lipids are not completely understood to accumulate.
However, lipid rafts are dynamic assemblies of proteins and lipids floating freely within the liquid-disordered bilayer of cellular membranes were cholesterol [35]. Indirect relationships that exist between cholesterol and sphingomyelin metabolism are strongly related to Aβ [36]. There are clues indicating that Aβ level changes in response to blood cholesterol content, and clinical progression of AD is commonly associated with hypercholesterolemia in the brain [37]. Lipids and proteins accumulation can transmute pHL optima outside 4.5–5.0, the functional conventional ranges. Outside the conventional range, the functional integrity of the acid hydrolases, necessary for synthesis and breakdown of biological materials along the clathrin-endolytic pathway (Fig. 3) is disrupted.
The cascade process of AD formation has well been studied. Because this cascade is necessary for understanding the diagnosis, the treatment and the management modes of AD, it deserves further attention. Unveiling the mechanism further is both fascinating and complex. Experimental and theoretical approaches such as in vivo and in vivo electrophysiology, numerical modelling, pharmacological and genetic have been useful. From the experimental and theoretical point of view, we proposed the role of pHL regulation and ML1/TPC2 cation-dependent homeostasis in the accumulation of macromolecules in AD (Figs. 2 and 3). Previously, we stated that accumulated biological materials potentially damages signaling. Accumulated Aβ42 and tau in Fig. 3 can affect the relay of ML1/TPC2-mediated Ca2+ signaling and pHL regulated functions. As shown in the model for pathological features of ML1/TPC2-mediated aberrant Ca2+ signaling (Fig. 2), it is therefore, appreciable that the phenotypes of this would include: depletion of signal transduction [38, 39], oxidative stress, inflammation [40], and transport defect.
With the identification of memory decline process in preclinical setting of HIV infection, where Aβ accumulation was known to cause memory impairment, the merits of this hypothesis are emerging. TRPs and TPCs are bypass for cation fluxes and are strategic for homeostasis of ionic milieu of the acidic organelles they confine to. By evoking Ca2+ efflux from the lysosome with ML1 potent agonist, the structure and magnitude of pHL (Fig. 1) was mechanized for and luminal acidification promoted [41]. This consequently cleared sphingomyelin and the accumulated Aβ from the lysosome. This in part is indicative of the model for pathological features of ML1/TPC2-mediated aberrant Ca2+ signaling in AD (Fig. 2) as a therapeutic target for improving memory decline through sphingomyelin and the Aβ clearance. However, this setting where Aβ predominantly accumulates intracellularly maybe distinct from core AD conditions where extracellular senile plaques recapitulate the feature.
It is underlying in the AD cascade presented in Fig. 2 that acid hydolases do not instantly become defective in the synthesis and breakdown of macromolecules including Aβ42. Abnormal pHL is the ‘stimulus’ for defective acid hydrolases, and regulation of this stimulus by ML1/TPC2 (Fig. 1) has been demonstrated [7, 24, 25]. This stimulus provides an excellent background upon which the famous amyloid hypothesis can be predicated, implying further a role of ML1/TPC2 in the cascade process of AD formation. It therefore beholds an era of lysosomal cation channel functions that will advance our current understanding of the cascade process leading to AD formation.
The Channels in Ca2+ Signaling
We opined the role of ML1/TPC2-mediated cation signaling in substrate accumulations within autophagosomal-endosomal-lysosomal system in AD for the first time. This sort of signaling has been reported in neurodegenerative LSD. We reflected on the seminal works that have reported ML1 and TPC2 mediated Ca2+ signaling in such processes. This is not to ‘reinvent the wheel’ but to signal awareness of lysosomal cation channels mediated homeostasis in AD. First for ML1, the biophysical properties of ML1 are being studied. However, the literature regarding their properties is somewhat conflicting. ML1 was said to be a nonselective outward rectifying monovalent cation channel [42]. In contrast, ML1 is a nonselective inward rectifying cation channel [43] for excellent review, [4, 5, 20] and thus permeable to Na+, K+, Ca2+, Mg2+, [31], Fe2+/Mn2+ [32], and Zn2+ . It opens and closes– gated, and is activated and localized in the lysosomes (Dong et al. [4, 5]). The localization and the biophysical nature of ML1 can be tangled with the distinct roles ML1 can perform. Understanding ML1 gating probability, permeability, and ML1 localization could be necessary for underpinning ML1 distinctive functions. Ca2+ signaling in itself is desperately a most influential requirement for many biological functions like the cell-cell communication, memory formation and/or loss, neuronal excitability and contraction and relaxation of muscles.
ML1 provides bypass for Ca2+ fluxes in cells and becomes a powerful strategic domain for juxta-organellar Ca2+ homeostasis necessary for gene transcription, apoptosis, and membrane trafficking in both normal and disease states including the Alzheimer’s. For instance, ML1 mutation causes mucolipidosis type IV (ML1V). ML1V is associated with enlarged vacuoles, lipid accumulations and altered Ca2+ homeostasis and membrane trafficking. To test that these phenotypes are associated with ML1V and to demonstrate the physiological importance of ML1 activated dependent Ca2+ signaling, [20], constructed a single wavelength lysosome-targeted genetically encoded Ca2+ indicator, GCaMP3-MLI for ML1. This construct was potentiated by a small chemical compound, a synthetic agonist for ML1, generated through high-throughput screening [44]. Using this construct and lysosomal whole-patch clamp [32, 45], that is now reliable for accessing the electrical behavior of lysosomes, Shen and Colleagues showed that Ca2+-dependent trafficking selectively compromised in NP was mediated by abnormal lipid storage that can inhibit ML1 Ca2+-mediated release.
This documentation precisely defined the source of Ca2+ and repudiates a previous report [22], that implicated only lysosomal Ca2+ stores as the source of Ca2+ signaling selectively compromised in NP. The source of the unidentified Ca2+ channels from the lumen of acidic organelles and vesicles [46], that can mediate Ca2+ release is the ML1 [4, 5, 34]. Though considering how difficult it may be to clamp intracellular Ca2+, a perfectly firm condition to rule out the participation of Ca2+ from the lysosomal Ca2+ stores may have not been adequately ensured in the [20] documentation. Perhaps, both sources may be necessary to provide Ca2+ clues required in the cell functions. Identification of the other factors is obligatory on ML1 genes expression and molecular identity of ML1 in membrane trafficking.
Membrane trafficking is the movement of membrane materials between endomembrane compartments. It is necessary for transport of proteins, lipids and other macromolecules in form of cargo sorting to various destinations during endolysosomal biogenesis. Membrane fusion and fission that characterize endolysosomal biogenesis is tightly regulated by both phosphoinositides (PIP2) and Ca2+ signaling [4, 5, 14, 16]. Cells that lack PI(3,5)P2 have enlarged vacuole as those that lack ML1. Using enlarged vacuoles, gotten from cells pretreated with vacuolin-1, [4, 5] demonstrated that ML1 regulates membrane trafficking by converting electrical signals regarding PI(3,5)P2 levels into specific and potent cues that modulate Ca2+ homeostasis. Consistently, a silent plasma membrane ML1 (ML1 exocytosed from the lysosome into the plasma membrane or inside-out membrane patch) was activated by direct decrease in PI(4,5)P2 levels or by direct increase in PI(3,5)P2 levels [14]. This suggests that PIP2 isoforms can either be an activator or an inhibitor of the ML1 mediated Ca2+ release in membrane trafficking.
Second, the ML1 counterpart, TPC2 as a lysosomal cation channel is not left out in the configurational frame work of intracellular Ca2+ dependent homeostasis. Unlike the ML1, TPC2 is a cation-selective channel. Like ML1, TPC2 is associated with acidic organelles in human cells [6, 47, 48]. Despite this information, the exact knowledge concerning the distribution and localization of TPC2 has remained yet unclear. This may be due to availability of inadequate TPCs antibodies that can distinguish subcellular localization of TPC2.
TPC2 is also important in clamping Ca2+ signaling in membrane trafficking, signal transduction, gene transcription and in AD. To elucidate the role of human lysosomal TPC2- mediate Ca2+ release, [6] used flash photolysis of caged-nicotinic acid adenine dinucleotide phosphate (NAADP), intracellular NAADP dialysis, fluorescence Ca2+ indicator— fluo3 and measurement of Ca2+-activated cation currents to demonstrate that lysosomal hTPC2 cell mediates Ca2+ release. Significantly, this activation was extremely potent at < 1 mM concentration of NAADP [49]. While this proposed NAADP dependent TPC2 activation, others [7, 14–16] proposed phosphoinositide isoforms dependent specific TPC2 activation. In the light of this controversy, [21] recently presented the first direct evidence that TPC2 can also be activated by NAADP as well as by PI(3,5)P2. Thus, confirming TPC2 potentiation by NAADP dependent Ca2+ release.
Conclusion
It is our conviction that the context of AD mechanism will be revolutionized by lysosomal cation channels in the near future, and the definition of the exact physiological functions of ML1 and TPC2 in the plethora of cellular functions they could perform is much closer to reality as intense research is ongoing towards underpinning their importance, physiological properties, and molecular identity. At present, their gating probability, activation mechanisms and subcellular localizations are not completely understood. Formation of antibodies with better immunogenicity to detect endogenously ML1/TPC2 channels would greatly help in defining their exact subcellular localizations. Also, identification of more of their genes and their synthetic agonists/antagonist can help towards addressing outstanding controversies and gap in knowledge about their physiological properties. ML1 and TPC2-mediated Ca2+ dependent signaling are necessary for but not limited to: neurodegeneration, membrane trafficking, neurotransmission, cell polarity, morphogenesis, migration apoptosis, gene transcription/regulation, autophagy, cancers including those of the skin, sensation to temperature and taste, ischemia/stroke and aging. The exact gradient of Ca2+ generated by ML1/TPC2 NAADP, PIP2, and GTPases-dependent Ca2+ release to potentiate the functions is poorly understood. This may be in part due to ubiquitous nature of Ca2+ signaling, effect of pHL, temperature and ionic strengths on the amount of intracellular Ca2+.
ML1 and TPC2 as road network for Ca2+ fluxes are also permeable to other cations and could provide a source for pHL regulation that maintains diverse biological functions of acid hydrolases. Abnormal function of these channels as a source of Ca2+-dependent regulatory machineries, abnormal accumulation of macromolecules through defective lysosome, defective hydrolase functions and alkalinized lysosomal lumen partly underpin features of neurodegenerative and LS diseases, ischemia/stroke, and aging.
References
Pillay CS, Elliott E, Dennison C (2002) Endolysosomal proteolysis and its regulation. Biochem J 363:417–429. doi:10.1042/0264-6021:3630417
Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S, Galione A (2002) NAADP mobilizes Ca2+ from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 111:703–708
Michelangeli F, Ogunbayo OA, Wootton LL (2005) A plethora of interacting organellar Ca2+ stores. Curr Opin Cell Biol 17:135–140
Dong XP, Wang X, Xu H (2010) TRP channels of intracellular membranes. J Neurochem 113:313–328
Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H (2010) PI (3,5)P 2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat Commun 1:11
Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, Zhu MX (2009) NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459:596–600
Cang C, Zhou Y, Navarro B, Seo Y, Aranda K, Shi L, Battaglia-Hsu S, Nissim I, Clapham ED, Ren D (2013) mTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell 152:778–790
Berridge MJ, Bootman MD, Roderick HL (2003) Calcium: calcium signalling: dynamics, homeostasis and remodeling. Nat Rev Mol Cell Biol 4:517–529
Zhang Z, Lu Y, Yue J (2013) Two pore channel 2 differentially modulates neural differentiation of mouse embryonic stem. PLos One 8:1–13
Mills E, Dong X, Wang F, Xu H (2010) Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders. Futur Med Chem 2(1):51
Pryor PR, Reimann F, Gribble FM, Luzio JP (2006) Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic 7:1388–1398
Thompson EG, Schaheen L, Dang H, Fares H (2007) Lysosomal trafficking functions of mucolipin-1 in murine macrophages. BMC Cell Biol 8:54
Yu W, Hill WG, Apodaca G, Zeide ML (2011) Expression and distribution of transient receptor potential (TRP) channels in bladder epithelium. Am J Physiol Renal Physiol 300:F49–F59
Zhang X, Li X, Xu, H (2012) Phosphoinositides isoforms determine compartment-specific ion channel activity PNAS 1–6
Wang X, Zhang X, Dong X, Samie M, Li X, Cheng X, Goschka A, Shen D, Zhou Y, Harlow J, Zhu MX, Clapham DE, Ren D, Xu H (2012) TPC proteins are phosphoinositide-activated sodium-selective ion channels in endosomes and lysosomes. Cell 151:372–383
Li X, Garrity AG, Xu H (2013) Regulation of membrane trafficking by signaling on endosomal and lysosomal membranes. J Phys 00:1–13
Chi X, Wang S, Huang Y, Stamnes M, Chen JL (2013) Roles of rho GTPases in intracellular transport and cellular transformation. Int J Mol Sci 14:7089–7108
Parkinson-Lawrence EJ et al (2010) Lysosomal storage disease: revealing lysosomal function and physiology. Physiology (Bethesda) 25:102–115
Vitner EB, Platt FM, Futerman AH (2010) Common and uncommon pathogenic cascades in lysosomal storage diseases. J Biol Chem 2(85):20423–20427
Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong X, Yu T, Lieberman AP, Showalter HD, Xu H (2012) Lipid storage disorders block lysosomal trafficking by inhibiting TRP channel and calcium release. Nat Commun 3:731
Jha A, Ahuja M, Patel S, Brailoiu E, Muallem S (2014) Convergent regulation of the lysosomal two-pore channel-2 by Mg2+, NAADP, PI(3,5)P2 and multiple protein kinases. EMBO J 33:501–511
Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM (2008) Niemann–Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 14:1247–1255
Morgan AJ, Galione A (2007) NAADP induces pH changes in the lumen of acidic Ca2+ stores. Biochem J 402:301–310
Bach G, Chen C, Pagano ER (1999) Elevated lysosomal pH in Mucolipidosis type IV cells. Clin Chim Acta 280:173–179
Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, Kiselyov K (2006) TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem 281:7294–7301
Forgac M (2007) Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8:917–929. doi:10.1038/nrm2272
Kornak U, Kasper D, Bo MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling W, Jentsch JT (2001) Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 104:205–215
Lange PF, Wartosch L, Jentsch TJ, Fuhrman JC (2006) ClC-7 requires Ostm1 as a b-subunit to support bone resorption and lysosomal function. Nature 440:220–223
Weinert S, Jabs S, Supanchart C, Schweizer M, Gimber N, Richter M, Rademann J, Stauber T, Kornak U, Jentsch TJ (2010) Lysosomal pathology and osteopetrosis upon loss of H + −Driven lysosomal Cl− accumulation. Science 328:1401
Graves AR, Curran PK, Smith CL, Mindell JA (2008) The Cl−/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature 453:788–792. doi:10.1038/nature06907
Xu H, Delling M, Li L, Dong X, Clapham DE (2007) Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. PNAS 104:18321–18326
Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, Xu H (2008) The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455:992–996
Liu R, Zhou Q, Ji S, Zhou Q, Feng D, Wu DY, Sui F (2010) Membrane localization of _-amyloid 1–42 in lysosomes a possible mechanism for lysosome labilization. J Biol Chem 285:19986–19996
Shen D, Wang X, Xu H (2011) Pairing phosphoinositides with calcium ions in endolysosomal dynamics. Bioessays 33:448–457
Simons K, Ehehalt R (2002) Cholesterol, lipid rafts, and disease. J Clin Investig 110:597–603
Grimm MO, Grimm HS, Patzold AJ et al (2005) Regulation of cholesterol and sphingomyelin metabolism by Amyloid-beta and presenilin. Nat Cell Biol 7:1118–1123
Abad-Rodriguez J, Ledesma MD, Craessaerts K et al (2004) “Neuronal membrane cholesterol loss enhances Amyloid peptide generation”. J Cell Biol 167:953–960
Wang H, Lee DHS, D’Andrea MR, Peterson PA, Shank RP, And Reitz AB (2000) β-Amyloid1–42 binds to α7 nicotinic acetylcholine receptor with high affinity. J Biol Chem 275:5626–5632
Hongpaisan J, Sun M, Daniel L (2011) Alkon1PKC _ activation prevents synaptic loss, a elevation, and cognitive deficits in Alzheimer’s Disease transgenic mice. J Neurosci 31:630–643
Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM (2000) Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s Disease. J Neurosci 20:5709–5714
Bae M, Patel N, Xu H, Lee M, Tominaga-Yamanaka K, Nath A, Geiger J, Gorospe M, Mattson MP, Haughey NJ (2014) Activation of TRPML1 clears intraneuronal Aβ in preclinical models of HIV infection. J Neurosci 34(34):11485–11503 11485
Kiselyov K, Chen J, Rbaibi Y, Oberdick D, Tjon-Kon-Sang S, Shcheynikov N, Muallem S, Soyombo A (2005) TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem 280:43218–43223
Clapham ED (2009) Transient receptor potential channels. Encycl Neurosci 9:1109–1133
Grimm C et al (2010) Small molecule activators of TRPML3. Chem Biol 17:135–148
Dong XP et al (2009) Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J Biol Chem 284:32040–32052
Hay JC (2007) Calcium: a fundamental regulator of intracellular membrane fusion? EMBO Rep 8:236–240
Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, Hooper R, Boulware MJ, Dun NJ, Marchant JS, Patel S (2009) Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol 186:201–209
Zong X, Schieder M, Cuny H, Fenske S, Gruner C, Rotzer K, Griesbeck O, Harz H, Biel M, Wahl-Schott C (2009) The two-pore channel TPCN2 mediates NAADP-dependent Ca2 + −release from lysosomal stores. Pflugers Arch 458:891–899
Zhu MX, Ma J, Parrington J, Galione A, Evans AM (2010) TPCs: endolysosomal channels for Ca2+ mobilization from acidic organelles triggered by NAADP. FEBS Lett 584:1966–1974
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ezeani, M., Omabe, M. A New Perspective of Lysosomal Cation Channel-Dependent Homeostasis in Alzheimer’s Disease. Mol Neurobiol 53, 1672–1678 (2016). https://doi.org/10.1007/s12035-015-9108-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9108-3