
Abstract
Although the transcription factor serum response factor (SRF) has been suggested to play a role in activity-dependent gene expression and mediate plasticity-associated structural changes in the hippocampus, no unequivocal evidence has been provided for its role in brain pathology, such as epilepsy. A genome-wide program of activity-induced genes that are regulated by SRF also remains unknown. In the present study, we show that the inducible and conditional deletion of SRF in the adult mouse hippocampus increases the epileptic phenotype in the kainic acid model of epilepsy, reflected by more severe and frequent seizures. Moreover, we observe a robust decrease in activity-induced gene transcription in SRF knockout mice. We characterize the genetic program controlled by SRF in neurons and using functional annotation, we find that SRF target genes are associated with synaptic plasticity and epilepsy. Several of these SRF targets function as regulators of inhibitory or excitatory balance and the structural plasticity of neurons. Interestingly, mutations in those SRF targets have found to be associated with such human neuropsychiatric disorders, as autism and intellectual disability. We also identify novel direct SRF targets in hippocampus: Npas4, Gadd45g, and Zfp36. Altogether, our data indicate that proteins that are highly upregulated by neuronal stimulation, identified in the present study as SRF targets, may function as endogenous protectors against overactivation. Thus, the lack of these effector proteins in SRF knockout animals may lead to uncontrolled excitation and eventually epilepsy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epilepsy is a chronic neurological disorder that affects approximately 1 % of the human population [1]. The development of epilepsy (epileptogenesis) involves progressive alterations in synaptic connections (aberrant plasticity), the molecular mechanisms of which are still poorly understood. The regulation of gene transcription by neuronal activity is an integral part of adaptive plasticity [2]. Therefore, the identification of gene expression programs that result from and control long-term synaptic changes is crucial for understanding the molecular mechanisms that underlie epilepsy [3].
Serum response factor (SRF) is a MADS-box protein that binds DNA at the CC(A/T)6GG consensus sequence, known as a CArG box or serum response element (SRE). SREs are found in promoters of actin cytoskeleton genes and immediate-early genes (IEGs; [4–6]). SRF-dependent transcription is activated by neurotrophins or in response to calcium influx in neurons [7–11]. The inactivation of SRF leads to a decrease in the expression of such plasticity-linked genes as Fos, Arc, and Egr1 and produces a deficiency in hippocampal synaptic plasticity and learning [12, 13]. The activity of SRF is also important during the development of the nervous system [14–16] and the regulation of structural plasticity [17, 18]. The molecules that are involved in physiological plasticity, such as SRF, may also be engaged in pathological or aberrant plasticity processes.
Although SRF has recently been suggested to play a role in plasticity and mediate structural changes in the hippocampus, no solid evidence has been provided for its role in brain pathology. Interestingly, the increased binding of SRF to DNA and upregulation of SRF protein levels were found in the hippocampus after pilocarpine-induced status epilepticus, and SRF accumulation and phosphorylation were observed after kainic acid-induced status epilepticus [19, 20]. These findings indicate that the transcriptional activity of SRF is enhanced during epileptogenesis but provide no explanation for its precise role in this brain pathology. Furthermore, many SRF-dependent genes that are important for synaptic plasticity likely still remain unidentified, and no global analysis of SRF-dependent gene expression in response to neuronal stimulation in the adult brain has yet been reported.
In the present study, we investigated the role of the SRF-dependent transcriptional program in epileptogenesis using brain-specific, inducible SRF gene knockout (KO) in mice. We found that SRF KO mice exhibited an increase in the susceptibility to spontaneous seizure development and more severe seizures. We also identified 378 activity-dependent SRF target genes, among which we distinguished a group with functions associated with epilepsy and synaptic plasticity that may be responsible for the observed phenotype. Furthermore, we identified several novel genes that are directly regulated by SRF in the hippocampus in vivo: Npas4, Gadd45g, and Zfp36.
Materials and Methods
Animals
Mice with the conditional deletion of Srf in forebrain neurons (Srf CaMKCreERT2; mutant mice; KO) were generated by crossing the Srf f/f mutant strain (stock nr: 006658; Jackson’s Laboratories, USA; [12]), carrying the loxP flanked Srf, with mice expressing Cre recombinase under the control of neuron-specific CaMKIIα promoter which is activated only in excitatory forebrain neurons (strain nr: EM:02125; The European Mouse Mutant Archive; [21]). To obtain time-specific control of the genetic process, Cre recombinase was originally fused with mutated ligand-binding domain of human estrogen receptor to obtain CreERT2 enzyme which is translocated to the nucleus and active only in response to tamoxifen (TAM) treatment. CreERT2-negative littermates were used as a control (Srf f/f; control mice; CTR). Adult mice, both genotypes, CTR and KO (2–6 months) were injected intraperitoneally with 1 mg of TAM twice daily for 5–10 days (Sigma). Experiments were performed at least 4 weeks after tamoxifen injections. For the ChIP experiments adult C57BL/6 mice were used. The animals were treated in accordance with the ethical standards of European and Polish regulations. All experimental procedures were approved by the Local Ethics Committee. The mice were bred in the Animal House of Nencki Institute of Experimental Biology, Warsaw. Animals were housed in individual cages under 12-h light/dark cycle with constant temperature (23–25 °C) and with food and water available ad libitum.
Immunohistochemistry
Mice were transcardially perfused with 4 % paraformaldehyde in PBS. Brains were further fixed in the same buffer overnight at 4 °C and cut on vibratome on 40-μm slices. Free floating brain slices were permeabilized, blocked in normal goat serum for 1 h at room temperature, washed, and incubated with anti-SRF antibody (1:500) (H-300; Santa Cruz Biotechnology) overnight at 4 °C. Next, immunochistochemistry was performed using standard procedure with Avidin/Biotin Complex (ABC) Kit Vectastain PK-6100 (Vector Labs) and visualized using SIGMAFAST™ DAB (Sigma). For Nissl staining, brain sections were air dried on slides. Next, the slides were stained with 0.1 % cresyl violet solution (containing 3 % acetic acid) for 5 min, washed, dehydrated, cleared in xylene, and coverslipped.
Western Blotting
Dentate gyrus of the hippocampus was dissected and homogenized in a lysis buffer containing 1 mM MgCl2, 5 mM HEPES (pH = 7.4), 320 mM sucrose, 1 mM Na2F, and protease inhibitor cocktail complete (Roche) using Dounce glass homogenizer. Protein concentration was measured using BCA Protein Assay Kit (Pierce). Fifteen microgram of homogenates was dissolved on 8 % polyacrylamide gels. Western blot was performed using a standard procedure using anti-SRF antibody (1:300) (H-300; Santa Cruz Biotechnology). Blots were reprobed with an anti-α-tubulin antibody (1:5000) (Sigma) to ensure equal total protein levels. Chemiluminescent detection method was used. For the quantification of individual bands, the scan of the photographic film was analyzed by densitometry using GeneTools software (Syngene).
Kainic Acid Treatments
CTR and KO animals were injected with 70 nl of 20 mM solution of kainic acid (KA, Nanocs) in 0.9 % NaCl or saline at the left CA1 area of the dorsal hippocampus. For intrahippocampal injection of kainic acid only males were used. For intraperitoneal injections mice were habituated to handling and injected with saline for 3 days before the experiments. Then, CTR and SRF KO were injected intraperitoneally with 35–50 mg/kg kainic acid or saline. For the microarrays profiling experiments only male mice were used. Confirmation of microarray results by qRT-PCR was performed on males and females. Six hours after kainic acid-induced status epilepticus (SE) or 6 h after saline injection, mice were sacrificed and brain hemispheres were dissected and incubated overnight in RNAlater solution in 4 °C (Ambion). Next, the dentate gyrus of the hippocampus was dissected and frozen in −80 °C for further RNA isolation. For ChIP assay only males were used. Animals 2 h after kainic acid-induced status epilepticus were sacrificed and the hippocampus dissected and used for chromatin immunoprecipitation.
Surgery and Video-Electroencephalography (EEG)
Surgical Procedures
Animals (adult males) were anesthetized by inhalation of 1.5–2 % isoflurane (Baxter) in oxygen and placed in a stereotaxic frame and on a heating pad in order to maintain a constant body temperature. Mice were injected with kainic acid (with rapidity of 50 nl/min) or saline (control animals) aimed at the left CA1 area of the dorsal hippocampus with following coordinates: AP −1.8 mm, ML +1.7 mm, and DV −2.3 mm from bregma.
Electrode Preparation, Implantation, Electroencephalography, and Video Recordings
Cortical electrodes were home-made: a screw was joined to socket contact by copper wire and welded (Bilaney Consultants Ltd.). Two electrodes were placed bilaterally into the scull over the frontal cortex and next two over the cerebellum (first as reference and second as ground electrode). Bipolar hippocampal electrode (Bilaney Consultants Ltd.) was implanted into the injected hippocampus using the coordinates AP −2.0 mm and ML +1.3 mm with bregma as reference point and DV −1.7 mm below dura. Electrodes were placed into pedestal (Bilaney Consultants Ltd.) and secured with dental acrylate (Duracryl Plus, Spofa Dental, Czech Republic). After operation, mice were connected to a digital acquisition system (TWin Clinical Software for EEG, Grass Technologies) and EEG activity of freely behaving animals was monitored for 18 days in an isolated room. The unit consisted of six cages.
Analysis of Video-EEG Monitoring
The occurrence of spontaneous seizures was studied by browsing through EEG record on a computer screen. All motion artifacts were excluded from analysis. An encephalographic seizure was specified as a high-amplitude (more than 2× baseline) discharge lasting more than 5 s. Several seizure parameters were characterized: behavioral severity (average score), seizure frequency, and average seizure duration. Behavioral severity was analyzed from matching video-EEG recording. Seizures were estimated based on modified Racine’s scale [22]: score 1—immobility, facial clonus; score 2—“wet-dog” shakes, head nodding; score 3—piano pose (bilateral clonus of forepaws); score 4—clonic seizures with falling; and score 5—generalized tonic-clonic seizures. Seizure frequency was specified as a total number of detected seizures divided by recording days, and duration was determined on the basis of EEG recordings. SE severity was analyzed during first 24 h after intrahippocampal injection of KA. Spontaneous seizures were defined as seizures appearing as early as 24 h after KA administration.
Gene Expression Profiling Using Microarrays
Total RNA was isolated from the dentate gyrus using RNeasy Mini Kit (Qiagen) as described by the manufacturer. Residual DNA was removed by digestion with DNase I (Qiagen). RNA concentration was calculated from the absorbance at 260 nm. Purity and integrity of RNA was determined using Agilent RNA 6000 Nano Chips on the Agilent 2100 Bioanalyzer System (Agilent Technologies). A starting amount of 200-ng high-quality total RNA was used to generate complementary DNA (cDNA) and complementary RNA (cRNA) with the Illumina TotalPrep RNA Amplification Kit (Illumina Inc., San Diego, CA, USA). The obtained cDNA served as a template for in vitro transcription with T7 RNA polymerase and biotin UTP to generate multiple copies of biotinylated cRNA. Each cRNA sample (1.5 μg) was hybridized overnight to MouseWG-6 BeadChip array (Illumina); subsequently, chips were washed, dried, and scanned with the BeadArray Reader (Illumina). A total of 22 microarrays were used (5–6 biological replicates per group). Analysis and quality control of microarrays were performed using BeadArray R package. After background subtraction, data was normalized using quantile normalization and then log2-transformed. The obtained signal was taken as the measure of mRNA abundance derived from the level of gene expression. Statistical analysis of the microarray results was performed using a two-way ANOVA (for the factors genotype and treatment) followed by the estimation of FDR (percent FDR; false discovery rate) using the Benjamini and Hochberg method. Microarray data are available in the NCBI Gene Expression Omnibus (GEO) under accession number GEO: GSE60772.
Gene ontology enrichment analysis was performed using DAVID database. Pathway and global functional analysis was performed using Ingenuity Pathway Analysis (IPA; Ingenuity® Systems, Qiagen). Additionally, manual analysis of genes’ functions and their role in neurons was assigned on the basis of published data (manual search of PubMed).
In Silico Analysis of Putative SRF-Binding Sites
Overrepresentation analysis of transcription factor binding sites (TFBS) in the group of identified genes was performed using the cREMaG database with default parameters [23].
Independently, using NGD database [24] we identified genes with at least two putative SRF-binding sites in the conserved noncoding sequences between mouse and human, identified with programs AVID-VISTA [25, 26] within the −10 kb/+10 kb from Transcription Start Site. Potential SRF-binding sites were predicted using all the motifs from the $SRFF family (Genomatix MatBase v. 8.4). Moreover, for the selected genes (Table 3) we compared previously identified potential CArG boxes in the human genome [27] to the mouse sequences. Among the putative SRF-binding sites identified using the three above approaches (cREMaG, NGD, CArG), for further analysis, we chose only the sites conforming exactly to the consensus sequence motif: CC(A/T)6GG or having a maximum two base pair mismatch (only one mismatch in the CC or GG) to the consensus sequence.
RNA Preparation and Quantitative Real-Time PCR
Total RNA was isolated from the dentate gyrus of the hippocampus using RNeasy Mini Kit (Qiagen) as described by the manufacturer. Residual DNA was removed by digestion with DNase I (Qiagen). RNA concentration was calculated from the absorbance at 260 nm and the purity of RNA was determined by the 260/280 nm absorbance ratio. RNA was reverse transcribed with the SuperScript III Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions. Quantitative real-time PCR reactions were performed using Fast SYBR Green Master Mix (Applied Biosystems) in the Applied Biosystems 7900HT Fast Real-Time PCR System using the following cycling conditions: 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Primer sequences are presented in Table 1. Fold changes in expression were determined using the ∆∆CT relative quantification method. Values were normalized to the relative amounts of ribosomal protein large P0 (Arbp) mRNA, which was found to be stable across different phases of KA-induced epilepsy in mice [28].
Chromatin Immunoprecipitation
C57BL/6 mice were injected with KA (35–50 mg/kg), and 2 h after KA-induced status epilepticus or saline injection, hippocampi were dissected, chopped (0.5-mm fragments), and fixed with 1.5 % formaldehyde for 30 min at room temperature, then glycine was added and incubated for 10 min to quench formaldehyde (hippocampi from each mouse were processed separately). Samples were put on ice, washed with ice-cold phosphate-buffered saline (PBS) supplemented with protease inhibitor cocktail (Roche) and 1 mM PMSF and briefly homogenized with an Eppendorf fitting pestle. Samples were centrifuged again, washed with PBS with protease inhibitors, and centrifuged (1.3 g/5 min/4 °C), and the pellet was resuspended in 2 ml of lysis buffer (5 mM PIPES, pH 8.0, 85 mM KCl, 0.5 % Nonidet P-40, with protease inhibitors), incubated for 10 min on ice, and disrupted using a glass–glass homogenizer. Crude nuclear extract was collected by centrifugation at 1.3 g for 10 min at 4 °C and frozen in −80 °C. Nuclear extracts were pooled and resuspended (3 hippocampi in 200 μl) in high-salt lysis buffer with protease inhibitors (1× PBS, 1 % Nonidet P-40, 0.5 % sodium deoxycholate; 1 % SDS). Samples were sonicated on ice 14 times at 25 % output for 20 s with 30 s breaks (on ice) using a Branson Sonifier 250 obtaining an average of 300–500 bp DNA fragments. Samples were diluted 10× with high-salt lysis buffer without SDS (final concentration of 0.1 % SDS), centrifuged for 15 min to remove cell debris, and precleared with sonicated salmon sperm DNA/protein A agarose (50 % slurry) at 4 °C for 30 min with rotation. The DNA content was measured spectrophotometrically, and equal amounts of DNA were immunoprecipitated overnight at 4 °C using 5-μg anti-SRF antibody (H-300, Santa Cruz) or 5 μg of normal rabbit IgG as negative control. Next day, salmon sperm DNA protein A-agarose beads (Milipore) were added for 2 h, then the beads were washed with low-salt wash buffer (0.1 % SDS, 1 % Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 150 mM NaCl), high-salt wash buffer (0.1 % SDS, 1 % Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 500 mM NaCl), LiCl wash buffer (0.25 M LiCl, 1 % Nonidet P-40, 1 % deoxycholate, 1 mM EDTA, 10 mM Tris–HCl, pH 8.1), and twice in TE. Antibody-DNA complexes were eluted with elution buffer (1 % SDS, 0.1 M NaHCO3), crosslinking was reversed, and proteins were removed by adding 5 M NaCl and proteinase K and incubation for 2 h at 42 °C, followed by incubation at 65 °C overnight, next DNA was purified using QIAquick Gel Extraction Kit (Qiagen). Immunoprecipitated chromatin was used as a template for real-time PCR reactions using Fast SYBR Green Master Mix (Applied Biosystems) in the Applied Biosystems 7900HT Fast Real-Time PCR System with specific primers located near the predicted SRF binding sites (Table 2). Obtained data was analyzed using the ∆∆CT relative quantification method and calculated as the percentage of the input.
Statistical Analysis
Data on the graphs are expressed as means ± standard errors of the means (SEM) from at least three independent experiments. The statistical analysis of the data was performed with GraphPad Prism software (GraphPad Software, Inc.). For comparison of two groups either unpaired t test or Mann–Whitney test (nonparametric) was used. For comparison of multiple groups, a two-way analysis of variance (ANOVA) with post hoc Bonferroni’s multiple comparisons test was used.
Results
Characterization of Srf f/f;CaMKCreERT2 Mice
To investigate the role of SRF in the development of temporal lobe epilepsy (TLE), Srf gene KO mice were used. Because of embryonic lethality in Srf-null mice, we used conditional mutants of Srf that were obtained by crossing mice in which the Srf gene was flanked by loxP sites (Srf f/f) with an CaMKCreERT2 line to ablate its expression exclusively in excitatory forebrain neurons [12, 21]. Additionally, to enable the time-specific induction of SRF deletion, adult mice (≥8 weeks old) were injected with tamoxifen, which stimulates the translocation of recombinant Cre recombinase to the nucleus. Eight weeks after mutation activation, homozygous adult Srf f/f mice (CTR) and Srf gene KO mice that carried a single copy of Cre recombinase Srf f/f;CaMKCreERT2 (KO) were used for the experiments.
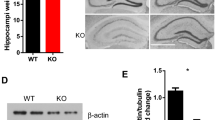
Immunohistochemical analysis of the brain slices obtained from KO animals (KO = Srf f/f;CaMKCreERT2) and control littermates (CTR = Srf f/f) revealed a specific decrease in SRF protein levels in the dentate gyrus (DG) and CA1 subfield of the hippocampus (Fig. 1a). Similar results that showed a large decrease in SRF protein levels in the DG in KO mice were obtained using Western blot (Fig. 1c). The remaining SRF staining may have resulted from its expression in astrocytes [29].

Conditional deletion of SRF in the adult hippocampus. a Immunohistochemical staining of SRF in the hippocampus in control (CTR; Srff/f) and knockout (KO; Srff/f;CaMKCreERT2) animals. Serum response factor elimination was observed in the dentate gyrus (DG) and CA1 subfield of the hippocampus. b Nissl staining of hippocampal sections from CTR and KO mice. No neuroanatomical differences were observed in the hippocampus in KO mice. Scale bar = a', b' 200 μm, c'–f' 400 μm. c Loss of two SRF isoforms (67 and 62 kDa) in protein extracts from DG in KO mice compared with CTR (Western blot and quantification of Western blot results), each line represents a single, independent animals, **p < 0.01 (Mann–Whitney test)
We found no evident neuroanatomical differences between CTR and KO animals’ brains. Nissl staining of hippocampal sections did not show any structural defects in KO animals (Fig. 1b). Similarly, the staining of nerve fibers with a zinc transporter member 3 (Znt3)-specific antibody revealed no significant differences in mossy fiber projections in CTR and KO adult brains (data not shown). Thus, the selective deletion of SRF in adult forebrain neurons did not cause general morphological abnormalities as a consequence of SRF deletion during the early stages of development [15, 14].
SRF Deficiency Enhances Kainic Acid-Induced Epileptogenesis
To test whether SRF deficiency in the adult brain is sufficient to cause spontaneous aberrant neuronal activity, electroencephalograms (EEGs) were recorded from sham-operated, saline-injected animals (n = 6 per group, males). EEG activity was monitored daily for 18 days, 24 h/day, beginning 7 days after electrode implementation. EEGs were recorded from hippocampal and cortical electrodes. Sham-operated CTR and KO animals did not develop spontaneous seizures under basal conditions (data not shown). However, in all of the experiments performed in CTR and KO mice, three of 40 female KO mice developed seizures after i.p. saline injection, and this phenomenon was not observed in KO males and in CTR mice.
To study the phenotype of KO animals in the context of epileptogenesis, we used the kainic acid model of TLE. Because SRF KO animals were created on a C57BL/6 genetic background and mice with this genetic background do not show neurodegeneration upon systematic administration of kainic acid (data not shown and [30]), epilepsy was induced by an intrahippocampal injection of kainic acid [30]. To detect seizures, animals (n = 6 per group) were monitored by EEG video continuously for 18 days, 24 h/day, starting just after the intrahippocampal kainic acid administration. During the 18 days of recordings, all of the CTR and KO mice developed seizures.
During the first 24 h, we did not observe any differences in latency, number of seizures, seizure frequency, and seizures score (Supplementary Fig. S1). Also, the latency to the first spontaneous seizure did not significantly differ between KO and CTR animals (55.50 ± 12.57 h vs. 49.17 ± 12.95 h; Student’s t test, p > 0.05; Fig. 2c). Importantly, however, the seizure frequency was higher in KO animals than in CTR animals (3.13 ± 0.80 seizures/day vs. 0.81 ± 0.19 seizures/day, respectively; Mann–Whitney test, p < 0.05; Fig. 2d). The behavioral analysis of spontaneous seizures revealed that KO animals developed more severe seizures than CTR animals (Mann–Whitney test, p < 0.01; Fig. 2e). However, average spontaneous seizure duration did not significantly differ between KO and CTR animals (35.7 ± 2.8 vs. 30.0 ± 2.6 s, respectively; Student’s t test, p > 0.05; Fig. 2f). Similar epileptic phenotypes were observed for another cohort of animals (n = 12 per group) that were monitored by EEG video continuously for 7 days, 24 h/day, 10 days after kainic acid administration (data not shown). Altogether, our findings indicate that the lack of SRF-enhanced epileptogenesis.

Lack of SRF increases number and duration of spontaneous seizures in the kainic acid model of TLE. a Schematic representation of the experimental design. b Representative EEG recordings of a spontaneous seizure in a CTR mouse and KO mouse, with a hippocampal (h) electrode and cortical (c) electrode (CTR, 5 days after intrahippocampal kainic acid injection, 33 s duration, behavioral severity on Racine scale = 3; KO, 6 days after intrahippocampal kainic acid injection, 36 s duration, behavioral severity on Racine scale = 5). The beginning and end of the seizures are indicated by arrows. c CTR and KO animals did not differ in the latency to the first spontaneous seizure. d Knockout animals developed a greater average number of seizures per day than CTR animals (Mann–Whitney test, p < 0.05). e Knockout animals showed more severe behavioral seizures measured according to Racine’s scale (Mann–Whitney test, p < 0.01). f The mean duration of spontaneous seizures was not significantly different in the KO group compared with the CTR group (Student’s t test, p > 0.05). All EEG recordings were conducted in males
Loss of SRF Leads to Specific Deficits in Activity-Induced Gene Expression
To investigate the molecular mechanisms that underlie the consequences of SRF deletion, we sought to identify SRF-dependent activity-induced genes. Global gene expression induced by intraperitoneal injection of kainic acid was analyzed by microarrays. We did not observe significant differences in seizures severity and latency to seizures (score 4 or 5) in CTR vs. KO animals (Supplementary Fig. S2). Six hours after seizure induction (i.e., seizure severity = 5, based on a modified Racine scale) or saline injection, CTR and KO animals were sacrificed, and the DG was microdissected and used for RNA isolation. To identify SRF-regulated genes, we used Illumina MouseWG-6 v. 2.0 Expression BeadChips that include 45,281 transcripts. We focused our microarray analysis on the DG because (i) it is not vulnerable to kainic acid-induced excitotoxicity, (ii) it undergoes aberrant plasticity, and (iii) DG circuitry controls seizure propagation into the hippocampus [31, 32]. Kainic acid induces recurrent seizures, and the selected time point (i.e., 6 h) after the induction of status epilepticus enabled us to observe not only late-response genes but also early-response genes (see Fig. 3; e.g., Fos, JunB, FosB, Egr2, Egr4).

Microarray analysis shows that SRF is an important regulator of gene transcription in response to kainic acid-induced status epilepticus. The figure shows the results of gene expression profiling performed on RNA isolated from the dentate gyrus of the hippocampus in control (CTR) and Srf conditional knockout (KO) mice 6 h after kainic acid-induced status epilepticus (KA; intraperitoneal injection) or saline injection (saline). Twenty-two animals (males) were analyzed (5/6 animals per each genotype/treatment combination). a Scatter plot showing fold change values for KA responding genes in CTR and SRF mutants. b Pool of 3241 probes which expression was significantly changed at least 1.5 fold after KA (fold change > 1.5 or < 0.66; Tukey’s test, p < 0.05). Red dots indicate population of genes (729 probes) differentially regulated in SRF KO as compared to CTR (fold change CTR KA vs. KO KA >1.5 or <0.66 and Tukey’s test, p < 0.05). The red lines indicate microarray probes changed by kainic acid at least 1.5 fold. c, d In the heatmaps, each column corresponds to one animal with the indicated genotype/treatment combination. Rows represent transcripts as indicated on the right. Colors indicate normalized expression values as shown by the scale at the bottom (log2 change). The results for the 77 transcripts with the highest changes after kainic acid-induced status epilepticus in KO vs. CTR are shown (fold change in response to kainic acid >2.5 or <0.4, genotype × treatment interaction, FDR < 1 %; fold change in KO vs. CTR after kainic acid >1.96 or <0.51). Transcripts are ordered by fold change in CTR animals treated with kainic acid vs. saline. The values correspond to the indicated fold change (linear scale). The transcripts that were chosen for further analysis are shown in bold. c Selected genes upregulated in CTR animals in response to kainate but not changed or upregulated to a lesser extent in KO animals after kainic acid. d Genes downregulated in response to kainic acid in CTR animals but not changed or inhibited to a lesser extent in KO animals
The statistical analysis of the microarray results showed no significantly downregulated transcripts under basal conditions (i.e., in saline-treated animals) in the DG in SRF KO mice, with the exception of Srf itself (Tukey’s test, p < 0.05; fold change < 0.66). Moreover, two transcripts (Arsi and Sstr1) were significantly upregulated in KO mice compared with CTR mice (Tukey’s test, p < 0.05; fold change > 1.5). This result was consistent with our EEG recording data, showing no difference in neuronal activity in CTR vs. KO animals under basal conditions (data not shown).
In contrast, comparisons of the gene expression profiles of the two genotypes (KO and CTR) in response to kainic acid-induced status epilepticus showed robust differences (Fig. 3a). Importantly, however, the loss of SRF in adult neurons did not cause general impairment in activity-dependent gene expression in the DG. Upon seizure induction, 3241 transcripts were significantly up- or downregulated in CTR animals (fold change > 1.5 or < 0.66; Tukey’s test, p < 0.05) (Fig. 3b). Among this group, only 729 probes were differentially regulated by KA in KO as compared to CTR (fold change > 1.5 or < 0.66; Tukey’s test, p < 0.05). The remaining 2512 probes (3241 − 729 = 2512; 77.5 %) did not meet the criteria (fold change CTR KA vs. KO KA: >1.5 or <0.66 and Tukey’s test, p < 0.05). Thus, SRF deficiency in adult neurons altered only a subset of genes, suggesting the specificity of the regulation. To further restrict our analysis and obtain a list of the most significantly changed transcripts, we performed a two-way analysis of variance (ANOVA) with false discover rate (FDR) correction at the 1 % level. Using this approach, we identified 431 probes (378 genes) with a significantly altered expression profile as a result of SRF knockdown after kainic acid (ANOVA, genotype × treatment interaction, p < 0.0005; correction FDR < 1 %; multiplicity of changes in CTR animals after the administration of kainic acid >1.5 or <0.66, Table S1).
Among those 378 genes, two main groups of genes were clearly distinguishable: (i) genes upregulated in CTR animals in response to kainic acid (216 genes) and not changed or upregulated to a lesser extent in KO animals after kainic acid and (ii) genes downregulated in response to kainic acid in CTR animals (162 genes) and not changed or inhibited to a lesser extent in KO animals after kainic acid. Genes with the most prominent differences in the expression profile between the two genotypes after kainic acid are shown in the heatmaps (Fig. 3a: 62 transcripts upregulated after kainic acid in CTR animals >2.5, fold change in KO vs. CTR after kainic acid < 0.51; Fig. 3b: 15 transcripts downregulated after kainic acid in CTR animals <0.4; fold change in KO vs. CTR after kainic acid >1.96). The probes are ordered by fold induction in CTR animals after kainic acid-induced status epilepticus. The genes that were chosen for further analysis (as described in the next paragraph; i.e., functions associated with the regulation of neuronal excitability and structural plasticity) are shown in bold. The values correspond to the indicated fold change (linear scale). To identify the overrepresentation of transcription factor binding sites (TFBSs) in the group of identified genes, we performed an in silico analysis using the cis-regulatory elements in the mammalian genome (cREMaG) database [23]. We found a significant overrepresentation of SRF binding sites in the group of 216 genes that were upregulated after kainic acid, suggesting the contribution of genes that are directly regulated by SRF (3.7-fold higher than expected by chance, p = 0.0194). Additionally, a significant overrepresentation of MEF2A, another MADS-box family transcription factor, was also found (3.7-fold higher than expected by chance, p = 0.0184).
Functional Classification of SRF-Dependent Genes
To functionally classify the identified SRF-dependent transcripts, lists of downregulated and upregulated genes were analyzed by Gene Ontology (GO, DAVID). In the group of genes with decreased abundance in KO animals, the overrepresentation of transcripts that are involved in Behavior (4.1-fold enrichment, p = 3.48E−6; e.g., Egr1, Egr2, Bdnf, Ntrk2, Cyr61, Nr4a2, and Nr4a3) and MAPK signaling pathway (6.3-fold enrichment, p = 9.71E−11; e.g., Ntrk2, Bdnf, Rps6ka3, Gadd45g, Map2k3, Mapkapk3, and Map3k5) was observed.
To further extend the functional analysis, Ingenuity Pathway Analysis (IPA; Ingenuity® Systems, Qiagen) was applied. The Diseases and Bio-Functions analysis revealed Neurological Disease as a top-associated category (68 molecules; p = 4.55E−25 to 3.58E−04). The results of the analysis showed a significant correlation with annotations: Epilepsy (35 genes; p = 8.51E−19; Neurological Disease category; e.g., Bdnf, Cacna1h, Fos, Gadd45g, Zfp36, Cyr61, Egr1, Egr2, and Egr4), Plasticity of Synapse (9 genes; p = 0.000103; nervous system development and function category; e.g., Bdnf, Ntrk2, Pcdh8, and Vgf), and Outgrowth of Neurites (21 genes; p = 0.0000411; Nervous System Development and Function category; e.g., Npas4, Bdnf, Ntrk2, and Gpr3). In addition to GO and IPA, a manual analysis of gene function was performed, and their role in neurons was assigned based on published data (manual search in PubMed). Several functional groups were found, including (i) transcription factors and other regulatory proteins (e.g., Fos, Npas4, Fosb, Junb, Egr1, Egr2, Egr4, Atf3, Sp7, and Cited2) and (ii) genes with functions associated with the regulation of neuronal excitability and structural plasticity of dendritic spines (e.g., Fos, Npas4, Bdnf, Syt4, Gadd45g, Acan, Lcn2, Pcdh8, Elmo1, Magoh, and Zfp36; Table 3).
The functional analysis of the group of SRF targets was consistent with the reported roles of SRF in the regulation of neurite outgrowth, synaptic plasticity, and behavior [15, 8, 12, 13, 48]. Thus, the identified SRF targets could explain the seizure-vulnerable phenotype observed in KO animals.
Identification of SRF Direct Targets
A group of genes with functions associated with the regulation of neuronal excitability and structural plasticity of spines was selected for validation. Microarray data were verified using quantitative real-time polymerase chain reaction (qRT-PCR; saline: n ≥ 5 for each genotype; kainic acid: n ≥ 7 for each genotype) for the selected transcripts. An increase in the expression level 6 h after kainic acid stimulation (intraperitoneal KA injection) and dependence on the transcription factor SRF was confirmed for FBJ osteosarcoma oncogene (Fos; p < 0.001), lipocalin 2 (Lcn2, NGAL; p < 0.01), neuronal PAS domain protein 4 (Npas4; p < 0.01), brain-derived neurotrophic factor (Bdnf; p < 0.05), aggrecan (Acan; p < 0.001), protocadherin 8 (Pcdh8; p < 0.05), zinc finger protein 36 (Zfp36; tristetraprolin [TTP]; p < 0.01), mago-nashi homolog, proliferation-associated (Magoh; p < 0.05), synaptotagmin IV (Syt4; p < 0.05), engulfment, and cell motility 1 (Elmo1; p < 0.001), and growth arrest and DNA-damage-inducible 45 gamma (Gadd45g; p < 0.05) two-way ANOVA genotype × treatment interaction, Bonferroni multiple-comparisons test (Fig. 4).

SRF is required for the activation of several plasticity genes in kainic acid-induced status epilepticus. The results obtained with microarray analysis were verified using qRT-PCR. qRT-PCR amplification of 11 transcripts from control (CTR) and knockout (KO) DG (saline, n ≥ 5 for each genotype; kainic acid, n ≥ 7 for each genotype; males and females) revealed an increase in mRNA levels in CTR animals in response to kainic acid stimulation (6 h after intraperitoneal injection of KA), which was abolished in KO animals. *p < 0.05, **p < 0.01, ***p < 0.001 (two-way ANOVA followed by Bonferroni post hoc test)
To determine which of the selected SRF-dependent genes are direct targets of SRF, we analyzed putative SRF binding sites within the evolutionary conserved regions between mouse and human, using two database tools, namely cREMaG [23] and NGD [24] (see “Materials and Methods” section for details). Additionally, we searched the human CArGome (according to the group of Miano; [27]) for potential CArG boxes that are conserved in mouse. Among the pool of potential SRF-binding sites identified with the above methods, only the motifs with a maximum of two mismatches to the CArG box consensus [CC(A/T)6GG] and with at most one mismatch in CC or GG were selected for the experimental validation.
To identify direct targets of SRF bound in vivo to the gene promoters in the hippocampus, we applied a model of kainic acid-induced status epilepticus. We investigated recruitment of the endogenous transcription factor SRF to the identified regions of selected genes using chromatin immunoprecipitation. Chromatin from the hippocampus in C57BL/6 mice that were treated with kainic acid (intraperitoneal kainic acid injection, 2 h after seizure onset) or naive mice was immunoprecipitated using an anti-SRF antibody or normal immunoglobulin G (IgG) to determine the background, followed by qRT-PCR amplification with specific primers (for the list of primers and potential CArG boxes, see Table 2). We observed the in vivo binding of SRF to the promoter of Fos under basal conditions (i.e., in naive animals), whereas a significant in vivo enrichment of SRF binding 2 h after seizure induction was observed for Npas4, Gadd45g, and Zfp36 (Fig. 5). The binding of SRF to the promoters of those genes indicated direct regulation by SRF. They also presented a full CArG box consensus (or one mismatch in the case of Zfp36) in the identified SRE sequences. In contrast, we did not observe significant enrichment in SRF occupancy at potential CArG box regions of other analyzed genes (Lcn2, Syt4, Bdnf, Magoh, and Pcdh8). Altogether, we identified a group of novel SRF targets in neurons, which could explain the epilepsy-vulnerable phenotype observed in KO animals (Table 3). We showed that three of those genes (Npas4, Gadd45g, and Zfp36) are novel direct targets of SRF in the hippocampus in vivo, whereas the other identified genes are likely to be indirectly regulated by SRF in neurons.

Endogenous SRF binds to the promoter regions of Fos, Npas4, Gadd45g, and Zfp36. The recruitment of the endogenous transcription factor SRF to the identified regions of selected genes was analyzed using chromatin immunoprecipitation with SRF-specific antibody, followed by qRT-PCR with primers surrounding predicted SRF-binding sites (for primer sequences and locations, see Table 2). Data are plotted as a percentage of input and are the averages of three independent experiments ± SEM. A fragment of Arbp exon was used as a negative control. The level of background was determined with normal IgG. Genes with SRF bound to their promoter fragments (Fos, Npas4, Zfp36, and Gadd45g) are marked with frames (>2-fold increase in precipitation level compared with Arbp exon and IgG controls). Kainic acid-induced status epilepticus (KA, intraperitoneal injection) enhanced the binding of SRF especially to the promoters of the Npas4, Zfp36, and Gadd45g genes. All ChIP analysis were conducted in males
Discussion
The present study assessed the behavioral and transcriptional effects of SRF deficiency in neurons in the context of epileptogenesis. Animals with adult, neuronal deletion of SRF developed a more severe seizure phenotype compared with CTR animals in a kainic acid model of TLE. Using genome-wide analysis of activity-induced genes, we identified 378 genes that are differentially expressed in the hippocampal dentate gyrus of SRF-deficient mice, including regulators of inhibitory/excitatory balance and structural plasticity of neurons, together with three new SRF direct targets (Npas4, Gadd45g, Zfp36).
SRF is an Important Transcription Factor in Epilepsy
Epilepsy is a clinically relevant form of hyperexcitability-associated brain pathology. Genes that are activated during seizures may contribute to epileptogenic processes via network reorganization that leads to hyperexcitability or via compensatory or protective mechanisms. In the present study, we uncovered SRF as an important transcription factor (TF) that regulates epileptogenesis. The deletion of SRF in the adult hippocampus increased the epileptic phenotype, manifested by more severe and frequent spontaneous seizures (Fig. 2), despite similar severity of acute seizures (Supplementary Fig. S1).
Activity-induced gene expression in neurons might also be regulated by the transcription factors CREB and MEF2. Despite partially overlapping patterns of gene expression, the inactivation of these TFs leads to divergent consequences. CREB deficiency decreases neuronal excitability and suppresses epileptogenesis [49–51]. MEF2 was shown to decrease the number of excitatory synapses and weaken neuronal strength [52, 53]. MEF2A binding sites, along with SREs, were identified in the present study as TF binding sites that were overrepresented in the promoters of genes regulated by SRF, suggesting that SRF and MEF2 can activate similar transcription programs in neurons.
Possible Role of SRF-Dependent Genes in the Development of Epilepsy
Epilepsy is associated with robust synaptic plasticity that occurs at the cellular level caused by changes in gene expression [54]. Previous studies on SRF-dependent transcription in neurons focused mainly on basal gene expression or were performed using in vitro models [55, 56]. Although previous studies investigated the program downstream of SRF (recently reviewed by [57]), SRF-dependent transcription in neurons was focused mainly on a basal gene expression or were performed using in vitro models [55, 56]. SRF is one of the major regulators of plastic changes [12, 13]. This large-scale study of SRF-controlled transcription programs under in vivo conditions provides more insights into the molecular mechanisms that lead to the development of pathology. As expected, we found a large number of SRF-dependent genes associated with synaptic plasticity and epilepsy, the expression of which was decreased in SRF KO mice after seizures.
Functional annotation revealed that many of the SRF targets we have identified are known as regulators of inhibitory/excitatory balance, structural plasticity, and mRNA translation (see Table 3). Control of inhibitory/excitatory balance in neurons can be regulated by Npas4, which is an activity-dependent TF that controls inhibitory synapse development and a number of γ-aminobutyric acid-releasing synapses on excitatory neurons [58, 59]; Bdnf, which controls the maturation and function of inhibitory synapses [60, 61]; Syt4, which regulates synaptic function and plasticity by modulating BDNF release [62]; and Gadd45g, a member of the Gadd45 family that is engaged in the activity-induced demethylation of Bdnf promoters and transcription [63]. Acan, as a component of perineuronal nets around inhibitory interneurons, stabilizes synapses and restrict their reorganization.
Another group of identified genes are those encoding proteins regulating structural and physiological plasticity of excitatory neurons. Lcn2, Pcdh8, and Elmo1 influence the electrophysiological properties of neurons by either decreasing dendritic spine density or changing their morphology [64–66]. These activity-induced proteins may suppress the number of spines to dampen synaptic function after elevated neuronal activity, similar to other neural activity-regulated molecules, such as MEF2 and PLK2 [53, 67].
A third group of genes are genes that encode RNA-binding proteins, such as Magoh and Zfp36, which regulate the metabolism of mRNAs by targeting them for degradation [68, 69].
The decreased production of proteins form any of mentioned above groups may explain the seizure-vulnerable phenotype observed in SRF KO animals. However, further studies are needed to address the specific role of SRF in the regulation of the balance between inhibition and activation in neurons.
We found an increase in epileptogenesis in SRF KO animals, which is consistent with the results of several studies that used animals with individual deletions of SRF-dependent genes. The ablation of such genes as Npas4, Fos, and Arc leads to increased seizure susceptibility [58, 70, 71]. The in vitro disruption of Acan causes seizure-like activity in hippocampal cultures [72]. Similarly, enhanced epileptiform responses were observed in slices from Syt4 KO animals [73]. Moreover, Lcn2-deficient neurons show increased excitability [64, 74]. These proteins are induced by an increase in neuronal activity and appear to play a role as endogenous inhibitors of epilepsy; thus, impairment of their expression in SRF KO mice could enhance the epileptic phenotype, as demonstrated by the present results.
SRF-Controlled Genes Associated with Human Pathology
Although no evidence for the role of SRF in human epilepsy was provided so far, it is important to note that either deletion or insertion in the regions of SRF target genes identified in our study was associated with human neurological disorders (Table 3). Single-nucleotide polymorphisms identified within or in close proximity to CArG boxes in humans were shown to be linked with neurological disorders, such as bipolar disorder, amyotrophic lateral sclerosis, and Alzheimer’s disease [27]. Several SRF-controlled genes that were identified in the present study (e.g., Cyr61, Bdnf, Zfp36, Fos, JunB) are upregulated in the cortex in patients who suffer from epilepsy. Statistically significant enrichment of SREs on the proximal promoters in this group of genes was observed [75].
In a group of genes that were downregulated in CTR animals after kainic acid-induced status epilepticus but not in KO animals (Fig. 3b), we identified Cacna1h (Cav3.2). Mutations in this gene are linked to a wide spectrum of idiopathic generalized epilepsies [76, 77] and influence neuronal excitability [78, 79]. The lack of downregulation of Cacna1h mRNA after status epilepticus in SRF KO mice may contribute to their enhanced epileptic phenotype.
Molecular Mechanisms of SRF-Dependent Gene Regulation
Our results suggest that SRF can regulate gene expression by two possible molecular mechanisms. The first mechanism is direct binding to the gene promoters. We found in vivo enrichment of SRF occupancy on promoters of Fos, Npas4, Gadd45g, and Zfp36. Although Fos has been previously reported to be regulated by SRF, this is the first report of which we are aware on the direct regulation of Npas4, Gadd45g, and Zfp36 by SRF in neurons. Significant enrichment of SRF binding to the new SRF targets was observed upon kainic acid stimulation in the present study. The binding of SRF to DNA can be constitutive, as observed for Fos, or inducible upon stimulation [80].
The second mechanism involves other transcription factors. Because of the relatively late time point analyzed in the microarray experiments (i.e., 6 h after seizure induction), SRF targets may be indirectly regulated through the activation of other genes that encode transcription factors (e.g., Fos, Npas4, Egr1), similar to our previous study that found that Mmp-9 was regulated in neurons by SRF through Fos [81]. The identification of several transcription factors, among SRF target genes, that are important for neuronal plasticity suggests that SRF may be a primary hub that can orchestrate the regulation of several aspects of synaptic plasticity.
Conclusions
Our data show that SRF is an important regulator of activity-induced gene expression in neurons and may be involved in the development of epilepsy. SRF regulates the expression of several plasticity genes that together may decrease hyperexcitation in response to a strong neuronal stimulation. The lack of these genes may lead to the development of a more severe TLE as a consequence of homeostatic imbalance. Still, further studies are needed to determine which of the identified SRF target genes are actually involved in the development of epilepsy and what is the molecular mechanism underlying this process. To address this question, identified genes need to be analyzed individually and their potential role in the context of epilepsy should be assessed.
References
Thurman DJ, Beghi E, Begley CE, Berg AT, Buchhalter JR, Ding D, Hesdorffer DC, Hauser WA, Kazis L, Kobau R, Kroner B, Labiner D, Liow K, Logroscino G, Medina MT, Newton CR, Parko K, Paschal A, Preux PM, Sander JW, Selassie A, Theodore W, Tomson T, Wiebe S (2011) Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia 52(7):2–26. doi:10.1111/j.1528-1167.2011.03121.x
Kaczmarek L, Chaudhuri A (1997) Sensory regulation of immediate-early gene expression in mammalian visual cortex: implications for functional mapping and neural plasticity. Brain Res Brain Res Rev 23(3):237–256
Pitkanen A, Lukasiuk K (2011) Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol 10(2):173–186. doi:10.1016/S1474-4422(10)70310-0
Treisman R (1987) Identification and purification of a polypeptide that binds to the c-fos serum response element. EMBO J 6:2711–2717
Prywes R, Roeder RG (1987) Purification of the c-fos enhancer-binding protein. Mol Cell Biol 7:3482–3489
Miano JM, Long X, Fujiwara K (2007) Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol 292(1):C70–81
Kalita K, Kharebava G, Zheng JJ, Hetman M (2006) Role of megakaryoblastic acute leukemia-1 in ERK1/2-dependent stimulation of serum response factor-driven transcription by BDNF or increased synaptic activity. J Neurosci 26(39):10020–10032
Wickramasinghe SR, Alvania RS, Ramanan N, Wood JN, Mandai K, Ginty DD (2008) Serum response factor mediates NGF-dependent target innervation by embryonic DRG sensory neurons. Neuron 58(4):532–545
Misra RP, Bonni A, Miranti CK, Rivera VM, Sheng M, Greenberg ME (1994) Calcium entry through L-type voltage sensitive calcium channels can activate transcription via the serum response factor. J Biol Chem 269:25483–25493
Morgan JI, Curran T (1986) Role of ion fluxes in the control of c-fos expression. Nature 322:552–555
Miranti CK, Ginty DD, Huang G, Chatila T, Greenberg ME (1995) Calcium activates serum response factor-dependent transcription by a Ras- and Elk-1-independent mechanism that involves a Ca2+/calmodulin-dependent kinase. Mol Cell Biol 15:3672–3684
Ramanan N, Shen Y, Sarsfield S, Lemberger T, Schutz G, Linden DJ, Ginty DD (2005) SRF mediates activity-induced gene expression and synaptic plasticity but not neuronal viability. Nat Neurosci 8(6):759–767, Epub 2005 May 2008
Etkin A, Alarcon JM, Weisberg SP, Touzani K, Huang YY, Nordheim A, Kandel ER (2006) A role in learning for SRF: deletion in the adult forebrain disrupts LTD and the formation of an immediate memory of a novel context. Neuron 50(1):127–143
Alberti S, Krause SM, Kretz O, Philippar U, Lemberger T, Casanova E, Wiebel FF, Schwarz H, Frotscher M, Schutz G, Nordheim A (2005) Neuronal migration in the murine rostral migratory stream requires serum response factor. PNAS 102(17):6148–6153
Knoll B, Kretz O, Fiedler C, Alberti S, Schutz G, Frotscher M, Nordheim A (2006) Serum response factor controls neuronal circuit assembly in the hippocampus. Nat Neurosci 9(2):195–204
Li CL, Sathyamurthy A, Oldenborg A, Tank D, Ramanan N (2014) SRF phosphorylation by glycogen synthase kinase-3 promotes axon growth in hippocampal neurons. J Neurosci 34(11):4027–4042. doi:10.1523/JNEUROSCI. 4677-12.2014
Stritt C, Knoll B (2010) Serum response factor regulates hippocampal lamination and dendrite development and is connected with reelin signaling. Mol Cell Biol 30(7):1828–1837
Kalita K, Kuzniewska B, Kaczmarek L (2012) MKLs: co-factors of serum response factor (SRF) in neuronal responses. Int J Biochem Cell Biol 44(9):1444–1447
Morris TA, Jafari N, Rice AC, Vasconcelos O, DeLorenzo RJ (1999) Persistent increased DNA-binding and expression of serum response factor occur with epilepsy-associated long-term plasticity changes. J Neurosci 19(19):8234–8243
Herdegen T, Blume A, Buschmann T, Georgakopoulos E, Winter C, Schmid W, Hsieh TF, Zimmermann M, Gass P (1997) Expression of activating transcription factor-2, serum response factor and cAMP/Ca response element binding protein in the adult rat brain following generalized seizures, nerve fibre lesion and ultraviolet irradiation. Neuroscience 81(1):199–212
Erdmann G, Schutz G, Berger S (2007) Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci 8:63
Racine RJ (1972) Modification of seizure activity by electrical stimulation I after-discharge threshold. Electroencephalogr Clin Neurophysiol 32(3):269–279
Piechota M, Korostynski M, Przewlocki R (2010) Identification of cis-regulatory elements in the mammalian genome: the cREMaG database. PLoS One 5(8):e12465
Krystkowiak I, Lenart J, Debski K, Kuterba P, Petas M, Kaminska B, Dabrowski M (2013) Nencki Genomics Database—Ensembl funcgen enhanced with intersections, user data and genome-wide TFBS motifs. Database (Oxford) 2013:bat069
Bray N, Dubchak I, Pachter L (2003) AVID: A global alignment program. Genome Res 13(1):97–102
Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, Pachter LS, Dubchak I (2000) VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16(11):1046–1047
Benson CC, Zhou Q, Long X, Miano JM (2011) Identifying functional single nucleotide polymorphisms in the human CArGome. Physiol Genomics 43(18):1038–1048
Pernot F, Dorandeu F, Beaup C, Peinnequin A (2010) Selection of reference genes for real-time quantitative reverse transcription-polymerase chain reaction in hippocampal structure in a murine model of temporal lobe epilepsy with focal seizures. J Neurosci Res 88(5):1000–1008
Paul AP, Pohl-Guimaraes F, Krahe TE, Filgueiras CC, Lantz CL, Colello RJ, Wang W, Medina AE (2010) Overexpression of serum response factor restores ocular dominance plasticity in a model of fetal alcohol spectrum disorders. J Neurosci 30(7):2513–2520. doi:10.1523/JNEUROSCI. 5840-09.2010
Schauwecker PE, Steward O (1997) Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. PNAS 94(8):4103–4108
Nedivi E, Hevroni D, Naot D, Israeli D, Citri Y (1993) Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature 363(6431):718–722
Hevroni D, Rattner A, Bundman M, Lederfein D, Gabarah A, Mangelus M, Silverman MA, Kedar H, Naor C, Kornuc M, Hanoch T, Seger R, Theill LE, Nedivi E, Richter-Levin G, Citri Y (1998) Hippocampal plasticity involves extensive gene induction and multiple cellular mechanisms. J Mol Neurosci 10(2):75–98
Rutherford LC, DeWan A, Lauer HM, Turrigiano GG (1997) Brainderived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci 17(12):4527–35
Hong Z, Li W, Qu B, Zou X, Chen J, Sander JW, Zhou D Serum brainderived neurotrophic factor levels in epilepsy. Eur J Neurol 21(1):57–64
Kokaia M, Ernfors P, Kokaia Z, Elmer E, Jaenisch R, Lindvall O (1995) Suppressed epileptogenesis in BDNF mutant mice. Exp Neurol 133(2):215–24
Croll SD, Suri C, Compton DL, Simmons MV, Yancopoulos GD, Lindsay RM, Wiegand SJ, Rudge JS, Scharfman HE (1999) Brain-derived neurotrophic factor transgenic mice exhibit passive avoidance deficits, increased seizure severity and in vitro hyperexcitability in the hippocampus and entorhinal cortex. Neuroscience 93(4):1491–506
He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO (2004) Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron 43(1):31–42
Vician L, Lim IK, Ferguson G, Tocco G, Baudry M, Herschman HR (1995) Synaptotagmin IV is an immediate early gene induced by depolarization in PC12 cells and in brain. PNAS 92(6):2164–8
Glisovic S, Glavan G, Saghafi MM, Zivin M (2007) Upregulation of synaptotagmin IV protein in kainate-induced seizures. Neuroreport 18(8):831–5
Yamauchi J, Miyamoto Y, Murabe M, Fujiwara Y, Sanbe A, Fujita Y, Murase S, Tanoue A (2007) Gadd45a, the gene induced by the mood stabilizer valproic acid, regulates neurite outgrowth through JNK and the substrate paxillin in N1E-115 neuroblastoma cells. Exp Cell Res 313(9):1886–96
McRae PA, Baranov E, Rogers SL, Porter BE (2012) Persistent decrease in multiple components of the perineuronal net following status epilepticus. Eur J Neurosci 36(11):3471–82
Kjeldsen L, Bainton DF, Sengelov H, Borregaard N (1993) Structural and functional heterogeneity among peroxidase-negative granules in human neutrophils: identification of a distinct gelatinase-containing granule subset by combined immunocytochemistry and subcellular fractionation. Blood 82(10):3183–91
Yamagata K, Andreasson KI, Sugiura H, Maru E, Dominique M, Irie Y, Miki N, Hayashi Y, Yoshioka M, Kaneko K, Kato H, Worley PF (1999) Arcadlin is a neural activity-regulated cadherin involved in long term potentiation. J Biol Chem 274(27):19473–9
Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, Al-Saad S, Ware J, Joseph RM, Greenblatt R, Gleason D, Ertelt JA, Apse KA, Bodell A, Partlow JN, Barry B, Yao H, Markianos K, Ferland RJ, Greenberg ME, Walsh CA (2008) Identifying autism loci and genes by tracing recent shared ancestry. Science 321(5886):218–23
Dibbens LM, Tarpey PS, Hynes K, Bayly MA, Scheffer IE, Smith R, Bomar J, Sutton E, Vandeleur L, Shoubridge C, Edkins S, Turner SJ, Stevens C, O'Meara S, Tofts C, Barthorpe S, Buck G, Cole J, Halliday K, Stevens C, O'Meara S, Tofts C, Barthorpe S, Buck G, Cole J, Halliday K, Jones D, Lee R, Madison M, Mironenko T, Varian J, West S, Widaa S, Wray P, Teague J, Dicks E, Butler A, Menzies A, Jenkinson A, Shepherd R, Gusella JF, Afawi Z, Mazarib A, Neufeld MY, Kivity S, Lev D, Lerman-Sagie T, Korczyn AD, Derry CP, Sutherland GR, Friend K, Shaw M, Corbett M, Kim HG, Geschwind DH, Thomas P, Haan E, Ryan S, McKee S, Berkovic SF, Futreal PA, Stratton MR, Mulley JC, Gecz J (2008) Xlinked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 40(6):776–81
Silver DL, Watkins-Chow DE, Schreck KC, Pierfelice TJ, Larson DM, Burnetti AJ, Liaw HJ, Myung K, Walsh CA, Gaiano N, Pavan WJ (2010) The exon junction complex component Magoh controls brain size by regulating neural stem cell division. Nat Neurosci 13(5):551–8
Giorgi C, Yeo GW, Stone ME, Katz DB, Burge C, Turrigiano G, Moore MJ (2007) The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell 130(1):179–91
Parkitna JR, Bilbao A, Rieker C, Engblom D, Piechota M, Nordheim A, Spanagel R, Schutz G (2010) Loss of the serum response factor in the dopamine system leads to hyperactivity. Faseb J 24(7):2427–2435
Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, Neve R, Poirazi P, Silva AJ (2009) CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci 12(11):1438–1443
Gruart A, Benito E, Delgado-Garcia JM, Barco A (2012) Enhanced cAMP response element-binding protein activity increases neuronal excitability, hippocampal long-term potentiation, and classical eyeblink conditioning in alert behaving mice. J Neurosci 32(48):17431–17441
Zhu X, Han X, Blendy JA, Porter BE (2012) Decreased CREB levels suppress epilepsy. Neurobiol Dis 45(1):253–263
Barbosa AC, Kim MS, Ertunc M, Adachi M, Nelson ED, McAnally J, Richardson JA, Kavalali ET, Monteggia LM, Bassel-Duby R, Olson EN (2008) MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. PNAS 105(27):9391–9396
Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME (2006) Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311(5763):1008–1012
Scharfman HE (2002) Epilepsy as an example of neural plasticity. Neuroscientist 8(2):154–173
Stritt C, Stern S, Harting K, Manke T, Sinske D, Schwarz H, Vingron M, Nordheim A, Knoll B (2009) Paracrine control of oligodendrocyte differentiation by SRF-directed neuronal gene expression. Nat Neurosci 12(4):418–427
Benito E, Valor LM, Jimenez-Minchan M, Huber W, Barco A (2011) cAMP response element-binding protein is a primary hub of activity-driven neuronal gene expression. J Neurosci 31(50):18237–18250
Benito E, Barco A (2014) The neuronal activity-driven transcriptome. Mol Neurobiol. doi:10.1007/s12035-014-8772-z
Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, Greenberg ME (2008) Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455(7217):1198–1204
Spiegel I, Mardinly AR, Gabel HW, Bazinet JE, Couch CH, Tzeng CP, Harmin DA, Greenberg ME (2014) Npas4 regulates excitatory-inhibitory balance within neural circuits through cell-type-specific gene programs. Cell 157(5):1216–1229
Marty S (2000) Differences in the regulation of neuropeptide Y, somatostatin and parvalbumin levels in hippocampal interneurons by neuronal activity and BDNF. Prog Brain Res 128:193–202
Vicario-Abejon C, Collin C, McKay RD, Segal M (1998) Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci 18(18):7256–7271
Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, Chapman ER (2009) Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat Neurosci 12(6):767–776
Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, Flavell RA, Lu B, Ming GL, Song H (2009) Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323(5917):1074–1077
Mucha M, Skrzypiec AE, Schiavon E, Attwood BK, Kucerova E, Pawlak R (2011) Lipocalin-2 controls neuronal excitability and anxiety by regulating dendritic spine formation and maturation. PNAS 108(45):18436–18441
Yasuda S, Tanaka H, Sugiura H, Okamura K, Sakaguchi T, Tran U, Takemiya T, Mizoguchi A, Yagita Y, Sakurai T, De Robertis EM, Yamagata K (2007) Activity-induced protocadherin arcadlin regulates dendritic spine number by triggering N-cadherin endocytosis via TAO2beta and p38 MAP kinases. Neuron 56(3):456–471
Kim JY, Oh MH, Bernard LP, Macara IG, Zhang H (2011) The RhoG/ELMO1/Dock180 signaling module is required for spine morphogenesis in hippocampal neurons. J Biol Chem 286(43):37615–37624
Pak DT, Sheng M (2003) Targeted protein degradation and synapse remodeling by an inducible protein kinase. Science 302(5649):1368–1373
Gehring NH, Lamprinaki S, Hentze MW, Kulozik AE (2009) The hierarchy of exon-junction complex assembly by the spliceosome explains key features of mammalian nonsense-mediated mRNA decay. PLoS Biol 7(5):e1000120
Singh KK, Wachsmuth L, Kulozik AE, Gehring NH (2013) Two mammalian MAGOH genes contribute to exon junction complex composition and nonsense-mediated decay. RNA Biol 10(8):1291–1298
Zhang J, Zhang D, McQuade JS, Behbehani M, Tsien JZ, Xu M (2002) c-fos regulates neuronal excitability and survival. Nat Genet 30(4):416–420
Peebles CL, Yoo J, Thwin MT, Palop JJ, Noebels JL, Finkbeiner S (2010) Arc regulates spine morphology and maintains network stability in vivo. PNAS 107(42):18173–18178
Vedunova M, Sakharnova T, Mitroshina E, Perminova M, Pimashkin A, Zakharov Y, Dityatev A, Mukhina I (2013) Seizure-like activity in hyaluronidase-treated dissociated hippocampal cultures. Front Cell Neurosci 7:149
Dean C, Liu H, Staudt T, Stahlberg MA, Vingill S, Buckers J, Kamin D, Engelhardt J, Jackson MB, Hell SW, Chapman ER (2012) Distinct subsets of Syt-IV/BDNF vesicles are sorted to axons versus dendrites and recruited to synapses by activity. J Neurosci 32(16):5398–5413
Skrzypiec AE, Shah RS, Schiavon E, Baker E, Skene N, Pawlak R, Mucha M (2013) Stress-induced lipocalin-2 controls dendritic spine formation and neuronal activity in the amygdala. PLoS One 8(4):e61046
Beaumont TL, Yao B, Shah A, Kapatos G, Loeb JA (2012) Layer-specific CREB target gene induction in human neocortical epilepsy. J Neurosci 32(41):14389–14401
Chen Y, Lu J, Pan H, Zhang Y, Wu H, Xu K, Liu X, Jiang Y, Bao X, Yao Z, Ding K, Lo WH, Qiang B, Chan P, Shen Y, Wu X (2003) Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol 54(2):239–243
Heron SE, Khosravani H, Varela D, Bladen C, Williams TC, Newman MR, Scheffer IE, Berkovic SF, Mulley JC, Zamponi GW (2007) Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann Neurol 62(6):560–568
Vitko I, Chen Y, Arias JM, Shen Y, Wu XR, Perez-Reyes E (2005) Functional characterization and neuronal modeling of the effects of childhood absence epilepsy variants of CACNA1H, a T-type calcium channel. J Neurosci 25(19):4844–4855
Eckle VS, Shcheglovitov A, Vitko I, Dey D, Yap CC, Winckler B, Perez-Reyes E (2014) Mechanisms by which a CACNA1H mutation in epilepsy patients increases seizure susceptibility. J Physiol 592(Pt 4):795–809
Esnault C, Stewart A, Gualdrini F, East P, Horswell S, Matthews N, Treisman R (2014) Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev 28(9):943–958
Kuzniewska B, Rejmak E, Malik AR, Jaworski J, Kaczmarek L, Kalita K (2013) Brain-derived neurotrophic factor induces matrix metalloproteinase 9 expression in neurons via the serum response factor/c-Fos pathway. Mol Cell Biol 33(11):2149–2162
Acknowledgments
This work was supported by a Marie Curie International Reintegration Grant within the 7th European Community Framework Programme under grant agreement no. 230992, EpiTarGene, and the Polish National Science Center grant (SONATA BIS 2) DEC-2012/07/E/NZ3/01814. The authors wish to thank Prof. Ryszard Przewlocki, Dr. Michal Korostynski, and Dr. Marcin Piechota from GenBioInfo and Institute of Pharmacology PAN for performing the microarray hybridization and statistical and bioinformatic analysis; Ewelina Szmajda for the technical assistance; the Laboratory of Microarrays Analysis, Institute of Biochemistry and Biophysics PAN, for bioinformatic analysis using IPA, Qiagen; and Dr. Katarzyna Lukasiuk for the help with EEG recordings. The manuscript was corrected by professional proofreader Michael Arends.
Author Contributions
K.K. designed and performed the experiments and analyzed the data. B.K. performed the experiments and analyzed the data. K.N. performed the EEG recordings and analyzed the data. M.D. and B.K performed the bioinformatic analysis of SRF binding sites. B.K. and K.K. wrote the paper. K.K. and L.K. supervised the project.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Fig. S1
Severity of status epilepticus (SE) in CTR and KO animals after intrahipocampal kainic acid injection. CTR and KO animals did not significantly differ in latency to first induced seizure (a), number of seizure (b), mean duration of seizure (c) and seizure behavioral score (d) during first 24 hours after intrahippocampal kainate injection (a, b, c, d; Student’s t-test, p > 0.05). All EEG recordings were conducted in males (GIF 6 kb)
Fig. S2
Severity of seizures in CTR and KO animals after intraperitoneal kainic acid injection. Results for CTR and KO animals did not significantly differ in seizure behavioral score (a) and latency behavioral seizure score 4 or 5 (b) after intraperitoneal administration of kainic acid. (a, b, Student’s t-test, p > 0.05). Analysis were conducted in males and females (GIF 4 kb)
Table S1
List of genes with a significantly altered expression profile as a result of SRF knockdown 6 hours after intraperitoneal kainic acid injections. (XLSX 117 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Kuzniewska, B., Nader, K., Dabrowski, M. et al. Adult Deletion of SRF Increases Epileptogenesis and Decreases Activity-Induced Gene Expression. Mol Neurobiol 53, 1478–1493 (2016). https://doi.org/10.1007/s12035-014-9089-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-9089-7