
Abstract
Lead (Pb)-induced neurodegeneration and its link with widespread neurobehavioral changes are well documented. Experimental evidences suggest that ethanol could enhance the absorption of metals in the body, and alcohol consumption may increase the susceptibility to metal intoxication in the brain. However, the underlying mechanism of ethanol action in affecting metal toxicity in brain cells is poorly understood. Thus, an attempt was made to investigate the modulatory effect of ethanol on Pb intoxication in PC12 cells, a rat pheochromocytoma. Cells were co-exposed to biological safe doses of Pb (10 μM) and ethanol (200 mM), and data were compared to the response of cells which received independent exposure to these chemicals at similar doses. Ethanol (200 mM) exposure significantly aggravated the Pb-induced alterations in the end points associated with oxidative stress and apoptosis. The finding confirms the involvement of reactive oxygen species (ROS)-mediated oxidative stress, and impairment of mitochondrial membrane potential, which subsequently facilitate the translocation of triggering proteins between cytoplasm and mitochondria. We further confirmed the apoptotic changes due to induction of mitochondria-mediated caspase cascade. These cellular changes were found to recover significantly, if the cells are exposed to N-acetyl cysteine (NAC), a known antioxidant. Our data suggest that ethanol may potentiate Pb-induced cellular damage in brain cells, but such damaging effects could be recovered by inhibition of ROS generation. These results open up further possibilities for the design of new therapeutics based on antioxidants to prevent neurodegeneration and associated health problems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The toxicological consequences due to heavy metal exposure from anthropogenic and environmental sources have been recognized as a major health concern globally [1, 2]. Among the group of heavy metals with proven toxicity in human, lead (Pb), a ubiquitous pollutant in the ecosystem, is known to be highly neurotoxic [3] and developmental neurotoxic [4]. The major Pb-induced neurological disorders reported in humans and experimental animals are behavioral abnormalities, learning impairment, decreased hearing, impaired cognitive function, etc. [5]. Despite several in vitro [6, 7], in vivo [8, 9] and epidemiological [10] studies, the exact mechanism(s) of Pb-induced neurotoxicity in both fetus/children and adults have still not been elucidated and are also not well understood. However, among the several proposed mechanisms of Pb-induced neurotoxicity, the oxidative stress-mediated apoptosis is the most prominent [11–13]. Pb exposure increases the levels of reactive oxygen species (ROS) and intracellular Ca2+, which in turn disturbs the mitochondrial membrane potential, leading to release of mitochondrial cytochrome-c in the cytoplasm by activating caspase cascade-mediated apoptotic cell death [14–16]. It also disturbs the antioxidant defense system via thiol binding [17, 18] and by its calcium mimicking ability [15]. Previous reports showed significant altered manifestations of Pb intoxication due to individual’s habits such as intake of total food, amount and type of fat, calcium and iron, etc. [19].
Alcohol consumption is one of the major lifestyle factors. After intake, alcohol is distributed throughout the body tissues and rapidly crosses the blood-brain barrier and damages the nervous system [20–23]. Teratogenic and developmental neurotoxic effects of ethanol are also well documented [24, 25]. Chronic exposure to ethanol has been reported to cause neuronal degeneration [26], cognitive impairment [27] with permanent structural damages in the brain [28, 29]. At cellular level, ethanol mediates neurodegeneration by altering the expression of markers (messenger (m)RNA and proteins) associated with myelin degeneration [30], insulin responsiveness [31], ROS-mediated oxidative stress [32] and apoptosis [33], and deregulated acetylcholine homeostasis [34], etc.
There are numerous studies demonstrating high vulnerability of human subjects towards Pb exposure, especially those who regularly consume alcohol [35]. Reports have shown the presence of high Pb levels in the blood of alcoholics when compared with non-alcoholics of same occupation and residing in the same vicinity [36]. Enhanced Pb intoxication in alcoholics has been suggested in part due to increased permeability of membranes and eventually increased burden of Pb and depletion of essential metals [37, 38]. Both Pb and alcohol are known neurotoxicants, and alcohol is known to cross the blood-brain barrier. As a consequence, this can result in increasing the neurotoxicity level further by facilitating the entry of Pb in the brain [39].
In this regard, the in vivo reports are available suggesting the additive/synergic toxic responses of co-exposure to ethanol and Pb [40]. However, detailed studies on the exact mechanism of ethanol-Pb co-exposure inducing neuronal impairment, oxidative stress, and subsequent apoptosis in controlled in vitro conditions using neuronal cells have not been reported so far. Thus, the present investigations were carried out to study the modulatory effect of ethanol concentrations on Pb-induced oxidative stress, apoptosis, and subsequent neurodegeneration in PC12 cells, one of the most widely studied cell line for neurobiology/neurotoxicity studies.
Materials and Methods
Reagents and Consumables
All the specified chemicals, reagents, diagnostic kits were purchased from Sigma Chemical Company. (St. Louis, MO, USA). Nutrient mixture F-12 Hams culture medium, antibiotics, fetal bovine serum (FBS), and horse serum (HS) were purchased from Gibco BRL, USA. Culture wares and other plastic consumables used in the study were purchased from Nunc, Denmark. All other chemicals were of analytical grade of purity and were procured locally.
Cell Culture
PC12, a rat pheochromocytoma cell line, was originally procured from National Centre for Cell Science, Pune, India and cultured in 82.5 % Nutrient Mixture (F-12 Hams), supplemented with 2.5 % heat-inactivated FBS, 15 % HS, 0.2 % sodium bicarbonate (NaHCO3), and antibiotic and anti-mycotic solution (100X, Invitrogen, Life Technologies, USA). Cells were maintained in a humidified atmosphere of 95 % air and 5 % CO2 at 37 °C in an incubator. The culture medium was replaced twice a week, and cell cultures were passaged at a ratio of 1:6 in a week. Prior to use the cells in the experiments, they were screened for their viability using trypan blue dye exclusion assay and batches showing more than 95 % viability were used in the study.
3-(4, 5-Dimethylthiazol-2-yl)-2, 5-Diphenyltetrazolium Bromide) Assay
Prior to use in the experiments, cytotoxic effect of Pb, ethanol, and their combination on PC12 cells were carried out using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) (MTT) assay as described earlier [41]. Briefly, 1 × 104 cells were allowed to adhere to poly-l-lysine-coated 96-well culture plates for 24 h under humid conditions in 5 % CO2 and 95 % atmospheric air at 37 °C. The medium was aspirated, and cells were incubated with medium containing graded series of Pb (0.1–200 μM) and ethanol (50–600 mM) for 24 h. To address the synergistic cytotoxic effect, PC12 cells were incubated with combined doses of Pb (10 μM) and ethanol (200 mM) for 24 h. After incubation, the cells were washed twice with phosphate-buffered saline (PBS), and 200 μl of culture medium containing 5 mg/ml of MTT dye was added to each well and incubated further for 4 h. The reaction mixture was carefully taken out and 200 μl of culture grade dimethylsulphoxide (DMSO) was added to each well and mixed by pipetting up and down several times until the content was homogenized. After 10 min, the color was read at 550 nm using multi-well microplate reader (Synergy HT, Bio-Tek, USA). The unexposed cells which served as control were also run simultaneously under identical conditions.
Measurement of Intracellular Calcium [Ca2+]i
The calcium levels have been demonstrated to play a key role in signal transduction and in various metabolic and physiological processes in the cells. Thus, even a small change in the level of intracellular calcium [Ca2+]i may induce major alterations in the cellular activities [14]. [Ca2+]i was measured by fluorometric analysis using molecular probes fura-2 acetoxymethyl ester pentapotassium (fura-2-AM; Sigma-Aldrich, USA). PC12 cells were loaded with 3 μmol/l fura-2-AM in loading solution containing 125 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2 mM CaCl2, 6 mM glucose, 25 mM Heps-NaOH (pH 7.4) buffer and incubated for 30 min at 37 °C in 5 % CO2 and 95 % atmospheric air. Unbound fura-2-AM was removed by rinsing the cells twice with titration solution and saline A containing NaCl, 8.182 g/l; KCl, 0.372 g/l; NaHCO3 0.336 g/l; and glucose, 0.9 g/l. The cells (1 × 106 ml−1) receiving Pb (10 μM) and/or ethanol (200 mM) or the unexposed control groups were represented in titration solution or saline A. Ca2+ fluorescence was measured by BMG FLUOstar Omega at an emission wave length of 510 nM using a pair of excitation length at 355 and 380 nM, and (Fo) excitation was obtained at the ratio of that generated by 355/380. Fmax was obtained after cells were lysed with 0.1 % Triton X-100. Fmin was obtained after the addition of EGTA (5 mM/l final concentration). Ca2+ fluorescence was examined simultaneously at time points 5, 15, 30, and 45 min and 1 h. The results shown are representative of three independent experiments carried out under identical experimental conditions. The concentration of intracellular calcium [Ca2+]i was calculated using the following formula:
where K d is the dissociation constant of fura-2-AM calcium complex and its value is 224 nmol/l. The results are expressed in terms of nanomoles of calcium.
ROS Generation
Pb- and/or ethanol-induced ROS generation in PC12 cells which was assessed using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; Sigma-Aldrich, USA) dye as the fluorescence agent [42]. Briefly, cells (5 × 104/well) were seeded in poly-l-lysine-pre-coated tissue culture slides flasks and allowed to adhere. Cells were then exposed to either of Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), NAC (10 μM), or Pb (10 μM) + ethanol (200 mM) + (10 μM) for 24 h. After exposure, cells were washed twice with PBS and incubated for 30 min at 37 °C in the dark in incomplete culture medium containing H2DCFH-DA (20 μM). Slides were washed twice again with PBS and mounted for microscopic analysis. Intracellular fluorescence was measured using an upright fluorescence microscope by grabbing the images (Nikon Eclipse 80i equipped with Nikon DS-Ri1 12.7 megapixel camera). Quantification of fluorescence was done using the image analysis software Leica Qwin 500, and data were expressed as the fold change of unexposed control cells. Cells exposed to H2O2 (100 μM) for 6 h under identical conditions were used as a positive control.
Glutathione Content Measurement
Intracellular glutathione content was estimated following the protocol of Akhtar et al. 2010 [43]. The exposure of cells to the test chemical and other conditions were identical to that of ROS generation. After exposure, cells were washed twice in cold PBS and lysed by four cycles of freeze thaw in distilled water containing deoxycholic acid plus sucrose and centrifuged at 10,000×g for 10 min at 4 °C. The protein estimation was done in supernatant. The supernatant protein was then precipitated with 1 % perchloric acid and centrifuged again at 10,000×g for 5 min at 4 °C. The precipitated protein (20 μl) was mixed with 160 μl of 0.1 M phosphate, 5 mM EDTA buffer (pH 8.3), and 20 μl o-phthalaldehyde (OPT, 1 mg/ml in methanol) in a black bottom 96-well plate. The plates were incubated for 3 h at room temperature in the dark, and fluorescence was measured at an emission/excitation wavelengths of 460/355 nm respectively, along with similarly prepared standards of glutathione (GSH) in 1 % perchloric acid. The results are expressed as nanomole of GSH per milligram of cellular protein. Cells exposed to H2O2 (100 μM) for 6 h under identical conditions were used as a positive control.
Lipid Peroxidation
Lipid peroxidation was evaluated by the thiobarbituric acid-reactive substance (TBARS) assay, which identifies mainly malondialdehyde (MDA), an end product of lipid peroxidation [44]. MDA was measured by following the protocol of Akhtar et al. 2010 [43]. Briefly, after exposure to Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), NAC (10 μM), and Pb (10 μM) + ethanol (200 mM) + NAC (10 μM) for 24 h, cells were washed twice by isotonic trace element-free Tris-HCl buffer (400 mM, pH 7.3). A 200-μl aliquot of cell suspension was subsequently mixed with 800 μl of lipid peroxidation (LPO) assay cocktail containing 0.4 % (w/v) thiobarbituric acid, 0.5 % (w/v) sodium dodecyl sulfate (SDS), and 5 % (v/v) acetic acid (pH 3.5) and incubated for 60 min at 95 °C. The sample was cooled using tap water and centrifuged at 2500×g for 5 min. The absorbance of the supernatant was read at 532 nm against a standard curve prepared using the MDA standard (10 mM) standard (10 mM 1,1,3,3-tetramethoxypropane in 20 mM Tris-HCl, pH 7.4). The results are calculated as nanomoles of MDA per milligram of protein. Cells exposed to H2O2 (100 μM) for 6 h under identical conditions were used as a positive control.
Mitochondrial Membrane Potential
Mitochondrial membrane potential is one of the earliest biomarkers to identify the induction of the oxidative stress and apoptosis. Flow cytometric analysis of apoptotic cell population was done by Mitolight™ Apoptosis Detection Kit (APT142, Chemicon, USA). Mitolight (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidazolyl carbocyanine chloride) is a mitochondrial dye that stains the mitochondria of living cells only. Briefly, cells after exposure to Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), NAC (10 μM), and Pb (10 μM) + ethanol (200 mM) + NAC (10 μM) for 24 h were re-suspended in 1 ml of pre-diluted Mitolight™ solution for 15 min at 37 °C. Cells were then immediately analyzed by flow cytometer (BD FACSCanto™) equipped with the FACSDiva Version 6.0.0. software. Cells exposed to H2O2 (100 μM) for 6 h under identical conditions were used as a positive control.
TUNEL Assay
Deoxynucleotide transferase dUTP nick end labeling (TUNEL) is a method for detecting DNA fragments during the cell cycle by labeling of the deoxynucleotide transferase dUTP nick end. This assay has been carried out using APO-BrdU™ TUNEL Assay Kit with Alexa Fluor® 488 anti-BrdU (Molecular Probes, Invitrogen Detection Technologies, USA, Catalog No. A23210) by flow cytometer (BD FACSCanto™, USA) equipped with BD FACSDiva, Version 6.1.2, software. Debris was excluded by forward and side-way light-scattering. Though, the positive controls provided with kit were used, but at the same time, cells receiving H2O2 (100 μM) exposure for 6 h were also used as positive controls.
Transcriptional Changes (Real-Time PCR)
Pb (10 μM) and/or ethanol (200 mM) induced alterations in the expression level of mRNA of marker genes involved in oxidative stress, and apoptosis-mediated pathways were studied using quantitative real-time PCR (RT-PCR). Total RNA was isolated from both exposed and unexposed control cells using Trizol reagent (Invitrogen). Total RNA (1 μg) was reverse transcribed into complementary (c)DNA by superscript 111 first strand cDNA synthesis kit (Catalog No 18080–05, Invitrogen Lifescience USA). RT-PCR was performed by SYBR Green dye (ABI, USA) using ABI PRISM® 7900HT sequence detection system (Applied Biosystem, USA). Real-time reactions were performed in triplicate for each sample. The specificity for each primer (Table 1) was assessed by melting curve analysis and running no template control (NITCs) for respective primers. β-Actin was used as internal control to normalize the data. Expressional changes (mRNA) are expressed in relative quantification (RQ).
Translational Changes (Western Blotting)
Alteration in the expression of marker proteins associated with oxidative stress (glutathione S-transferase A4-4 (GSTA4-4), HSP70) and apoptosis (CYP-2E1, Bax, Bcl2, PARP, p53, Cytochrome-c, and activated Caspase-9 and 3) was studied using Western blot analysis. In brief, after exposure of the cells to Pb (10 μM) and/or ethanol (200 mM), cells were pelleted and lysed using CelLytic M cell lysis reagent (Sigma) in the presence of protein inhibitor cocktail (Sigma). After protein estimation by Bradford’s reagent (Fermentas Inc., Glen Burnie, MD), equal amounts (50 μg/well) of denatured proteins were resolved onto Tricine–SDS/SDS-PAGE gel and blotted onto a polyvinylidene fluoride membrane (Millipore). After blocking, the membranes were incubated overnight at 4 °C with primary antibodies of p53, CYP-2E1 (1:1000, Millipore, USA), activated caspase-9 and 3 (1:200, Millipore, USA), GSTA4-4 (1:3000, Abcam, USA), PARP, Cytochrome-c (1:1000, CST, USA), Bax, Bcl2 (1:1000, Santa Cruz, USA), and β-actin (1:500, Millipore, USA). The membranes were washed three times with PBS and then re-incubated with horseradish peroxidase-conjugated secondary antibodies (Calbiochem, USA). The blots were developed using luminol (Catalog No. 34080, Thermo Scientific, USA), and densitometry for protein-specific bands was done in Gel Documentation System (Alpha Innotech, USA) using AlphaEase™ FC StandAlone V. 4.0.0 software. β-actin was used as internal control to normalize the data. The data are expressed in terms of fold change comparing the control.
Translocation Studies
Translocation of Bax protein from cytosol to mitochondria and cytochrome-c from mitochondria to cytosol due to exposure-induced mitochondrial membrane permeabilization was studied by Western blotting and immunocytochemical analysis using specific anti-Bax (Santa Cruz, USA) and anti-Cytochrome-c primary antibodies (CST, USA). After exposure of cells to Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), Pb (10 μM) + ethanol (200 mM) + NAC (10 μM), and NAC (10 μM) for 24 h, cells were harvested and processed for isolation of mitochondrial and cytosolic fraction using the protocols of Ghosh et al. 2002 [45] and Waterhouse et al. 2004 [46]. The cross-contamination of cytosolic protein in the mitochondrial fraction and vice versa was also assessed by running separate blots using antibodies specific to mitochondria and cytosolic proteins. Translocation proteins between cytosol and mitochondria were also confirmed by immunocytochemistry using anti-Bax- and anti-Cytochrome-c-specific monoclonal antibodies. For immunocytochemical analysis studies, cells were seeded in PLL-pre-coated eight-well chamber slides at a density of 1 × 104 cells/well. After the exposures, the cells were washed with PBS (100 μl/well) and stained with Mitotracker dye (100 nM) for 1 h at 37 °C, then fixed in 4 % paraformaldehyde. Cells were then washed twice with PBS and incubated for 15 min in PBS containing 0.02 % Triton × 100 and 0.1 % BSA to block the nonspecific binding sites. The cells were then incubated with primary antibodies of Cytochrome-c and Bax for 24 h. The antibodies were diluted in PBS containing 0.02 % Triton × 100 and 0.1 % BSA. The cells were then washed three times with PBS and re-incubated for 2 h at room temperature with secondary anti-primary immunoglobulin G (IgG) conjugated with rhodamine fluorescence dye (Calbiochem, USA). Cells were visualized under upright microscope (Nikon Eclipse 80i equipped with Nikon DS-Ri1 12.7 megapixel camera, Japan) and quantified by measuring the change in percent area of protein expression with the help of Leica Qwin 500 Image Analysis Software (Leica, Germany).
Caspase-9/3 Activity
Caspase-9/3 activity was assessed as per the protocols of the manufacturer (Biovision, Catalog no. K119 and K106, USA). After exposure to Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), NAC (10 μM), and Pb (10 μM) + ethanol (200 mM) + NAC (10 μM) for 24 h, cells were pelleted, re-suspended in pre-chilled extraction buffer (50 μl), and incubated on ice for 10 min. The samples were then centrifuged for 5 min at 500×g, and supernatant (50 μl/well) was transferred to 96-well culture plates. Assay buffer (50 μl) and substrate conjugate (5 μl) of caspase-9 and caspase-3 were added, mixed well, and incubated for 1 h at 37 °C in dark. Upon completion of incubation period, cells were mixed thoroughly and read absorbance was read at 405 nm for both caspases. The values of exposed groups were compared with unexposed control sets, and the data were expressed as a percentage of control. Cells exposed to H2O2 (100 μM) for 6 h under identical conditions were used as a positive control.
Statistical Analysis
Results were expressed as mean ± standard error of mean (SEM) for the values obtained from at least three independent experiments. Statistical analysis was performed using one-way analysis of variance (ANOVA) and Dunnett’s multiple comparison test using GraphPad Prism (Version 5.0) software. The valves */# p < 0.05 were considered as significant, **/## p < 0.01 more significant, and ***/### p < 0.001 highly significant.
Results
Cytotoxicity Studies
A concentration-dependent decrease in the percent cell viability was observed in PC12 cells exposed to Pb (10–200 μM) for 24 h. Pb concentration at 10 μM was found to be non-cytotoxic. The higher concentrations of Pb, i.e., 50, 100, and 200 μM, have shown statistically significant reduction in percent cell viability (Fig. 1a). Exposure to ethanol up to 100 mM did not elicit any cytotoxic response. Thereafter, there was a statistically significant dose-dependent decrease in the percent cell viability (Fig. 1b). Based on the cytotoxicity data of individual toxicants, further experiments were carried out to evaluate the effect of co-exposure to Pb (10 μM) and ethanol (200 mM) in PC12 cells. The co-exposure to Pb and ethanol showed an additive effect on the loss of cell viability in comparison to individual exposure to Pb and ethanol at the same concentration (Fig. 1c).

Percent cell viability assessed by MTT assay in PC 12 cells a Cells exposed to Pb (0.1 to 200 μM) for 24 h. b cells exposed to ethanol (50–600 mM) for 24 h. *p < 0.05, **p < 0.01, ***p < 0.001. c Cells exposed to Pb (10 μM) and/or ethanol (200 mM) for 24 h. */# p < 0.05, **/## p < 0.01, ***/### p < 0.001;*p < 0.05, **p < 0.01, *** p < 0.001 (unexposed control vs. experimental group); # p < 0.05, ## p < 0.01, ### p < 0.001 (compared between treated groups)
Intracellular Calcium Levels [Ca2+]i
Both Pb and ethanol induce a significant increase in the levels of [Ca2+]i at each time point studied, with maximum levels at 30 and 45 min for Pb and ethanol exposure, respectively. There was an additive effect in cells co-exposed to Pb and ethanol, and this additive effect increased gradually till 45 min. At 60 min, the trend was similar but the magnitude of increase in the levels of [Ca2+]i was lower than that at 45 min. In general, the level of increase was statistically significant in all the cases, i.e., Pb (p < 0.05), ethanol (p < 0.05), and co-exposure to Pb and ethanol (p < 0.001) (Fig. 2).

Measurement of intracellular Ca2+ levels (nmol/l) in PC 12 cells. Cells exposed to Pb (10 μM) and/or ethanol (200 mM) at various time periods from 5 to 60 min. *p < 0.05, **p < 0.01, ***p < 0.001 (unexposed control vs. experimental group)
ROS Generation
Results of Pb- and/or ethanol-induced ROS generation in PC12 cells are depicted in Fig. 3. Fluorescence microscopy studies showed that PC12 cells receiving co-exposures of Pb 10 μM + ethanol 200 mM have a significantly high (p < 0.05) ROS production in comparison to the individual exposures to Pb (10 μM) and ethanol (200 mM). Similarly, the positive control (H2O2 100 μM) also induced significant (p < 0.01) ROS generation as compared to control. However, NAC (10 μM) (a known antioxidant) exposure showed a significant (p < 0.05) restoration in ROS levels, when compared with Pb 10 μM + ethanol 200 mM exposure group (Fig. 3a, b).

ROS generation measurement by fluorescence microscope using 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) dye. a Cells (1 × 106) received exposure of Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), NAC (10 μM), and Pb (10 μM) + ethanol (200 mM) + NAC (10 μM) for 24 h. Following the exposure, cells were incubated with DCFH-DA (20 μM) for 30 min at 37 °C, and then, microphotographs were taken by Nikon phase contrast cum fluorescence microscope (model 80i), and for fluorescence, quantification was done using Leica Q win500 image analysis Software. b Relative quantification of fold induction in ROS generation by intracellular image analysis, receiving the exposure of Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), NAC (10 μM), and Pb (10 μM) + ethanol (200 mM) + NAC (10 μM) at 24 h. Cells exposed to H2O2 (100 μM) for 6 h under identical experimental conditions and served as positive control. */# p < 0.05, **/## p < 0.01, ***/### p < 0.001,* = p < 0.05; **p < 0.01, ***p < 0.001 (unexposed control vs. experimental group); # p < 0.05, ## p < 0.01, ### p < 0.001 (compared between treated groups)
Glutathione Levels
A significant reduction in the levels of GSH was observed in all groups receiving individual and co-exposures to Pb and ethanol when compared with unexposed control sets. Ethanol 200 mM showed significant (p < 0.01) additive effect in the Pb (10 μM)-induced alterations in GSH levels, while NAC treatment showed significant (p < 0.05) restoration in GSH levels when compared with Pb 10 μM + ethanol 200 mM exposure group. As anticipated, a highly significant (p < 0.01) reduction in GSH levels was recorded in cells exposed to the positive control (H2O2 100 μM) group (Fig. 4a).

Analysis of oxidative stress marker glutathione and lipid peroxidation. a Levels of glutathione (GSH) expressed in nanomoles per milligram protein following the exposure of Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), Pb (10 μM) + ethanol (200 mM) + NAC (10 μM), and NAC (10 μM) after 24 h. b Levels of lipid peroxidation (LPO) expressed in nanomoles per 10 mg protein following the exposure of Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), Pb (10 μM) + ethanol (200 mM) + NAC (10 μM), and NAC (10 μM) after 24 h. */# p < 0.05, **/## p < 0.01, ***/### p < 0.001, *p < 0.05; **p < 0.01, ***p < 0.001 (unexposed control vs. experimental group); # p < 0.05, ## p < 0.01, ### p < 0.001 (compared between treated groups)
Lipid Peroxidation
The results indicate that co-exposure to Pb (10 μM) + ethanol (200 mM) significantly adds to the individual exposure to Pb (10 μM) or ethanol (200 mM) induced increase in LPO. But, the level of LPO was reduced following the exposure to Pb (10 μM) + ethanol (200 mM) with antioxidant (NAC 10 μM) in comparison to LPO induction due to Pb (10 μM) + ethanol (200 mM) exposure. Moreover, cells exposed to H2O2 (10 μM) for 6 h have also shown highly significant increase in the levels of LPO (Fig. 4b).
Mitochondrial Membrane Potential
Flow cytometric analysis clearly showed that co-exposure to Pb (10 μM) and ethanol (200 mM) induced maximum reduction (19.3 %) in mitochondrial membrane potential (MMP) compared with individual exposures to Pb 10 μM (15.7 %) and ethanol 200 mM (7.4 %), respectively. On the other hand, NAC (10 μM) reduced the adverse effect of Pb (10 μM) + ethanol (200 mM) by 15.8 % when given together. Cells exposed to H2O2 (100 μM) for 6 h have also shown a significant reduction (17.0 %) in MMP (Fig. 5)

Apoptotic analysis by Mitolight™ Apoptosis Detection Kit (APT142, Chemicon, USA). a Unstained cells. b Experimental positive control (H2O2 100 μM). c Control. d Pb 10 μM. e Ethanol 200 mM. f Pb 10 μM + Ethanol 200 mM. g. Pb 10 μM + Ethanol 200 mM + NAC 10 μM. h NAC 10 μM. Briefly, following the exposure of Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), Pb (10 μM) + ethanol (200 mM) + NAC (10 μM), and NAC (10 μM) for 24 h, cells were re-suspended in 1 ml of pre-diluted Mitolight™ solution for 15 min at 37 °C. Cells were then immediately analyzed by flow cytometer (BD FACSCanto™) equipped with the FACSDiva Version 6.0.0. software
TUNEL Assay
Pb- and/or ethanol-induced apoptosis was also assessed using the TUNEL assay with the help of FACS analysis (Fig. 6). Exposure to Pb and ethanol separately was found to induce apoptosis of 7.9 and 3.9 %. These changes were synergistic in case of co-exposure to Pb and ethanol, i.e., 12 %. Pb and ethanol induced apoptotic changes, which could be reverted back towards the basal level (6.8 %) in cells simultaneously exposed to NAC (10 μM) (Fig. 6)

DNA damage analysis in PC12 cells using APO-BrdU™ TUNEL (deoxynucleotide transferase dUTP nick end labeling) Assay Kit with Alexa Fluor® anti-BrdU (Molecular Probes, Invitrogen detection Technologies, USA, Cat No.# A23210) by a flow cytometer (BD FACSCanto™, USA) equipped with BD FACSDiva, Version 6.1.2, software. a Positive control (human lymphoma cells) provided with the kit. b Experimental positive control (H2O2 100 μM). c Control. d Pb 10 μM. e. Ethanol 200 mM. f Pb 10 μM + Ethanol 200 mM. g Pb 10 μM + Ethanol 200 mM + NAC 10 μM. h NAC 10 μM
Transcriptional Changes
Pb and/or ethanol exposure-induced alterations in the mRNA expression of marker genes associated with oxidative stress and apoptosis and are presented in Fig. 7. Though, the individual exposures to Pb and ethanol have shown significant upregulation of oxidative stress and pro-apoptotic genes and downregulation of anti-apoptotic genes, the magnitude of these alterations was significantly increased in the cells co-exposed to Pb (10 μM) + ethanol (200 mM), i.e., p53 4.14-, Bax 3.33-, caspase-9 9.69-, caspase-3 4.15-, CYP-2E1 7.95-, GSTA4-4 10.84-, Hsp70 3.75-, and Bcl2 0.376-fold of control, respectively.

mRNA expression profiling of marker genes associated with oxidative stress DNA damage and apoptosis in PC12 cells following the exposure of Pb (10 μM) and/or ethanol (200 mM) for 24 h. Quantitative real-time PCR (RT-PCRq) was performed in triplicate by SYBR Green dye using ABI PRISM® 7900HT Sequence Detection System (Applied Biosystems, USA). β-Actin was used as internal control to normalize the data, and changes in mRNA expression are expressed in relative quantity compared with respective unexposed control groups
Translational Changes
The results of Western blot analysis to evaluate the altered expressions of marker proteins of oxidative stress and apoptosis are presented in Fig. 8. Co-exposure to Pb and ethanol effectively upregulated the protein expression of p53 (2.78-fold), cytochrome-c (2.26-fold), Bax (1.87-fold), activated caspase-9 (1.68-fold,) activated caspase-3 (1.67-fold), CYP-2E1 (1.93-fold), GSTA4-4 (1.97-fold), and HSP-70 (1.81-fold) and downregulated Bcl2 (0.51-fold) of respective controls. Protein expression for poly (ADP-ribose) polymerase (PARP) showed the fragments of cleavage of 116 and 89 kDa proteins exposed to toxic insult, which was higher in co-exposure to Pb and ethanol in comparison to its individual groups. There was linearity in the data at both transcriptional and translational levels in co-exposure as well as in individual exposure groups.

Protein expression profiling of marker genes associated with oxidative stress, DNA damage, and apoptosis in PC12 cells following the exposure Pb (10 μM) and/or ethanol (200 mM) for 24 h. β-Actin was used as internal control to normalize the data. Lanes A untreated control, B Pb 10 μM, C Ethanol 200 mM, and D Pb 10 μM + ethanol 200 mM, respectively. Molecular weight of protein studied: p53 (53 kDa), Cytochrome-c (12 kDa), Bax (23 kDa), Bcl2 (29 kDa), activated caspase-9 (37 kDa), activated caspase-3 (17 kDa), CYP-2E1 (50 kDa), HSP70 (70 kDa), PARP-Cleavage (116 and 89 kDa), and β-actin (42 kDa) for normalization. Quantification was done in Gel Documentation System (Alpha Innotech, USA) with the help of AlphaEase™ FC StandAlone V.4.0 software
Translocation of Bax and Cytochrome-c
The involvement of mitochondria-mediated apoptotic pathways in PC12 cells co-exposed to Pb and ethanol was confirmed by the translocation of Bax protein from cytosol to mitochondria and cytochrome-c protein from mitochondria to cytoplasm using the Western blot analysis and immunocytochemical localization. There was a significantly higher translocation of proteins between mitochondria and cytoplasm in cells co-exposed to Pb and ethanol, when compared with magnitude of translocation following individual exposure to Pb or ethanol. The NAC treatment was found to be significantly effective in reducing the Pb and ethanol co-exposure-mediated translocation of Bax and cytochrome-c between mitochondria and cytoplasm. The high purity of both cytosolic and mitochondrial fractions could be ascertained as there was no cross-contamination of the cytosol- and mitochondria-specific proteins. The observations recorded through Western blot analysis and immunocytochemistry were found to have similar trends and linearity (Fig. 9a, b).

a Translocation of cytoplasmic Bax protein to mitochondria and mitochondrial cytochrome-c protein to cytoplasm in PC12 cells following the exposure of Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), Pb (10 μM) + ethanol (200 mM) + NAC (10 μM), and NAC (10 μM) for 24 h. A Untreated control. B Pb 10 μM. C Ethanol 200 mM. D Pb 10 μM + ethanol 200 m. E Pb 10 μM + ethanol 200 mM + NAC 10 μM. F NAC 10 μM. β-Actin and V-DAC were used to assess the purity of cytosolic and mitochondrial fraction. b Representative microphotographs showing translocation of cytoplasmic Bax protein to mitochondria and mitochondrial cytochrome-c protein to cytoplasm in PC12 cells following the exposure of Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), Pb (10 μM) + ethanol (200 mM) + NAC (10 μM), and NAC (10 μM) for 24 h. A Untreated control. B Pb 10 μM. C Ethanol 200 mM. D Pb 10 μM + ethanol 200 mM. E Pb 10 μM + ethanol 200 mM + NAC 10 μM. F NAC 10 μM. The images were snapped by Nikon DS-Ri1 (12.7 megapixel) camera using upright phase contrast plus fluorescence microscope (Nikon 80i, Japan) at ×10 × 100 oil immersion magnification. Mitochondria stained with MitoTracker green (Mt-green) and nuclei stained with DAPI
Activity of Caspase-9/3
Highlights of Pb- and/or ethanol-induced alterations in the activity of caspase-9/3 and ameliorative responses of NAC are shown in Fig. 10a, b. The activity of both caspase-9 and caspase-3 was found to be increased significantly in all the exposure groups when compared with the control group. The magnitude of induction was significantly higher (p < 0.01) in cells co-exposed to Pb and ethanol. Similarly, the positive control (H2O2 100 μM) also induced the activity of caspase-9 and caspase-3 significantly (p < 0.01) in comparison to control. NAC was found to be effective in ameliorating the activity of caspase activity significantly (p < 0.05), as NAC (10 μM) significantly brought down the caspase-9 and caspase-3 activity in cells co-exposed to Pb and ethanol, which was comparable to cells receiving individual exposure of either of Pb or ethanol (Fig. 10a, b).

Induction in the activity of caspase-9/3 in PC12 cells following the exposure of Pb (10 μM), ethanol (200 mM), Pb (10 μM) + ethanol (200 mM), NAC (10 μM), and Pb (10 μM) + ethanol (200 mM) + NAC (10 μM) for 24 h. a Induction in the activity of caspase-9. b Induction in the activity of caspase-3. Cells exposed to H2O2 (100 μM) for 6 h under identical conditions were used as a positive control. */# p < 0.05, **/## p < 0.01, ***/### p < 0.001,*p < 0.05; **p < 0.01, ***p < 0.001 (unexposed control vs. experimental group); # p < 0.05, ## p < 0.01, ### p < 0.001 (compared between treated groups)
Discussion
The working hypothesis for the present investigation was to study, whether the co-exposure of non-cytotoxic concentrations of Pb and ethanol leads to oxidative stress which further precedes apoptotic condition in PC12 cells, a most representative cell line used in neurotoxicity studies. The reason for selecting the combination of Pb and ethanol was due to available reports on the influence of alcohol consumption on Pb intoxication [39] and more precisely to neurotoxicity and developmental neurotoxicity [4]. The association between Pb exposure and alcohol consumption in industrial workers has already been demonstrated [47]. It is also reported that oxidative stress is one of the molecular mechanisms for Pb toxicity. Pb increases the levels of ROS along with intracellular calcium [Ca2+]i which in turn causes a fall in the mitochondrial potential leading to apoptosis via cytochrome-c release [14–16]. Chronic alcohol exposure has been reported to induce cognitive impairment with permanent structural brain damage by affecting the neuronal cells of both central and peripheral nervous systems [27–29]. Such alcohol-mediated neurodegeneration results in loss of neurons and oligodendroglia by decreasing the expression of genes associated with myelination, insulin-responsiveness and mitochondrial functions, and increased indices of apoptosis, oxidative stress, mitochondrial dysfunction, lipid peroxidation, DNA damage, and deregulated acetylcholine homeostasis [30–33].
We observed an increase in the production of [Ca2+]i, ROS, and associated oxidative stress in cells receiving either Pb or ethanol individually. However, cells co-exposed to Pb and ethanol demonstrated more pronounced levels of [Ca2+]i, ROS production, and oxidative stress. Pb is reported to induce oxidative stress by inhibiting the enzymatic activity by its binding to the active thiol site, while ethanol-induced oxidative stress results due to conversion of ethanol into acetaldehyde [48]. Based on the results, it may be concluded that Pb and ethanol synergistically induce the adverse effects. The synergistic response for oxidative stress under the influence of co-exposure to Pb and ethanol in experimental animals has been reported to be associated with increased production of ROS [49]. ROS are oxygen-containing molecules, such as hydrogen peroxide (H2O2), superoxide anion (O2 −), and hydroxyl radical (HO•) that are involved in redox signaling. To confirm the ROS redox signaling under identical experimental conditions, PC12 cells were exposed to H2O2 (100 μM for 6 h), which served as positive control. H2O2-induced ROS production was similar as in the case of Pb and/or ethanol, suggesting involvement of similar signal transduction pathways. The significant reduction in ROS in the presence of a known antioxidant, NAC, with co-exposure to Pb and ethanol in PC12 cells, further confirms the involvement of ROS in toxicity. NAC works as a GSH precursor, and is an important cellular antioxidant and plays a major role in protecting the cells against oxidative stress through direct scavenging of ROS [50]. NAC treatment provided a sufficiently strong antioxidant defense system, resulting in restoring LPO and GSH of cells exposed to a combination of Pb and ethanol. A similar kind of effect had also been noticed in SHSY5Y cell line, which was treated with the antioxidant NAC against arsenic and dopamine [51]. Thus, the present study demonstrated that the cytotoxicity induced by the co-exposure to Pb and ethanol could be attenuated by antioxidant NAC (data not shown), suggesting that increased ROS generation and reduction in antioxidant defense system are responsible for the decreased cell viability.
The abundant amount of oxygen supply to the brain and high lipid contents in the cells make brain cells more susceptible to oxidative stress-mediated toxicity of Pb and ethanol. The limited regeneration and repair capacities of neuronal cells also add to that. A dose-dependent loss in cell viability was observed after independent exposure to Pb and ethanol. The magnitude of viability loss was significantly increased upon co-exposure to both Pb and ethanol. This increase in the loss of cell viability could be correlated well with the increased production of ROS and subsequently associated events of oxidative stress and apoptotic end points. The thiol binding affinity of Pb is one among the important underlying mechanisms responsible for its adverse effect on different cellular and sub-cellular molecules. GSH, a low-molecular-weight non-enzymatic thiol containing antioxidant compound which is synthesized in the cells in abundance, is known to be affected by Pb exposure [52]. GSH plays a key role in protecting cells from oxidative damage and toxicity of xenobiotics and maintaining redox homeostasis. Though, we observed depletion of GSH in all the experimental groups receiving Pb and/or ethanol, but the magnitude of depletion was statistically greater in cells co-exposed to both Pb and ethanol. Ethanol is known to deplete GSH via acetaldehyde-mediated adduct formation. So, the depletion of GSH levels in PC12 cells observed in the present investigation could be attributed to the increased utilization of GSH-GPx in detoxification of H2O2 generated by Pb and ethanol.
Though, the brain is capable of handling of increased levels of ROS by a number of cellular defense systems to lower down the concentration of free radical species and repair oxidative cellular damage. But, increased production of ROS may cause permeabilization of blood-brain barrier (BBB), which subsequently inhibits the mitochondrial respiration and known to induce progressive lipid peroxidation [38, 49]. In the present investigations, a significant increase in LPO and associated reduction in mitochondrial membrane potential was noticed in the cells exposed to Pb and/or ethanol. Similar to other end points, co-exposure to Pb and ethanol was showing a synergism in the increased lipid peroxidation and reduction in MMP.
The association of oxidative stress and apoptosis induction in the brain has been reported as indicator of neurotoxic response to both Pb and alcohol [53]. The results of TUNEL assay with labeled cells confirming DNA fragmentation also support our finding of ROS-induced apoptosis. We and others have already reported the involvement of mitochondrial chain complexes in ROS-induced apoptotic changes in cytoplasm [54]; however, such apoptotic changes are known to follow different pathways [55–59]. We observed that exposure to Pb and/or ethanol significantly upregulated the expression (both mRNA and protein) of caspase-3, caspase-9, Bax, p53, and Cytochrome-c and downregulated Bcl2. In general, transcriptional changes were well coordinated with translational changes and with physiological activity in the case of caspase-3 and caspase-9. Recent literature has shown that upregulation of cytosolic p53 protein interacts with mitochondria, thereby promoting mitochondrial membrane permeabilization [60], and plays a key role in the regulation of apoptosis [61]. This cytosolic p53 protein has been suggested to induce pro-apoptotic members of the Bcl2 family such as Bax and its displacement with anti-apoptotic Bcl2 protein [55]. However, induction of nuclear p53 protein is involved in preventing genotoxicity by inducing transcriptional reprogramming, which finally leads to controlled cell death [62, 63]. Thus, the alterations in the expression profile of marker genes in this study indicate that cytosolic p53 triggers the mitochondrial apoptotic cascade in PC12 cells exposed to Pb and/or ethanol. Moreover, activation of PARP also suggests the activation of DNA repair by nuclear p53 in cells exposed to Pb and/or ethanol.
In the present investigations, a synergistic increase in the translocation of Bax protein from cytosol to mitochondria and cytochrome-c protein from mitochondria to cytosol was observed in cells exposed to Pb and ethanol, when compared with exposures to Pb or ethanol alone. Altogether, these findings confirm the induction of cytosolic p53 and its role in triggering the pathway of mitochondrial membrane permeability in PC12 cells as an important phase in the induction of mitochondria-mediated apoptosis [64]. In pro-apoptotic phase, embedment of Bax in the mitochondrial membrane creates permeable channels which permit larger protein molecules to pass through them. This event leads to mitochondrial leakage from inner membrane followed by greater ROS generation, uncoupling of oxidation phosphorylation, and breakdown of mitochondrial membrane potential [65]. These observations are in agreement with the earlier reported studies with Pb and alcohol individually, but the present investigations provide the first experimental evidence of synergistic response of co-exposure to Pb and ethanol in PC12 cells.
Release of cytochrome-c [66] from mitochondria in the cytosol activates caspase-9 [67], which further activates caspase-3. Such activated caspase-3 executes the catabolic caspase cascade that eventually results in cell death. Activation of caspase cascade may occur through two different pathways, i.e., either by binding of procaspase-9 with Apaf-1 to form the apoptosome complex after the release of cytochrome-c from damaged mitochondria [68] or by activation of caspases through the OMI and SMACs molecules [69]. The expression of these changes also correlates with activity of caspase-9 and caspase-3 obtained in the present investigations which are indicative of mitochondria-mediated apoptosis that operates through caspase cascade.
Ethanol is reported to induce impairment in blood-brain barrier (BBB) [22]; hence, synergistic response of co-exposure to Pb and ethanol may be due to accumulation of Pb in the brain due to impaired BBB. Ethanol and its metabolites are also known to stimulate inositol 1,4,5-triphosphate receptor [IP(3)R)-operated intracellular calcium (Ca(2+)] release, which ultimately leads deficiency of calcium [70]. Ethanol is also reported to alter the distribution and metabolism of Pb [39]. Though, the mechanisms may be different but both Pb and ethanol induce oxidative stress and apoptosis in PC12 cells.
The activation of cytochrome P450s and their interaction with mitochondrial chain complexes have been suggested in chemical-induced apoptosis [55, 57]. We observed a significant upregulation in the expression of CYP-2E1 after exposure to Pb and/or ethanol, which was found to be up-streamed to ROS generation in PC12 cells. Such upregulated CYP-2E1 might have played an important role in the production of reactive oxygenated molecules (ROMs), which are known to induce ROS generation [71], LPO [72], and GSTs [73] and finally leads to apoptosis [74]. In the present investigations, apoptosis induction and oxidative stress were found to be associated with induction of CYP-2E1. Induced CYP-2E1 was found to cause oxidative stress by reduction of the intracellular GSH levels [75], activation of the p38 MAP kinase pathway, and induction of the transcription factor Nrf2 [76], in human hepatoma cell line HepG2. The role of CYP-2E1 has been suggested in alcohol-induced oxidative DNA damage in liver of null mice [77]. Pb- and ethanol-induced alterations in the expression profile of CYP-2E1 could be correlated with an earlier study showing CYP-2E1 induction by 4-hydroxynonenal (4-HNE), a product of lipid peroxidation [78], and organophosphate pesticide-monocrotophos in PC12 cells [79]. GSTs, a family of phase II enzymes, participate in detoxification of xenobiotics from cells. 4-HNE, a by-product of lipid peroxidation, conjugates with GSH, specifically by glutathione S-transferase A4-4 (GSTA4-4), which has been shown as a major pathway for 4-HNE neutralization [78]. There are reports demonstrating the protective role of GSTA4-4 in various cell systems having toxic insults due to a variety of environmental chemicals and metabolites [80, 81]. We also observed a similar pattern of elevated levels of GSTA4-4 in PC12 cells exposed to Pb and/or ethanol. Moreover, upregulation of GSTA4-4 expression may be the defense mechanism to protect against damage caused by 4-HNE and other by-products formed via increased lipid peroxidation induced by Pb and/or ethanol treatment. Heat shock proteins (HSPs) contain several different families of proteins, and their expressions are induced in response to wide variety of physiological and environmental insults. One such protein is HSP70, which is induced under high stress condition. HSP70 protects cells from stress-induced caspase-dependent apoptosis and facilitates the survival of cells against toxic stimuli [82]. Our study shows that Pb or ethanol significantly increased HSP70 expression both at mRNA and protein levels; however, combined treatment with both chemicals resulted in more pronounced HSP70 induction, suggesting greater magnitude of stress in the cells.
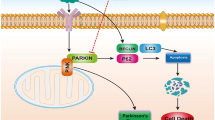
Based on the findings, we propose a schematic model on the synergism in apoptotic effects of co-exposure to Pb and ethanol to cells (Fig. 11). The co-exposure to Pb and ethanol first triggers intrinsic generation of [Ca2+]i and ROS, which subsequently initiates oxidative damage. The increased ROS levels and genotoxicity upregulate p53 and attenuate Bcl2 protein, leading to a declined ratio of Bcl-2/Bax. The alteration in Bcl2/Bax ratio results in the release of cytochrome-c. Finally, the release of cytochrome-c activates caspase-9 and subsequently triggers the caspase-3 cascade leading to apoptosis in PC12 cells. The present investigations conclude that PC12 cells exposed to both Pb and ethanol show more pronounced increase in oxidative stress and apoptosis than to the independent exposures to Pb and ethanol.

Schematic flow diagram to represent apoptotic pathway involved in PC12 cells exposed to Pb and/or ethanol
References
Briggs D (2003) Environmental pollution and the global burden of disease. Br Med Bull 68:1–24
Tong S, von Schirnding YE, Prapamontol T (2000) Environmental lead exposure: a public health problem of global dimensions. Bull World Health Organ 78:1068–1077
Florea AM, Busselberg D (2006) Occurrence, use and potential toxic effects of metals and metal compounds. Biometals 19:419–427
Giordano G, Costa LG (2012) Developmental neurotoxicity: some old and new issues. ISRN Toxicol 2012:814795
Zheng W, Aschner M, Ghersi-Egea JF (2003) Brain barrier systems: a new frontier in metal neurotoxicological research. Toxicol Appl Pharmacol 192:1–11
Kolesarova A, Roychoudhury S, Slivkova J, Sirotkin A, Capcarova M, Massanyi P (2010) In vitro study on the effects of lead and mercury on porcine ovarian granulosa cells. J Environ Sci Health A Tox Hazard Subst Environ Eng 45:320–331
Mrugesh T, Dipa L, Manishika G (2011) Effect of lead on human erythrocytes: an in vitro study. Acta Pol Pharm 68:653–656
Conterato GM, Augusti PR, Somacal S, Einsfeld L, Sobieski R, Torres JR, Emanuelli T (2007) Effect of lead acetate on cytosolic thioredoxin reductase activity and oxidative stress parameters in rat kidneys. Basic Clin Pharmacol Toxicol 101:96–100
Dewanjee S, Sahu R, Karmakar S, Gangopadhyay M (2013) Toxic effects of lead exposure in Wistar rats: involvement of oxidative stress and the beneficial role of edible jute (Corchorus olitorius) leaves. Food Chem Toxicol 55:78–91
Hu H, Shih R, Rothenberg S, Schwartz BS (2007) The epidemiology of lead toxicity in adults: measuring dose and consideration of other methodologic issues. Environ Health Perspect 115:455–462
Lidsky TI, Schneider JS (2003) Lead neurotoxicity in children: basic mechanisms and clinical correlates. Brain 126:5–19
Lu X, Jin C, Yang J, Liu Q, Wu S, Li D, Guan Y, Cai Y (2013) Prenatal and lactational lead exposure enhanced oxidative stress and altered apoptosis status in offspring rats’ hippocampus. Biol Trace Elem Res 151:75–84
Sanders T, Liu Y, Buchner V, Tchounwou PB (2009) Neurotoxic effects and biomarkers of lead exposure: a review. Rev Environ Health 24:15–45
Chen L, Yang X, Jiao H, Zhao B (2003) Tea catechins protect against lead-induced ROS formation, mitochondrial dysfunction, and calcium dysregulation in PC12 cells. Chem Res Toxicol 16:1155–1161
Flora SJ, Saxena G, Mehta A (2007) Reversal of lead-induced neuronal apoptosis by chelation treatment in rats: role of reactive oxygen species and intracellular Ca(2+). J Pharmacol Exp Ther 322:108–116
He L, Poblenz AT, Medrano CJ, Fox DA (2000) Lead and calcium produce rod photoreceptor cell apoptosis by opening the mitochondrial permeability transition pore. J Biol Chem 275:12175–12184
Aimo L, Oteiza PI (2006) Zinc deficiency increases the susceptibility of human neuroblastoma cells to lead-induced activator protein-1 activation. Toxicol Sci 91:184–191
Patra RC, Rautray AK, Swarup D (2011) Oxidative stress in lead and cadmium toxicity and its amelioration. Vet Med Int 2011:457327
Hernandez M, Margalida A (2009) Assessing the risk of lead exposure for the conservation of the endangered Pyrenean bearded vulture (Gypaetus barbatus) population. Environ Res 109:837–842
Brust JC (2010) Ethanol and cognition: indirect effects, neurotoxicity and neuroprotection: a review. Int J Environ Res Public Health 7:1540–1557
Crews FT, Braun CJ, Hoplight B, Switzer RC 3rd, Knapp DJ (2000) Binge ethanol consumption causes differential brain damage in young adolescent rats compared with adult rats. Alcohol Clin Exp Res 24:1712–1723
Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y (2005) Alcohol-induced oxidative stress in brain endothelial cells causes blood–brain barrier dysfunction. J Leukoc Biol 78:1223–1232
Singh AK, Jiang Y, Gupta S, Benlhabib E (2007) Effects of chronic ethanol drinking on the blood brain barrier and ensuing neuronal toxicity in alcohol-preferring rats subjected to intraperitoneal LPS injection. Alcohol Alcohol 42:385–399
Costa LG, Guizzetti M, Burry M, Oberdoerster J (2002) Developmental neurotoxicity: do similar phenotypes indicate a common mode of action? A comparison of fetal alcohol syndrome, toluene embryopathy and maternal phenylketonuria. Toxicol Lett 127:197–205
Goodlett CR, Horn KH, Zhou FC (2005) Alcohol teratogenesis: mechanisms of damage and strategies for intervention. Exp Biol Med (Maywood) 230:394–406
Mulholland PJ, Self RL, Stepanyan TD, Little HJ, Littleton JM, Prendergast MA (2005) Thiamine deficiency in the pathogenesis of chronic ethanol-associated cerebellar damage in vitro. Neuroscience 135:1129–1139
Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, Chandler LJ (2012) Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS ONE 7:e37541
Ridley NJ, Draper B, Withall A (2013) Alcohol-related dementia: an update of the evidence. Alzheimers Res Ther 5:3
Tiwari V, Kuhad A, Chopra K (2009) Suppression of neuro-inflammatory signaling cascade by tocotrienol can prevent chronic alcohol-induced cognitive dysfunction in rats. Behav Brain Res 203:296–303
de la Monte SM, Kril JJ (2014) Human alcohol-related neuropathology. Acta Neuropathol 127:71–90
Flanagan DE, Pratt E, Murphy J, Vaile JC, Petley GW, Godsland IF, Kerr D (2002) Alcohol consumption alters insulin secretion and cardiac autonomic activity. Eur J Clin Invest 32:187–192
Wang X, Ke Z, Chen G, Xu M, Bower KA, Frank JA, Zhang Z, Shi X, Luo J (2012) Cdc42-dependent activation of NADPH oxidase is involved in ethanol-induced neuronal oxidative stress. PLoS ONE 7:e38075
Ramachandran V, Watts LT, Maffi SK, Chen J, Schenker S, Henderson G (2003) Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J Neurosci Res 74:577–588
Kamens HM, Phillips TJ (2008) A role for neuronal nicotinic acetylcholine receptors in ethanol-induced stimulation, but not cocaine- or methamphetamine-induced stimulation. Psychopharmacology (Berl) 196:377–387
Probst-Hensch N, Braun-Fahrlaender C, Bodenmann A, Ackermann-Liebrich U (1993) Alcohol consumption and other lifestyle factors: avoidable sources of excess lead exposure. Soz Praventivmed 38:43–50
Telisman S, Cvitkovic P, Jurasovic J, Pizent A, Gavella M, Rocic B (2000) Semen quality and reproductive endocrine function in relation to biomarkers of lead, cadmium, zinc, and copper in men. Environ Health Perspect 108:45–53
Flora SJ, Kumar D, Sachan SR, Das Gupta S (1991) Combined exposure to lead and ethanol on tissue concentration of essential metals and some biochemical indices in rat. Biol Trace Elem Res 28:157–164
Flora SJ, Mittal M, Mehta A (2008) Heavy metal induced oxidative stress & its possible reversal by chelation therapy. Indian J Med Res 128:501–523
Gupta V, Gill KD (2000) Influence of ethanol on lead distribution and biochemical changes in rats exposed to lead. Alcohol 20:9–17
Kupraszewicz E, Brzoska MM (2013) Excessive ethanol consumption under exposure to lead intensifies disorders in bone metabolism: a study in a rat model. Chem Biol Interact 203:486–501
Agrawal M, Kumar V, Kashyap MP, Khanna VK, Randhawa GS, Pant AB (2011) Ischemic insult induced apoptotic changes in PC12 cells: protection by trans resveratrol. Eur J Pharmacol 666:5–11
Agrawal M, Kumar V, Singh AK, Kashyap MP, Khanna VK, Siddiqui MA, Pant AB (2013) trans-Resveratrol protects ischemic PC12 Cells by inhibiting the hypoxia associated transcription factors and increasing the levels of antioxidant defense enzymes. ACS Chem Neurosci 4:285–294
Akhtar MJ, Ahamed M, Kumar S, Siddiqui H, Patil G, Ashquin M, Ahmad I (2010) Nanotoxicity of pure silica mediated through oxidant generation rather than glutathione depletion in human lung epithelial cells. Toxicology 276:95–102
Siddiqui MA, Kashyap MP, Kumar V, Al-Khedhairy AA, Musarrat J, Pant AB (2010) Protective potential of trans-resveratrol against 4-hydroxynonenal induced damage in PC12 cells. Toxicol In Vitro 24:1592–1598
Ghosh S, Patel N, Rahn D, McAllister J, Sadeghi S, Horwitz G, Berry D, Wang KX, Swerdlow RH (2007) The thiazolidinedione pioglitazone alters mitochondrial function in human neuron-like cells. Mol Pharmacol 71:1695–1702
Waterhouse NJ, Steel R, Kluck R, Trapani JA (2004) Assaying cytochrome C translocation during apoptosis. Methods Mol Biol 284:307–313
Cezard C, Demarquilly C, Boniface M, Haguenoer JM (1992) Influence of the degree of exposure to lead on relations between alcohol consumption and the biological indices of lead exposure: epidemiological study in a lead acid battery factory. Br J Ind Med 49:645–647
Molina MF, Sanchez-Reus I, Iglesias I, Benedi J (2003) Quercetin, a flavonoid antioxidant, prevents and protects against ethanol-induced oxidative stress in mouse liver. Biol Pharm Bull 26:1398–1402
Flora SJS, Gautam P, Kushwaha P (2012) Lead and ethanol co-exposure lead to blood oxidative stress and subsequent neuronal apoptosis in rats. Alcohol and alcoholism:agr152
Suntres ZE (2002) Role of antioxidants in paraquat toxicity. Toxicology 180:65–77
Shavali S, Sens DA (2008) Synergistic neurotoxic effects of arsenic and dopamine in human dopaminergic neuroblastoma SH-SY5Y cells. Toxicol Sci 102:254–261
Bolin CM, Basha R, Cox D, Zawia NH, Maloney B, Lahiri DK, Cardozo-Pelaez F (2006) Exposure to lead and the developmental origin of oxidative DNA damage in the aging brain. FASEB J 20:788–790
Fazakas Z, Lengyel Z, Nagymajtenyi L (2005) Combined effects of subchronic exposure to lead, mercury and alcohol on the spontaneous and evoked cortical activity in rats. Arhiv za higijenu rada i toksikologiju 56:249–256
Kashyap MP, Singh AK, Siddiqui MA, Kumar V, Tripathi VK, Khanna VK, Yadav S, Jain SK, Pant AB (2010) Caspase cascade regulated mitochondria mediated apoptosis in monocrotophos exposed PC12 cells. Chem Res Toxicol 23:1663–1672
Galluzzi L, Morselli E, Kepp O, Tajeddine N, Kroemer G (2008) Targeting p53 to mitochondria for cancer therapy. Cell Cycle 7:1949–1955
Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87:99–163
Namazi MR (2009) Cytochrome-P450 enzymes and autoimmunity: expansion of the relationship and introduction of free radicals as the link. J Autoimmune Dis 6:4
Poplawski T, Pawlowska E, Wisniewska-Jarosinska M, Ksiazek D, Wozniak K, Szczepanska J, Blasiak J (2009) Cytotoxicity and genotoxicity of glycidyl methacrylate. Chem Biol Interact 180:69–78
Segui B, Legembre P (2010) Redistribution of CD95 into the lipid rafts to treat cancer cells? Recent Pat Anticancer Drug Discov 5:22–28
Moll UM, Wolff S, Speidel D, Deppert W (2005) Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol 17:631–636
Yu J, Zhang L (2005) The transcriptional targets of p53 in apoptosis control. Biochem Biophys Res Commun 331:851–858
Bargonetti J, Manfredi JJ (2002) Multiple roles of the tumor suppressor p53. Curr Opin Oncol 14:86–91
Cui Q, Yu JH, Wu JN, Tashiro S, Onodera S, Minami M, Ikejima T (2007) P53-mediated cell cycle arrest and apoptosis through a caspase-3- independent, but caspase-9-dependent pathway in oridonin-treated MCF-7 human breast cancer cells. Acta Pharmacol Sin 28:1057–1066
Saleh AM, Vijayasarathy C, Masoud L, Kumar L, Shahin A, Kambal A (2003) Paraoxon induces apoptosis in EL4 cells via activation of mitochondrial pathways. Toxicol Appl Pharmacol 190:47–57
Dejean LM, Martinez-Caballero S, Guo L, Hughes C, Teijido O, Ducret T, Ichas F, Korsmeyer SJ, Antonsson B, Jonas EA, Kinnally KW (2005) Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol Biol Cell 16:2424–2432
Wang S, Yan-Neale Y, Cai R, Alimov I, Cohen D (2006) Activation of mitochondrial pathway is crucial for tumor selective induction of apoptosis by LAQ824. Cell Cycle 5:1662–1668
Galluzzi L, Blomgren K, Kroemer G (2009) Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci 10:481–494
Zhao M, Zhang Y, Wang C, Fu Z, Liu W, Gan J (2009) Induction of macrophage apoptosis by an organochlorine insecticide acetofenate. Chem Res Toxicol 22:504–510
Wang X, Zhu C, Hagberg H, Korhonen L, Sandberg M, Lindholm D, Blomgren K (2004) X-linked inhibitor of apoptosis (XIAP) protein protects against caspase activation and tissue loss after neonatal hypoxia-ischemia. Neurobiol Dis 16:179–189
Petersen OH, Tepikin AV, Gerasimenko JV, Gerasimenko OV, Sutton R, Criddle DN (2009) Fatty acids, alcohol and fatty acid ethyl esters: toxic Ca2+ signal generation and pancreatitis. Cell Calcium 45:634–642
Dostalek M, Hardy KD, Milne GL, Morrow JD, Chen C, Gonzalez FJ, Gu J, Ding X, Johnson DA, Johnson JA, Martin MV, Guengerich FP (2008) Development of oxidative stress by cytochrome P450 induction in rodents is selective for barbiturates and related to loss of pyridine nucleotide-dependent protective systems. J Biol Chem 283:17147–17157
Lupp A, Kuhn UD, Karge E, Adam G, Fleck C (2006) In vitro investigations on the differential pro-oxidant and/or antioxidant properties of cyclosporin A and tacrolimus in human and rat liver microsomes. Int J Clin Pharmacol Ther 44:225–232
Mari M, Cederbaum AI (2001) Induction of catalase, alpha, and microsomal glutathione S-transferase in CYP2E1 overexpressing HepG2 cells and protection against short-term oxidative stress. Hepatology 33:652–661
Hewitt R, Forero A, Luncsford PJ, Martin FL (2007) Enhanced micronucleus formation and modulation of BCL-2:BAX in MCF-7 cells after exposure to binary mixtures. Environ Health Perspect 115(Suppl 1):129–136
Cederbaum AI (2006) CYP2E1—biochemical and toxicological aspects and role in alcohol-induced liver injury. Mt Sinai J Med 73:657–672
Gong P, Cederbaum AI (2006) Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology 43:144–153
Bradford BU, Kono H, Isayama F, Kosyk O, Wheeler MD, Akiyama TE, Bleye L, Krausz KW, Gonzalez FJ, Koop DR, Rusyn I (2005) Cytochrome P450 CYP2E1, but not nicotinamide adenine dinucleotide phosphate oxidase, is required for ethanol-induced oxidative DNA damage in rodent liver. Hepatology 41:336–344
Raza H, John A (2006) 4-Hydroxynonenal induces mitochondrial oxidative stress, apoptosis and expression of glutathione S-transferase A4-4 and cytochrome P450 2E1 in PC12 cells. Toxicol Appl Pharmacol 216:309–318
Kashyap MP, Singh AK, Kumar V, Tripathi VK, Srivastava RK, Agrawal M, Khanna VK, Yadav S, Jain SK, Pant AB (2011) Monocrotophos induced apoptosis in PC12 cells: role of xenobiotic metabolizing cytochrome P450s. PLoS ONE 6:e17757
Engle MR, Singh SP, Czernik PJ, Gaddy D, Montague DC, Ceci JD, Yang Y, Awasthi S, Awasthi YC, Zimniak P (2004) Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: generation and analysis of mGsta4 null mouse. Toxicol Appl Pharmacol 194:296–308
Wang L, Yang Y, Dwivedi S, Xu Y, Chu ET, Li J, Fitchett K, Boor PJ (2006) Manipulating glutathione-S-transferases may prevent the development of tolerance to nitroglycerin. Cardiovasc Toxicol 6:131–144
Li CY, Lee JS, Ko YG, Kim JI, Seo JS (2000) Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J Biol Chem 275:25665–25671
Acknowledgments
The authors are grateful to the Director, IITR, Lucknow, India, for his keen interest in the study. Financial support from Department of Biotechnology, Ministry of Science & Technology, Government of India, New Delhi, India [Grant No. 102/IFD/SAN/PR1524/2010–2011]; Department of Science and Technology, Ministry of Science & Technology, Government of India, New Delhi, India [Grant No. SR/SO/Z 36/2007/91/10]; and Council of Scientific & Industrial Research, Government of India, New Delhi, India [Grant No. BSC0111/INDEPTH/ CSIR Network Project] is acknowledged. Indian Council of Medical Research, (ICMR), New Delhi, India, is acknowledged for providing the fellowship to Dr. Vivek Kumar. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Conflict of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kumar, V., Tripathi, V.K., Jahan, S. et al. Lead Intoxication Synergies of the Ethanol-Induced Toxic Responses in Neuronal Cells—PC12. Mol Neurobiol 52, 1504–1520 (2015). https://doi.org/10.1007/s12035-014-8928-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8928-x