
Abstract
Atonal genes are basic helix-loop-helix transcription factors that were first identified as regulating the formation of mechanoreceptors and photoreceptors in Drosophila. Isolation of vertebrate homologs of atonal genes has shown these transcription factors to play diverse roles in the development of neurons and their progenitors, gut epithelial cells, and mechanosensory cells in the inner ear and skin. In this article, we review the molecular function and regulation of atonal genes and their targets, with particular emphasis on the function of Atoh1 in the development, survival, and function of hair cells of the inner ear. We discuss cell-extrinsic signals that induce Atoh1 expression and the transcriptional networks that regulate its expression during development. Finally, we discuss recent work showing how identification of Atoh1 target genes in the cerebellum, spinal cord, and gut can be used to propose candidate Atoh1 targets in tissues such as the inner ear where cell numbers and biochemical material are limiting.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Identification of Atonal and Its Function in Drosophila
Proneural genes play essential roles in the development of the peripheral nervous system (PNS) in Drosophila [1]. There are three major sensory organs in Drosophila PNS: external sensory organs, chordotonal organs, and multidendritic neurons. Members of the achaete-scute complex (AS-C) are proneural genes required for the formation of all external sensory organs and some multidendritic neurons [2–5] but not chordotonal organs [6]. In order to identify proneural genes that regulate the formation of chordotonal organs, atonal was isolated by polymerase chain reaction (PCR) amplification of Drosophila genomic DNA using degenerate primers designed from shared features between members of the AS-C complex [7]. Atonal is a single-exon gene which encodes a protein of 312 amino acids with a size of 32 kDa. The protein product of atonal contains a basic helix-loop-helix (bHLH) motif at its very C-terminus [7]. The bHLH sequence is a ∼60-amino acid protein structural motif characterized by two conserved domains: an N-terminal basic domain that binds to DNA consensus sequences called E-boxes (with the core sequence CANNTG) and a C-terminal HLH domain composed of two helices connected by a loop that can form heterodimers with other bHLH proteins [8]. The bHLH motif of atonal shares a high degree of similarity with those of other bHLH proteins: 46 % identity with scute and 30 % with daughterless, although its location within the protein can vary—for example, AS-C proteins do not have the bHLH motif at the C-terminus. Electrophoretic mobility shift assays showed that the atonal protein can form a heterodimer with the ubiquitously expressed bHLH protein daughterless to bind to E-boxes [7].
Chordotonal organs are sensory organs widely distributed throughout the body of adult and developing Drosophila, where they function in detecting movements and stretch of the limbs and body wall. In the second segment of the antenna, a large chordotonal organ, termed Johnston’s organ, also serves to detect sound, gravity, and air movements in the environment [9]. In situ hybridization showed that atonal transcripts were expressed in the regions of the embryo and developing imaginal discs which give rise to chordotonal organs. In these areas, atonal was initially expressed in patches of epidermal cells, followed by a more restricted and stronger expression in the sensory organ precursors (SOPs) of each cluster. Chordotonal organs and some multidendritic neurons are absent from the embryos of atonal mutant flies, but external sensory organs are not affected [7]. Gain-of-function experiments in Drosophila generated ectopic chordotonal organs observed after global mis-expression of atonal [7], suggesting atonal is not only necessary but also sufficient for chordotonal organ development in Drosophila. In light of the expression of atonal in chordotonal organ precursors and its necessity and sufficiency in the development of chordotonal organs, we can conclude atonal is a proneural gene specific for chordotonal organ formation [10].
Besides the areas forming the future chordotonal organs, atonal expression is also observed in the developing eye in Drosophila [7, 11]. In the Drosophila eye imaginal disc, atonal expression initiates on the anterior edge of the morphogenetic furrow, and then becomes restricted in regularly spaced cells which will differentiate into R8, the first photoreceptor formed in each Drosophila ommatidium. Loss- and gain-of-function experiments together suggest atonal is both necessary and sufficient for R8 selection during Drosophila eye development [11]. Although atonal is not directly involved in the development of other photoreceptors (R1–R7), their formation still relies on R8 induction [12]. Therefore, atonal also acts as a proneural gene in the formation of Drosophila photoreceptors. Atonal and amos are also necessary for the formation of olfactory and gustatory sensory organ precursors in larval olfactory organs [13].
The Evolution of Atonal Homologs
bHLH transcription factors can be found in a wide range of eukaryotes from yeast to humans and play important roles in a large number of developmental processes. Two types of bHLH proteins have been shown to function in neurogenesis. Class I bHLH proteins (also known as E-proteins) are broadly expressed, including E12, E47, HEB, and E2-2 in vertebrates and daughterless in Drosophila. They can form homo- or heterodimers to bind E-box sequences and regulate transcription. Class II bHLH proteins are expressed in tissue-specific patterns and usually heterodimerize with class I members to bind E-boxes [8]. In Drosophila, both atonal and AS-C members belong to the class II bHLH family.
Atonal homologs show evidence of duplication from an ancestral diploblast atonal gene [14]. In Drosophila, two atonal-related genes were isolated by PCR using degenerate primers based on homology to the atonal bHLH motif. These genes were named absent multidendritic and olfactory sensilla (amos) [15] and cousin of atonal (cato) [16]. The bHLH region of amos and cato are 74 and 64 % identical to that of atonal, respectively. Both amos and cato are involved in the development of Drosophila sensory organs [15, 16]. Amos is a proneural gene that regulates the development of two classes of olfactory neurons and a class of multidendritic neurons [15, 17, 18]. cato, which is widely expressed in PNS after the selection of neural precursors and before their terminal differentiation, is required for proper sensory neuron morphology [16, 19].
Vertebrate atonal homologs were originally identified as eight members of atonal homolog (Atoh) family. Three of them—Atoh1, Atoh7, and Atoh8—have now been designated as Atoh family members, with the rest of the vertebrate atonal homologs having been re-assigned to a closely related bHLH family, the Neurogenin (Neurog)/Neurogenic differentiation factor (NeuroD) family (Fig. 1). Mouse Atoh1 (formerly called Math1) plays essential roles in the development of different types of cells, including neurons in the brain [22–26] and the dorsal spinal cord [27, 28], intestinal secretory cells [29, 30], Merkel cells [31], and inner ear hair cells [32–34]. The expression of mouse Atoh7 (formerly called Math5) is mostly restricted to the developing retina. Atoh7 is critical for the formation of retinal ganglion cells and optic nerves [35, 36]. Atoh8 (formerly called Math6) is widely expressed in multiple organs and is involved in the development of brain, kidney, liver, pancreas, retina, and skeletal muscle [37–40].

Unlike the Atoh family, Neurog/NeuroD family genes have been lost in some eukaryote lineages, such as arthropods (including insects such as Drosophila) [14]. Neurog1 (also known as NeuroD3) is a key regulator that promotes neurogenesis in the CNS and sensory organs [41–45]. Neurog2 (also known as Atoh4) participates broadly in neurogenesis in the cortex, cerebellum, olfactory system, and cranial ganglia [46–49]. Although Neurog3 (also known as Atoh5) is expressed mainly in the pancreas and plays an important role in the differentiation of endocrine cells [50], it has also been shown to affect dendrite morphology and the ratio of excitatory/inhibitory synaptic inputs in the hippocampus [51] and is also expressed in the developing hypothalamus [52]. In general, NeuroD subfamily members tend to be expressed in more mature neuronal progenitors or in post-mitotic neurons. NeuroD1 plays critical roles in the development of both the nervous system and the pancreas [53–57]. NeuroD2 is expressed in the post-mitotic neurons [58] and is involved in synapse development [59–61]. NeuroD4 (also known as Atoh3) is expressed in the brain, retina, and cranial ganglions and is involved in the development of cortical projection neurons, retinal amacrine cells, and branchiomotor neurons [62–64]. NeuroD6 (also known as Atoh2) was shown to be a key regulator of the development of projection neurons of the neocortex and amacrine cells in the retina [65, 66].
The Discovery of Atoh1 and Its Functions in Cell Type Specification
Mouse Atoh1 was isolated by PCR using degenerate primers corresponding to the conserved sequence within the bHLH motif of Drosophila atonal [67, 68]. Atoh1 is also a single-exon gene encoding a protein of 351 amino acids with a size of 38 kDa. ATOH1 shares ∼70 % identity with atonal in its bHLH motif. However, no significant similarity is found between other regions of the two proteins. Unlike Drosophila atonal whose bHLH motif is located at the very end of C-terminus, the bHLH motif of ATOH1 is in the middle of the protein (Fig. 2a). In addition, ATOH1 is rich in prolines which may facilitate protein-protein interactions. The C-terminus of Atoh1 is also rich in serine residues, raising the possibility that phosphorylation may be involved in the regulation of Atoh1 protein function [67]. Comparison of the protein sequences of ATOH1 from different vertebrates showed high degrees of identity in the bHLH motif and C-terminal serine-rich domain. The N-terminus, which is also suggested to be essential for the transcriptional activity, is only strongly conserved among mammals (Fig. 2b) [69]. To determine whether mouse Atoh1 and Drosophila atonal genes were also functional homologs, Atoh1 was expressed in atonal mutant flies and was shown to rescue chordotonal organ and photoreceptor development [70, 71]. In a complementary experiment in which atonal was knocked into the Atoh1 locus, the defects of Atoh1 null mice, including respiration and development of the cerebellum, spinal cord, inner ear, and gut, were rescued by a single allele of Drosophila atonal [71]. Therefore, despite the differences present in the sequences of the Drosophila and mammalian homologs, atonal and Atoh1 are functionally equivalent.

A diagram of the Atoh1 autoregulatory enhancer, which is located at the 3′ of Atoh1 gene and comprised two conserved elements. E-box, N-box, and verified transcription factor-binding sites are marked
In mouse, Atoh1 is expressed in various organs, including the developing brain, dorsal spinal cord, small intestine, Merkel cells of the skin, and the inner ear [34, 67, 68, 72], but little is known about how its expression is regulated. Some of the first studies of regulation of the Atoh1 locus identified an enhancer located at the 3′ of Atoh1 coding region. This enhancer comprised two evolutionarily conserved sequences which contain both E-box and N-box sites (Fig. 3). The enhancer can be recognized and bound by Atoh1 itself and is responsible for the autoregulation of Atoh1 expression [73]. Transgenic mice carrying a green fluorescent protein (GFP) reporter [72] or Cre/CreEr transgenes [74, 75] driven by this enhancer can recapitulate Atoh1 expression in most tissues, albeit with some ectopic regions of expression signals [72].

a Schematic diagrams of Drosophila atonal and mouse ATOH1 proteins showing the different positions of the bHLH domain and a serine-rich domain that may be a target for post-translational modification. b Cross-species comparison of the amino acid sequence of atonal homologs. The bHLH motif is conserved in Drosophila and vertebrates (highlighted in green). The C-terminal serine-rich domain is only conserved in vertebrates (highlighted in yellow). Conserved serine residues are highlighted in red. The alignment was created using MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/)
Gain- and loss-of-function studies in mice over the last 15 years have revealed essential roles for Atoh1 in the organ systems in which it is expressed [22, 24–26, 28–34, 76]. In the following sections, we describe the role of Atoh1 in the central nervous system (CNS), gut, and skin, and in the following section, we focus in depth on the role of Atoh1 in the developing inner ear.
The Function of Atoh1 in the Central Nervous System
In the developing mouse CNS, Atoh1 is first expressed in a restricted region of the dorsal neural tube adjacent to the roof plate, extending posteriorly from the midbrain/hindbrain boundary along the whole length of the neural tube. As the embryo develops, Atoh1 expression in the posterior portion of the developing metencephalon becomes restricted to the rhombic lip [67, 68]. In the rhombic lip, Atoh1 is expressed in the precursors of cerebellar granule cells. Atoh1 expression persists in these precursors as they migrate to the external germinal layer (EGL) of the developing cerebellum and is eventually lost from these cells as they differentiate and further migrate inwardly to form the internal granule layer (IGL) [26]. Cerebellar granule cells are the smallest but also the most abundant neurons in the mammalian brain, comprising over half of all brain neurons. As the only excitatory neurons in the cerebellar cortex, they play essential roles in the complex neuronal network of the cerebellum. When Atoh1 null mice were first generated, it was observed that the cerebellum in these mice was smaller in size compared with their wild-type and heterozygous littermates, and the EGL in these mice was completely missing [25]. Further examination of these mutant mice suggested that the absence of the cerebellar EGL was either due to the loss of granule cell precursors or the failure of granule cell proliferation [26], indicating the crucial role of Atoh1 in the development of cerebellar granule cells.
Atoh1 null mice die shortly after birth due to the respiratory failure with a slowed central respiratory rhythm. Examination of the hindbrain nuclei involved in the respiratory rhythm generation showed that Atoh1 is expressed in the retrotrapezoid nucleus (RTN) which regulates neonatal breathing and automatic breathing that maintains a constant level of plasma CO2 [24]. In the absence of Atoh1, RTN neurons fail to properly develop and establish connections with the pre-Bötzinger complex (preBötC), the respiratory rhythm generating center in mammals [24]. A subsequent study using mice with conditional deletion of Atoh1 specifically in RTN neurons suggests Atoh1 is not required for the specification or maintenance of RTN neurons but is critical for their migration and projection to the preBötC [22].
In the developing dorsal spinal cord, Atoh1 is expressed in a specific subset of neurons, the D1 commissural interneurons which give rise to the spinocerebellar tracts. Unlike cerebellar granule cells, in which Atoh1 is only present in the proliferating precursors, the expression of Atoh1 in the dorsal spinal cord neurons spans both the proliferating and differentiating phases [27]. Studies of Atoh1 null mice suggested that Atoh1 is required for the fate specification and ventral migration of the D1 interneuron precursors. In the absence of Atoh1, some D1 precursors acquire roof plate properties [28]. The role of Atoh1 in the dorsal spinal cord development was further examined and compared with another bHLH factor Neurog1 expressed in the adjacent precursors of a related class of commissural interneurons, the D3A interneurons. Deletion of Atoh1 up-regulates Neurog1 in a subset of D1 interneuron progenitors, the D1A progenitors, while deletion of Neurog1 up-regulates Atoh1 in D3A progenitors, suggesting there is a spatial cross-inhibition between the two genes [43].
The Function of Atoh1 in the Intestine
Atoh1 is expressed in the mouse intestinal epithelium from embryonic to adult stages [30, 67]. Atoh1 function is necessary for the differentiation of the secretory lineages in the intestine, which include the goblet, Paneth, and enteroendocrine cells. In the absence of Atoh1, mice fail to form intestinal secretory cells and the cells destined for the secretory lineages undergo a fate switch and become absorptive cells [29, 30]. In complementary experiments, over-expressing Atoh1 leads to ectopic secretory cells at the expense of absorptive cells [77]. The differentiation of different intestinal cell types is also regulated by an interplay between Atoh1 and Notch signaling and its downstream Hes family effectors. During the fate decision between secretory and absorptive lineages, Atoh1 and Notch/Hes play opposing roles: Hes genes activated by Notch signaling repress Atoh1 expression and therefore promote the absorptive fate, while cells in which Notch is not activated continue to express Atoh1 and differentiate into secretory cells [78].
The Function of Atoh1 in Merkel Cells of the Skin
Atoh1 is also required for the development of Merkel cells, the epidermis-derived cells essential for mediating light touch responses [31, 79, 80]. Merkel cell-neurite complexes are sensitive touch receptors which mediate slowly adapting type I (SAI) responses [81]. In the skin, Atoh1 is observed to be specifically expressed in Merkel cells. Conditional deletion of Atoh1 using Hoxb1-Cre mice, in which Cre recombinase is expressed throughout the dermis and epidermis of skin in the trunk but not head, results in the absence of Merkel cells all over the body except for the whisker pads [31]. Electrical recordings also showed a loss of SAI responses in the epidermis of these mutant mice [31].
Functions of Atoh1 in the Inner Ear
Atoh1 is the first transcription factor to be expressed in developing inner ear hair cells [34, 72]. One of the striking phenotypes of Atoh1 null mice is the complete absence of hair cells in both cochlear and vestibular sensory organs [32, 34, 82]. Since the essential role of Atoh1 in hair cell development was first discovered, subsequent studies have made great progress in understanding how this gene functions in hair cell development.
Atoh1 Expression in the Inner Ear
In the development of Drosophila chordotonal organs and photoreceptors, atonal functions as a proneural gene: it is broadly expressed in the proneural clusters before the selection of SOPs and is necessary and sufficient for the development of SOPs [7, 11]. Does vertebrate Atoh1 also function as a proneural gene? In zebrafish, atonal homologs appear to function as proneural genes essential for inner ear development. There are two atonal homologs in zebrafish, atoh1a and atoh1b, which likely arose by large-scale duplication events in the zebrafish genome [83, 84]. The two zebrafish atoh1 genes have different expression patterns with some overlaps. atoh1b is expressed broadly and early in the otic placode and is then confined to the future sensory epithelia before the overt differentiation of hair cells [84]. atoh1a expression starts later in the progenitors of the sensory epithelia, with expression up-regulated in the hair cells as they differentiate. Morpholino knockdown of atoh1a and atoh1b suggests the two atoh1 genes together regulate zebrafish hair cell development: atoh1b is necessary for specifying sensory precursor groups and regulating the development of the earliest hair cells or tether cells, while atoh1a is necessary for the formation of later-forming hair cells [84]. It was also shown that atoh1a over-expression can induce ectopic hair cells and Notch signaling negatively regulates both atoh1 genes [84]. The initial broad expression of atoh1b and the necessity and sufficiency of atoh1a in regulating hair cell differentiation suggest they act as proneural genes.
At present, there is no definitive consensus on whether Atoh1 acts as a proneural gene in the mammalian inner ear. Although Atoh1 has been shown to be both necessary and sufficient for hair cell development [32, 34, 76, 82, 85–88], the major obstacle to determine whether mammalian Atoh1 is a proneural gene is uncertainty as to whether Atoh1 is broadly expressed in the progenitors that can give rise to different types of cells, as in Drosophila and zebrafish, or is restricted to the progenitors that are already committed to a hair cell fate. Different techniques have been applied to characterize Atoh1 expression in the cochlea and have yielded inconsistent results. First, examination of mice in which the Atoh1-coding region is replaced with a LacZ gene encoding β-galactosidase suggested that the Atoh1 promoter was activated broadly all along the cochlea at E13.5 [33]. However, in situ hybridization for Atoh1 showed Atoh1 transcripts could be detected only in the basal turn of the cochlea at around E13, within a narrow band of cells which extend from the basilar membrane to the apical surface thought to be nascent hair cells [89, 90]. Moreover, immunostaining experiments using anti-ATOH1 antibody revealed the first ATOH1-expressing cells in the cochlear duct between E13.5 and E14.5 as a column of cells spanning the cochlear epithelium in a similar pattern to Atoh1 transcripts [32, 91]. The variability of these results has made it difficult to define what cell populations Atoh1 is initially expressed in. However, recent studies have made use of a knock-in mouse line, Atoh1 A1GFP/A1GFP, in which the endogenous Atoh1 gene is replaced with an Atoh1 EGFP fusion gene [24]. Examination of these mice showed that although ATOH1 expression precedes hair cell differentiation, initial ATOH1 expression is restricted to a subset of post-mitotic precursors and is not broadly expressed in the prosensory domain of the cochlea [92].
In two fate-mapping studies, the fate of Atoh1-expressing cells in the ear was performed with Atoh1 Cre knock-in mice or Atoh1 CrePR knock-in mice [90, 91]. When crossed with Cre reporter lines, some labeled supporting cells were observed in both the vestibular and cochlear sensory organs, suggesting Atoh1 may be initially expressed in progenitors that can turn into both hair cells and supporting cells [90, 91]. However, the number of labeled supporting cells in these studies varied greatly in different sensory organs, suggesting Atoh1 is not broadly expressed in all precursor cells that will give rise to the future organ of Corti [90, 91]. One interpretation of these data is that Atoh1 is expressed in hair cell progenitors immediately before they commit to a hair cell fate, but that some of these cells can be diverted to a supporting cell fate, most likely by Notch-mediated lateral inhibition. Based on the restricted Atoh1 expression observed in both Atoh1 GFP and Atoh1 Cre knock-in mice, we conclude that in the mammalian inner ear, Atoh1 may not completely fulfill the strict criteria of a proneural gene. However, it is still possible that Atoh1 is expressed broadly in the prosensory region of the cochlea at extremely low levels. It is notable that in zebrafish, atonal homologs precede the expression of Sox2 and are both necessary and sufficient for hair cell formation [84, 93], whereas in amniotes, Sox2 precedes Atoh1 expression and is necessary and sufficient for hair cell formation [94–99]. This suggests the possibility that the proneural roles of Sox2 and Atoh1 may have been transposed in the amniote (bird, reptile, and mammal) and anamniote (fish and amphibian) lineages [100].
Atoh1 expression begins at the mid-basal region of the cochlea. As cochlea grows, Atoh1 expression progresses to both base and apex [32, 72, 89]. It initially appears in a subset of precursor cells in the prosensory domain, and then is restricted and up-regulated in differentiated hair cells [32, 92]. After birth, Atoh1 expression in mouse cochlear hair cells starts to decrease. The down-regulation of Atoh1 also follows a basal-to-apical gradient. By postnatal day 3, Atoh1 transcripts can barely be detected along the cochlear duct [89]. Interestingly, in Atoh1 A1GFP/A1GFP knock-in mice, ATOH-EGFP fusion protein is initially expressed diffusely in the cytoplasm of hair cell precursors; however, in differentiated hair cells expressing Myosin6, ATOH1-EGFP is mostly localized to the nucleus [92]. Immunostaining using a newly described anti-ATOH1 antibody [91] confirmed this observation of a subcellular change of ATOH1 as hair cells undergo differentiation, suggesting that the redistribution is not the consequence of the EGFP tag fused to ATOH1 [92]. It is intriguing that the redistribution of ATOH1 proteins coincides with the expression of hair cell markers and the temporal requirement for ATOH1 in hair cell survival [32, 88, 92]. Many transcription factors undergo cytoplasmic-nuclear shuttling, and the shuttling is often associated with their activities [101–103]. Therefore, the ATOH1 redistribution we observed at the first appearance of hair cell markers may be related to its function in regulating hair cell differentiation and survival. The redistribution of ATOH1 proteins might be a result of clearance of cytosolic ATOH1 or active transport of ATOH1 into the nucleus by chaperones or transcriptional partners of ATOH1. Although fascinating, the exact mechanism and function of this redistribution is not yet clear.
The Function of Atoh1 in Hair Cell Survival and Development
In the Atoh1 null cochlea, precursor cells in the prosensory domain undergo apoptosis in a basal-to-apical gradient, similar to the hair cell differentiation gradient seen in the wild-type cochlea. Although Atoh1 expression in the cochlea starts no later than E13.5, mutant cells do not die until E15.5, the time when early hair cell markers such as Myosin6 start to be expressed in the wild-type cochlea [32]. This suggests precursor cells can survive without Atoh1 for about 2 days and the trigger of cell death may be their failure to correctly differentiate in the absence of Atoh1. It was shown that transient Atoh1 expression in mice in which Atoh1 is deleted by Atoh1 Cre (a Cre transgene driven by Atoh1 autoregulatory enhancer) is not sufficient to stop the death of most hair cells and cannot support the proper function of the remaining hair cells, suggesting that the level and duration of Atoh1 expression is critical for the hair cell survival and development [88]. Thus, do hair cells require continuous Atoh1 expression to maintain their viability? We have recently developed another Atoh1 conditional knockout (CKO) mouse model in which Atoh1 can be removed at any time during hair cell development [92]. We observed a critical time window of about 2 days after the initial expression of Atoh1 in which Atoh1 is absolutely required for hair cell survival [92]. Within this critical time window, removal of Atoh1 results in the death of hair cells followed by the death of the surrounding supporting cells. However, if Atoh1 is deleted 3 days or more after its initial expression, no immediate death of hair cells and supporting cells is observed [92]. However, besides regulating hair cell survival, Atoh1 also participates in the maturation process of hair cells. Removing Atoh1 from differentiated hair cells can result in disorganized hair bundles and delayed death of the hair cells [92]. Moreover, re-introduction of Atoh1 into hair cells with noise-induced damaged hair bundles can repair or regenerate the stereocilia [104]. How Atoh1 regulates hair bundle development is not clear, but since Atoh1 has been shown to have targets involved in a wide variety of biological processes in the cerebellum, including regulation of the cytoskeleton [105], it is possible that Atoh1 might directly or indirectly regulate proteins associated with cytoskeletal networks in hair cell bundles.
The loss of Atoh1 does not only affect hair cells directly but also disrupts the development of the surrounding supporting cells. In the Atoh1 null cochlea, the expression of most supporting cell markers is abolished or dramatically decreased [33, 82, 92]. Mis-expression of Atoh1 can also induce active Notch signaling and supporting cell formation adjacent to the ectopic hair cells [33]. The mechanism of how Atoh1 affects supporting cell differentiation and survival is still unclear. Since Atoh1 is not expressed in supporting cells, the regulation that supporting cells receive from Atoh1 may be cell non-autonomous and may involve Notch signaling-mediated lateral inhibition. Supporting this idea is the observation that conditional deletion of Atoh1 in hair cells leads to an up-regulation of Atoh1 promoter activity in supporting cells after hair cell death [92].
The Role of Atoh1 in Hair Cell Regeneration
Hair cell damage or hair cell loss is one of the main causes of hearing loss in humans. Hearing loss can be caused by aging or environmental factors, such as exposure to loud noise and use of ototoxic drugs such as aminoglycoside antibiotics or platinum-containing chemotherapy drugs. It is known that most non-mammalian vertebrates examined, such as fish and birds, can regenerate their hair cells and restore auditory and vestibular function. In these animals, hair cell turnover continually occurs in the vestibular organs and lateral line system and robust hair cell re-generation can also occur after acoustic trauma [106–110]. This re-generation is accomplished by cell proliferation of supporting cells and/or their trans-differentiation into hair cells [111, 112]. In birds, hair cell re-generation starts with direct trans-differentiation of supporting cells, followed by mitotic re-generation [113]. In both zebrafish and birds, re-generation of hair cells is accompanied by a re-expression of Atoh1 in the nascent hair cells [108, 111, 112, 114]. A small degree of hair cell re-generation can be observed in the cochlea of embryonic and neonatal mice [115–117]. However, mammalian hair cells rapidly lose their regenerative ability as animals get older [118]. For example, spontaneous hair cell re-generation can no longer occur when hair cells are damaged in mice that are 1-week old or older [115]. As a result, deafness in adult mammals caused by hair cell damage or loss is permanent.
Since the first demonstration over 25 years ago that birds and fish can regenerate hair cells, many studies have attempted to induce hair cell re-generation in mammals. One of the most promising strategies is Atoh1 gene therapy. Atoh1 alone is sufficient to induce ectopic hair cell formation when mis-expressed in the cochlea of embryonic or neonatal rodents [33, 76, 85, 86, 119, 120]. However, the induction of new hair cells by Atoh1 is also greatly reduced with age. Two recent studies demonstrated that the efficiency of hair cell re-generation by Atoh1 over-expression in mice is progressively decreased at postnatal ages and is largely lost when the mice reach 2 weeks of age [86, 87], around the time of the onset of hearing. Although some studies have reported successful hair cell re-generation in adult rodent cochlea by reactivating Atoh1 expression with viruses [121, 122], in these studies more detailed examination is required to clarify whether the “new” hair cells observed came from hair cell re-generation or the repair of previously damaged hair cells [104], and whether more severe lesions render the cochlear epithelium refractory to Atoh1 activity [123]. Moreover, recent work has shown that a failure to down-regulate Atoh1 from differentiating hair cells ultimately leads to their death [86, 87, 119]. Moreover, the hair cells generated in these experiments are typically immature, lacking markers such as prestin, oncomodulin, and vGlut3 expression, as well as having electrophysiological properties of immature hair cells. These results suggest that a transient pulse of Atoh1 is required to generate viable, mature hair cells, and that additional factors may be required to promote correct maturation of hair cells.
The idea of using Atoh1 gene therapy for hair cell re-generation in humans is to activate Atoh1 expression in cochlear supporting cells, so that the supporting cells can trans-differentiate into functional hair cells. However, this strategy will be of little use in clinical applications if adult supporting cells cannot respond to Atoh1 and become hair cells, as has been shown in mice [86, 87]. Thus, one important question is why Atoh1 expression in older supporting cells is unable to induce trans-differentiation into hair cells. It is assumed that the reason why older supporting cells lose the competence to trans-differentiate is because Atoh1 fails to correctly regulate some or all of its downstream targets and switch on the hair cell differentiation program in mature supporting cells. Several factors may affect Atoh1’s transcriptional activity in cells. First, the epigenetic state of supporting cells may change with age. Many forms of epigenetic regulation have been identified, including DNA methylation and histone post-translational modifications. For individual genes, their epigenetic state can vary depending on cell type, cell age, or even by exposure to different environmental conditions. As supporting cells get older, the epigenetic state of Atoh1 targets that are essential for hair cell differentiation may be converted to a state consistent with transcriptional repression. Alternatively, factors such as Sox2 that may regulate “stem-ness” in supporting cells may also become epigenetically modified with age [124, 125]. In consequence, hair cell target genes may lose their responsiveness to Atoh1 regulation with age and fail to be regulated appropriately in the presence of Atoh1. Comparison of the histone marks in neonatal and adult supporting cells using approaches such as histone chromatin immunoprecipitation-sequencing (ChIP-seq) can provide a global view of epigenetic state change in the whole genome. However, without more information about what genes Atoh1 regulates during hair cell development, it is difficult to interpret such data.
A second reason for the age-dependent decline in the ability of ATOH1 to induce new hair cells is that co-factors which are required for ATOH1 to regulate its downstream targets may be absent in adult supporting cells. As a class II bHLH protein, ATOH1 needs to form a heterodimer with an E-protein for DNA binding and proper function [8]. In Drosophila, atonal can form a heterodimer with daughterless and senseless [7, 126]. In mouse, it was also shown that ATOH1 can form dimers with different E-proteins and the activity of these ATOH1/E-protein heterodimers may vary depending on the cellular context [67, 127]. These data suggest different transcriptional co-factors may be involved in assisting individual ATOH1/E-protein heterodimers to differentially regulate downstream targets. However, little is known about the co-factors that are critical for ATOH1 activity in hair cells, with only a few factors, such as TCF3, GATA3, and GFI1 having been shown to co-operate with ATOH1 or atonal [126, 128–130]. ATOH1 immunoprecipitation followed by mass spectrometry may be a way to identify these co-factors.
Mechanisms of Atoh1 Regulation
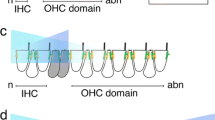
Several signaling pathways are known to be involved in regulating Atoh1 expression, such as Notch [131], Sonic Hedgehog (Shh) [132, 133], Wnts [134–136], and bone morphogenetic proteins (BMPs) [136–139]. Among these signaling pathways, Notch is the best studied, especially in the inner ear. Lateral inhibition mediated by the Notch receptor and its downstream Hes and Hey targets represses Atoh1 expression in supporting cells and thus stops supporting cells from differentiating into hair cells (Fig. 4) [131]. Notch inhibition by different approaches—such as mutant mice for various Notch components [140–145] or administration of Notch signaling inhibitors [146–149]—was shown to be able to release the lateral inhibition between hair cells and supporting cells and generate supernumerary hair cells at the expense of supporting cells.

The regulation of Atoh1 in cochlear development. a In prosensory cell, Eya1, Six1, and Sox2 together induce Atoh1 expression. Atoh1 expression can be repressed by Neurog1, Id genes, and Shh signaling. b After differentiation, Atoh1 in hair cells up-regulates the Notch ligands Jag2 and Dll1, which activate Notch signaling in adjacent supporting cells to prevent them from developing as hair cells. Meanwhile, Sox2 also represses Atoh1 expression in supporting cells
Recently, Shh signaling has been implicated to be involved in the regulation of the timing of hair cell differentiation in the cochlea (Fig. 4). Shh is expressed in the spiral ganglion which innervates cochlear hair cells [132, 150]. In the spiral ganglion-specific Shh knockout mice, the cochlear duct is shortened, prosensory cells exit cell cycle prematurely, and both Atoh1 expression and hair cell differentiation follow an apical-to-basal gradient which is opposite to the basal-to-apical direction observed in the wild-type cochlea [132]. Smoothened (Smo) is a G protein-coupled receptor in Shh signaling pathway. Conditional deletion of Smo in the prosensory domain results in premature differentiation of cochlear hair cells, a decrease in hair cell number, and hearing impairment. Conversely, conditional activation of Smo leads to disrupted differentiation of hair cells and supporting cells [133]. Both studies suggest that Shh signaling is critical for the proper expression of Atoh1 and proper timing of differentiation of cochlear hair cells.
Using biochemical assays or in silico predictions, several transcription factors have been suggested be able to directly interact with the Atoh1 enhancers and regulate Atoh1 transcriptional activity. These studies were mostly done in Atoh1-expressing tissues other than hair cells, such as the cerebellum, intestine and spinal cord. Some of these factors enhance Atoh1 expression (e.g., β-catenin, Cdx2 and Hnf1) [134, 136, 151], while others repress Atoh1 (e.g., Zic1 and Hic1) [137, 152] (Fig. 3). The Atoh1-related bHLH gene Neurog1 can negatively regulate Atoh1 expression (Fig. 4). In the inner ear, Neurog1 and Atoh1 are required for neural and sensory epithelia development, respectively. Neurog1 null mice show expanded Atoh1 expression and over-production of hair cells in the utricle. Conversely, there is a dramatic increase of Neurog1+ neural precursors in Atoh1 null utricle and saccule [44]. Thus, mutual antagonism between Neurog1 and Atoh1 plays a role in the neuronal versus hair cell fate decision [100]. Another Atoh1-related bHLH factor NeuroD1 has been suggested as a mediator of the cross-regulation of Atoh1 expression by Neurog1 (Fig. 4). Conditional deletion of NeuroD1 in the inner ear results in the ectopic hair cell formation in the sensory ganglia, suggesting that NeuroD1 prevents the sensory neurons from developing as hair cells [153]. NeuroD1 conditional knockouts also show aberrant expression of prosensory markers and a transformation of OHCs into IHCs in apical cochlea, indicating NeuroD1 also regulates differentiation of subtypes of cochlear hair cells [153]. Finally, Atoh1 can also be negatively regulated by inhibitor of differentiation genes (Id1-3). Id proteins contain a helix-loop-helix (HLH) domain which can dimerize with other bHLH proteins. It has been shown that Id proteins can bind and sequester E-proteins with high affinity, thus inhibiting the formation of functional bHLH heterodimers [154]. In the cochlea, Id genes are regulated by BMP signaling (Fig. 4) [139], and ectopic expression of Ids in the cochlea reduces Atoh1 expression and prevents hair cell differentiation [139, 155].
In Atoh1 GFP transgenic reporter mice, reporter signals in hair cells are abolished when the reporter is bred onto the Atoh1 null background, suggesting the expression of the GFP reporter is dependent on the continuing presence of Atoh1 in hair cells [44]. In addition, the onset of this reporter in the developing cochlea is slightly slower than the endogenous Atoh1 transcripts and proteins. The earliest stage that the reporter can be detected in cochlear hair cells is around E14.5 [72], compared with endogenous Atoh1 expression which can be observed between E13 and E13.5 [89, 90, 92]. Thus, before Atoh1 autoregulation is initiated, what signals turn on the initial expression of Atoh1? One recent study suggested that two candidate upstream genes regulating Atoh1’s initial expression are Eya1 and Six1. In the mouse cochlea, Eya1 and Six1 together are sufficient to induce the expression of Atoh1, and this induction can be synergistically enhanced by the presence of Sox2 (Fig. 4). All these three molecules are broadly expressed ahead of Atoh1 in the prosensory domain and both Six1 and Sox2 are shown to directly bind the Atoh1 enhancer region [96, 156, 157]. However, it is not clear how the broad expression of Eya1, Six1, and Sox2 can lead to the restricted pattern of hair cell-specific expression of Atoh1. Intriguingly, Sox2 plays a dual function in hair cell development. On one hand, Sox2 promotes sensory formation, in which Sox2—together with Eya1 and Six1—up-regulates Atoh1 expression [96, 156]. On the other hand, Sox2 prevents hair cell differentiation by inhibiting Atoh1 expression [158]. Down-regulation of Sox2 is shown to be necessary for hair cell formation [158]. It has been suggested that Sox2 may inhibit Atoh1 expression by activating Neurog1 and NeuroD, Hes/Hey factors in Notch signaling, and Id genes in an incoherent feed-forward loop (Fig. 4) [96, 97, 100]. Besides regulation at the transcriptional level, some studies also showed Atoh1 function may also be controlled through post-transcriptional or post-translational mechanisms. ATOH1 turnover was accelerated with BMP treatment in the granule neuron progenitor (GNP)-like medulloblastoma cells [138]. In vitro cell culture experiments also showed ATOH1 stability was increased or decreased when E47 or Id1/Id2 was co-expressed, respectively [138]. Moreover, in human colon cancer cells, ATOH1 was found to be degraded by glycogen synthase kinase 3β (GSK3β)-mediated proteolysis, which is regulated by Wnt signaling [135]. Since ATOH1 expression is strongly promoted by autoregulation, it is possible that post-translational down-regulation or degradation of ATOH1 protein may be crucial in breaking this autoregulatory loop.
It has been suggested that Atoh1 in Merkel cells is also directly regulated by the transcription factor Sox2. Ablation of Sox2 in Merkel cells in vivo leads to a decrease of Atoh1 expression and loss of Merkel cells. Intriguingly, Sox2 expression in Merkel cells is controlled by the Polycomb repressor complex, a multi-unit protein complex with histone methyltransferase activity that is involved in transcriptional silencing. Deletion of the key Polycomb subunits Ezh1 and Ezh2 from adult skin de-represses Sox2 and promotes Atoh1 expression and Merkel cell differentiation [159]. These data provide a novel example of how epigenetic regulation of gene expression plays a role in tissue homeostasis.
Identifying Atoh1 Target Genes
Although Atoh1 plays essential roles in the development of different cell types and tissues, its molecular function is poorly understood. The reason for this is a lack of knowledge about direct transcriptional targets of ATOH1. Using gene expression profiling combined with alignment of conserved E-boxes, a few genes have been identified as direct targets of ATOH1, such as BarHl1, Pou4f3, Hes6, Chrna1, and Nr2f6 [160–164]. In order to investigate Atoh1 function in depth, researchers have started to identify ATOH1 direct targets using genome-wide approaches, such as RNA-sequencing (RNA-seq) and ChIP-seq. A recent genome-wide study was performed in the mouse cerebellum to identify the ATOH1 “targetome” in cerebellar granule cells [105]. Three approaches were used in this study, including (1) ATOH1 ChIP-seq to identify Atoh1 binding cites, (2) Histone-seq to identify the global histone H3 lysine 4 (H3K4) methylation status in the cerebellum, and (3) RNA-seq to compare the expression differences between wild-type and Atoh1 null cerebella. With all three sets of data combined together, a total of 601 genes were identified as potential ATOH1 target genes in mouse cerebellar granule cells [105]. These targets are involved in various biological processes, including cell proliferation and differentiation, cell adhesion and migration, chromosomal organization, and cell metabolism. About one sixth of the target genes are associated with most of the known signaling pathways, such as Shh, Notch, transforming growth factor-beta (TGF-β), retinoic acid (RA), Wnt, and mitogen-activated protein kinase (MAPK) signaling. In this study, an extended E-box sequence composed of ten nucleotides was identified as the unique binding motif of ATOH1 by compiling large numbers of ATOH1 binding sites from ChIP-seq experiments. One or more of such extended Atoh1 E-Box-associated motifs (AtEAMs) are present within 5 kb of the transcriptional start sites of 64 % of the ATOH1 targets identified in the cerebellum, and it is possible that other AtEAMs are located further away from other ATOH1 targets [105]. This extended ATOH1-binding motif identified in mouse cerebellum is close, but not identical, to the one identified for Drosophila atonal. This extended ATOH1-binding motif is unlikely to be shared with other closely related bHLH factors, such as NEUROG1. Although ATOH1 and NEUROG1 have high similarity in sequences and bind to core E-boxes that differ by only one nucleotide [165], Neurog1 cannot fully substitute for Atoh1. In knock-in mice in which Atoh1 coding sequence is replaced by Neurog1, the cochlea develops patches of organ of Corti cells with microvillar structures but few hair cells [166]. The less-severe cochlear phenotype of these replacement knock-in mice compared with that in Atoh1 null mice suggests Neurog1 may only be able to regulate a subset Atoh1 targets. Therefore, NEUROG1 may possess its own DNA binding motif that is similar to, but distinct from the Atoh1 binding site AtEAM.
To date, only a few ATOH1 direct targets have been identified in inner ear hair cells [162, 163]. Therefore, a systematic study of ATOH1 targets in hair cells will be one of the ways to study the roles Atoh1 plays during hair cell development and re-generation. However, ChIP-seq experiments using purified hair cells have several technical obstacles. Unlike cerebellar granule cells, in which several million cells are present in each cerebellum and account for over 50 % of the cells in the neonatal cerebellum, cochlear hair cells are present in very limited numbers (only ∼3,000 hair cells per mouse cochlea) and only constitute a small proportion of total cells in the whole cochlea. As a result, it is difficult to enrich and collect enough cochlear hair cells to perform a ChIP-seq experiment with current approaches, although recent advances in amplification of small amounts of ChIP-seq material may make these experiments feasible with relatively small numbers of cells. Cross-referencing gene expression sets with the ChIP-seq data obtained from cerebellum, Johnson and colleagues were able to predict the direct targets of ATOH1 in dorsal spinal cord D1 interneurons. Using this strategy, five new genes were identified as the in vivo direct targets of ATOH1 in D1 interneurons [167]. The success of ATOH1 target identification in the dorsal spinal cord suggests this strategy may also be applied to the inner ear for identifying ATOH1 targets in hair cells. That said, a comparison of the transcriptomes of different Atoh1-expressing cell types, such as hair cells, gut epithelial cells, cerebellar granule cells, and Merkel cells is likely to only reveal a minority of genes common to all cell types. This is apparent upon consideration of the different properties of each cell type—for example, Atoh1-expressing cerebellar granule progenitors are proliferative, migratory, and need to extend neuronal processes as they mature, whereas Atoh1-expressing hair cells are post-mitotic, non-migratory, and do not elaborate axons or dendrites, although they still form synapses with sensory neurons. It will be of great interest to compare direct ATOH1 target genes in these different cell types to understand how higher order chromatin regulation and epigenetic modification are deployed to make different sets of ATOH1 targets available for transcription during the differentiation of different cell lineages.
References
Jan YN, Jan LY (1990) Genes required for specifying cell fates in Drosophila embryonic sensory nervous system. Trends Neurosci 13(12):493–498
Skeath JB, Carroll SB (1991) Regulation of achaete-scute gene expression and sensory organ pattern formation in the Drosophila wing. Genes Dev 5(6):984–995
Romani S, Campuzano S, Macagno ER, Modolell J (1989) Expression of achaete and scute genes in Drosophila imaginal discs and their function in sensory organ development. Genes Dev 3(7):997–1007
Cubas P, de Celis JF, Campuzano S, Modolell J (1991) Proneural clusters of achaete-scute expression and the generation of sensory organs in the Drosophila imaginal wing disc. Genes Dev 5(6):996–1008
Martin-Bermudo MD, Martinez C, Rodriguez A, Jimenez F (1991) Distribution and function of the lethal of scute gene product during early neurogenesis in Drosophila. Development 113(2):445–454
Dambly-Chaudière C, Ghysen A (1987) Independent subpatterns of sense organs require independent genes of the achaete-scute complex in Drosophila larvae. Genes Dev 1(3):297–306
Jarman AP, Grau Y, Jan LY, Jan YN (1993) Atonal is a proneural gene that directs chordotonal organ formation in the Drosophila peripheral nervous system. Cell 73(7):1307–1321
Massari ME, Murre C (2000) Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol 20(2):429–440
Boekhoff-Falk G, Eberl DF (2014) The Drosophila auditory system. Wiley Interdiscip Rev Dev Biol 3(2):179–191
Hassan BA, Bellen HJ (2000) Doing the MATH: is the mouse a good model for fly development? Genes Dev 14(15):1852–1865
Jarman AP, Grell EH, Ackerman L, Jan LY, Jan YN (1994) Atonal is the proneural gene for Drosophila photoreceptors. Nature 369(6479):398–400
Banerjee U, Zipursky SL (1990) The role of cell-cell interaction in the development of the Drosophila visual system. Neuron 4(2):177–187
Grillenzoni N, de Vaux V, Meuwly J, Vuichard S, Jarman A, Holohan E, Gendre N, Stocker RF (2007) Role of proneural genes in the formation of the larval olfactory organ of Drosophila. Dev Genes Evol 217(3):209–219
Fritzsch B, Eberl DF, Beisel KW (2010) The role of bHLH genes in ear development and evolution: revisiting a 10-year-old hypothesis. Cell Mol Life Sci 67(18):3089–3099
Goulding SE, zur Lage P, Jarman AP (2000) Amos, a proneural gene for Drosophila olfactory sense organs that is regulated by lozenge. Neuron 25(1):69–78
Goulding SE, White NM, Jarman AP (2000) Cato encodes a basic helix-loop-helix transcription factor implicated in the correct differentiation of Drosophila sense organs. Dev Biol 221(1):120–131
Huang ML, Hsu CH, Chien CT (2000) The proneural gene amos promotes multiple dendritic neuron formation in the Drosophila peripheral nervous system. Neuron 25(1):57–67
zur Lage PI, Prentice DRA, Holohan EE, Jarman AP (2003) The Drosophila proneural gene amos promotes olfactory sensillum formation and suppresses bristle formation. Development 130:4683–4689
zur Lage PI, Jarman AP (2010) The function and regulation of the bHLH gene, cato, in Drosophila neurogenesis. BMC Dev Biol 10:34
Dereeper A, Audic S, Claverie JM, Blanc G (2010) BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol 10:8
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36(Web Server issue):W465–469
Huang WH, Tupal S, Huang TW, Ward CS, Neul JL, Klisch TJ, Gray PA, Zoghbi HY (2012) Atoh1 governs the migration of postmitotic neurons that shape respiratory effectiveness at birth and chemoresponsiveness in adulthood. Neuron 75(5):799–809
Maricich SM, Xia A, Mathes EL, Wang VY, Oghalai JS, Fritzsch B, Zoghbi HY (2009) Atoh1-lineal neurons are required for hearing and for the survival of neurons in the spiral ganglion and brainstem accessory auditory nuclei. J Neurosci 29(36):11123–11133
Rose MF, Ren J, Ahmad KA, Chao HT, Klisch TJ, Flora A, Greer JJ, Zoghbi HY (2009) Math1 is essential for the development of hindbrain neurons critical for perinatal breathing. Neuron 64(3):341–354
Wang VY, Rose MF, Zoghbi HY (2005) Math1 expression redefines the rhombic lip derivatives and reveals novel lineages within the brainstem and cerebellum. Neuron 48(1):31–43
Ben-Arie N, Bellen HJ, Armstrong DL, McCall AE, Gordadze PR, Guo Q, Matzuk MM, Zoghbi HY (1997) Math1 is essential for genesis of cerebellar granule neurons. Nature 390(6656):169–172
Helms AW, Johnson JE (1998) Progenitors of dorsal commissural interneurons are defined by MATH1 expression. Development 125(5):919–928
Bermingham NA, Hassan BA, Wang VY, Fernandez M, Banfi S, Bellen HJ, Fritzsch B, Zoghbi HY (2001) Proprioceptor pathway development is dependent on Math1. Neuron 30(2):411–422
Shroyer NF, Helmrath MA, Wang VY, Antalffy B, Henning SJ, Zoghbi HY (2007) Intestine-specific ablation of mouse atonal homolog 1 (Math1) reveals a role in cellular homeostasis. Gastroenterology 132(7):2478–2488
Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY (2001) Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 294(5549):2155–2158
Maricich SM, Wellnitz SA, Nelson AM, Lesniak DR, Gerling GJ, Lumpkin EA, Zoghbi HY (2009) Merkel cells are essential for light-touch responses. Science 324(5934):1580–1582
Chen P, Johnson JE, Zoghbi HY, Segil N (2002) The role of Math1 in inner ear development: uncoupling the establishment of the sensory primordium from hair cell fate determination. Development 129(10):2495–2505
Woods C, Montcouquiol M, Kelley MW (2004) Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat Neurosci 7(12):1310–1318
Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ, Lysakowski A, Zoghbi HY (1999) Math1: an essential gene for the generation of inner ear hair cells. Science 284(5421):1837–1841
Wang SW, Kim BS, Ding K, Wang H, Sun D, Johnson RL, Klein WH, Gan L (2001) Requirement for math5 in the development of retinal ganglion cells. Genes Dev 15(1):24–29
Brown NL, Patel S, Brzezinski J, Glaser T (2001) Math5 is required for retinal ganglion cell and optic nerve formation. Development 128(13):2497–2508
Inoue C, Bae SK, Takatsuka K, Inoue T, Bessho Y, Kageyama R (2001) Math6, a bHLH gene expressed in the developing nervous system, regulates neuronal versus glial differentiation. Genes Cells 6(11):977–986
Ross MD, Martinka S, Mukherjee A, Sedor JR, Vinson C, Bruggeman LA (2006) Math6 expression during kidney development and altered expression in a mouse model of glomerulosclerosis. Dev Dyn 235(11):3102–3109
Lynn FC, Sanchez L, Gomis R, German MS, Gasa R (2008) Identification of the bHLH factor Math6 as a novel component of the embryonic pancreas transcriptional network. PLoS One 3(6):e2430
Yao J, Zhou J, Liu Q, Lu D, Wang L, Qiao X, Jia W (2010) Atoh8, a bHLH transcription factor, is required for the development of retina and skeletal muscle in zebrafish. PLoS One 5(6):e10945
Ma Q, Chen Z, del Barco BI, de la Pompa JL, Anderson DJ (1998) Neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron 20(3):469–482
Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, Fan G, Greenberg ME (2001) Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell 104(3):365–376
Gowan K, Helms AW, Hunsaker TL, Collisson T, Ebert PJ, Odom R, Johnson JE (2001) Crossinhibitory activities of Ngn1 and Math1 allow specification of distinct dorsal interneurons. Neuron 31(2):219–232
Raft S, Koundakjian EJ, Quinones H, Jayasena CS, Goodrich LV, Johnson JE, Segil N, Groves AK (2007) Cross-regulation of Ngn1 and Math1 coordinates the production of neurons and sensory hair cells during inner ear development. Development 134(24):4405–4415
Cau E, Casarosa S, Guillemot F (2002) Mash1 and Ngn1 control distinct steps of determination and differentiation in the olfactory sensory neuron lineage. Development 129(8):1871–1880
Nieto M, Schuurmans C, Britz O, Guillemot F (2001) Neural bHLH genes control the neuronal versus glial fate decision in cortical progenitors. Neuron 29(2):401–413
Henke RM, Savage TK, Meredith DM, Glasgow SM, Hori K, Dumas J, MacDonald RJ, Johnson JE (2009) Neurog2 is a direct downstream target of the Ptf1a-Rbpj transcription complex in dorsal spinal cord. Development 136(17):2945–2954
Roybon L, Deierborg T, Brundin P, Li JY (2009) Involvement of Ngn2, Tbr and NeuroD proteins during postnatal olfactory bulb neurogenesis. Eur J Neurosci 29(2):232–243
Fode C, Gradwohl G, Morin X, Dierich A, LeMeur M, Goridis C, Guillemot F (1998) The bHLH protein NEUROGENIN 2 is a determination factor for epibranchial placode-derived sensory neurons. Neuron 20(3):483–494
Gradwohl G, Dierich A, LeMeur M, Guillemot F (2000) Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A 97(4):1607–1611
Salama-Cohen P, Arevalo MA, Grantyn R, Rodriguez-Tebar A (2006) Notch and NGF/p75NTR control dendrite morphology and the balance of excitatory/inhibitory synaptic input to hippocampal neurones through Neurogenin 3. J Neurochem 97(5):1269–1278
Sommer L, Ma Q, Anderson DJ (1996) Neurogenins, a novel family of atonal-related bHLH transcription factors, are putative mammalian neuronal determination genes that reveal progenitor cell heterogeneity in the developing CNS and PNS. Mol Cell Neurosci 8(4):221–241
Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ (1997) Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev 11(18):2323–2334
Liu M, Pereira FA, Price SD, Chu MJ, Shope C, Himes D, Eatock RA, Brownell WE, Lysakowski A, Tsai MJ (2000) Essential role of BETA2/NeuroD1 in development of the vestibular and auditory systems. Genes Dev 14(22):2839–2854
Pennesi ME, Cho JH, Yang Z, Wu SH, Zhang J, Wu SM, Tsai MJ (2003) BETA2/NeuroD1 null mice: a new model for transcription factor-dependent photoreceptor degeneration. J Neurosci 23(2):453–461
Miyata T, Maeda T, Lee JE (1999) NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev 13(13):1647–1652
Boutin C, Hardt O, de Chevigny A, Core N, Goebbels S, Seidenfaden R, Bosio A, Cremer H (2010) NeuroD1 induces terminal neuronal differentiation in olfactory neurogenesis. Proc Natl Acad Sci U S A 107(3):1201–1206
McCormick MB, Tamimi RM, Snider L, Asakura A, Bergstrom D, Tapscott SJ (1996) NeuroD2 and neuroD3: distinct expression patterns and transcriptional activation potentials within the neuroD gene family. Mol Cell Biol 16(10):5792–5800
Wilke SA, Hall BJ, Antonios JK, Denardo LA, Otto S, Yuan B, Chen F, Robbins EM, Tiglio K, Williams ME, Qiu Z, Biederer T, Ghosh A (2012) NeuroD2 regulates the development of hippocampal mossy fiber synapses. Neural Dev 7:9
Yang Y, Kim AH, Yamada T, Wu B, Bilimoria PM, Ikeuchi Y, de la Iglesia N, Shen J, Bonni A (2009) A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science 326(5952):575–578
Ince-Dunn G, Hall BJ, Hu SC, Ripley B, Huganir RL, Olson JM, Tapscott SJ, Ghosh A (2006) Regulation of thalamocortical patterning and synaptic maturation by NeuroD2. Neuron 49(5):683–695
Inoue T, Hojo M, Bessho Y, Tano Y, Lee JE, Kageyama R (2002) Math3 and NeuroD regulate amacrine cell fate specification in the retina. Development 129(4):831–842
Ohsawa R, Ohtsuka T, Kageyama R (2005) Mash1 and Math3 are required for development of branchiomotor neurons and maintenance of neural progenitors. J Neurosci 25(25):5857–5865
Mattar P, Langevin LM, Markham K, Klenin N, Shivji S, Zinyk D, Schuurmans C (2008) Basic helix-loop-helix transcription factors cooperate to specify a cortical projection neuron identity. Mol Cell Biol 28(5):1456–1469
Bormuth I, Yan K, Yonemasu T, Gummert M, Zhang M, Wichert S, Grishina O, Pieper A, Zhang W, Goebbels S, Tarabykin V, Nave KA, Schwab MH Neuronal basic helix-loop-helix proteins Neurod2/6 regulate cortical commissure formation before midline interactions. J Neurosci 33 (2):641–651
Cherry TJ, Wang S, Bormuth I, Schwab M, Olson J, Cepko CL NeuroD factors regulate cell fate and neurite stratification in the developing retina. J Neurosci 31 (20):7365–7379
Akazawa C, Ishibashi M, Shimizu C, Nakanishi S, Kageyama R (1995) A mammalian helix-loop-helix factor structurally related to the product of Drosophila proneural gene atonal is a positive transcriptional regulator expressed in the developing nervous system. J Biol Chem 270(15):8730–8738
Ben-Arie N, McCall AE, Berkman S, Eichele G, Bellen HJ, Zoghbi HY (1996) Evolutionary conservation of sequence and expression of the bHLH protein atonal suggests a conserved role in neurogenesis. Hum Mol Genet 5(9):1207–1216
Mulvaney J, Dabdoub A (2012) Atoh1, an essential transcription factor in neurogenesis and intestinal and inner ear development: function, regulation, and context dependency. J Assoc Res Otolaryngol 13(3):281–293
Ben-Arie N, Hassan BA, Bermingham NA, Malicki DM, Armstrong D, Matzuk M, Bellen HJ, Zoghbi HY (2000) Functional conservation of atonal and Math1 in the CNS and PNS. Development 127(5):1039–1048
Wang VY, Hassan BA, Bellen HJ, Zoghbi HY (2002) Drosophila atonal fully rescues the phenotype of Math1 null mice: new functions evolve in new cellular contexts. Curr Biol 12(18):1611–1616
Lumpkin EA, Collisson T, Parab P, Omer-Abdalla A, Haeberle H, Chen P, Doetzlhofer A, White P, Groves A, Segil N, Johnson JE (2003) Math1-driven GFP expression in the developing nervous system of transgenic mice. Gene Expr Patterns 3(4):389–395
Helms AW, Abney AL, Ben-Arie N, Zoghbi HY, Johnson JE (2000) Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development 127(6):1185–1196
Machold R, Fishell G (2005) Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron 48(1):17–24
Matei V, Pauley S, Kaing S, Rowitch D, Beisel KW, Morris K, Feng F, Jones K, Lee J, Fritzsch B (2005) Smaller inner ear sensory epithelia in Neurog 1 null mice are related to earlier hair cell cycle exit. Dev Dyn 234(3):633–650
Zheng JL, Gao WQ (2000) Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat Neurosci 3(6):580–586
VanDussen KL, Samuelson LC (2010) Mouse atonal homolog 1 directs intestinal progenitors to secretory cell rather than absorptive cell fate. Dev Biol 346(2):215–223
Noah TK, Shroyer NF (2013) Notch in the intestine: regulation of homeostasis and pathogenesis. Annu Rev Physiol 75:263–288
Maksimovic S, Nakatani M, Baba Y, Nelson AM, Marshall KL, Wellnitz SA, Firozi P, Woo SH, Ranade S, Patapoutian A, Lumpkin EA (2014) Epidermal Merkel cells are mechanosensory cells that tune mammalian touch receptors. Nature 509(7502):617–621
Owens DM, Lumpkin EA (2014) Diversification and specialization of touch receptors in skin. Cold Spring Harb Perspect Med 4(6)
Wellnitz SA, Lesniak DR, Gerling GJ, Lumpkin EA (2010) The regularity of sustained firing reveals two populations of slowly adapting touch receptors in mouse hairy skin. J Neurophysiol 103(6):3378–3388
Pan N, Jahan I, Kersigo J, Kopecky B, Santi P, Johnson S, Schmitz H, Fritzsch B (2011) Conditional deletion of Atoh1 using Pax2-Cre results in viable mice without differentiated cochlear hair cells that have lost most of the organ of Corti. Hear Res 275(1–2):66–80
Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL, Westerfield M, Ekker M, Postlethwait JH (1998) Zebrafish hox clusters and vertebrate genome evolution. Science 282(5394):1711–1714
Millimaki BB, Sweet EM, Dhason MS, Riley BB (2007) Zebrafish atoh1 genes: classic proneural activity in the inner ear and regulation by Fgf and Notch. Development 134(2):295–305
Gubbels SP, Woessner DW, Mitchell JC, Ricci AJ, Brigande JV (2008) Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature 455(7212):537–541
Kelly MC, Chang Q, Pan A, Lin X, Chen P (2012) Atoh1 directs the formation of sensory mosaics and induces cell proliferation in the postnatal mammalian cochlea in vivo. J Neurosci 32(19):6699–6710
Liu Z, Dearman JA, Cox BC, Walters BJ, Zhang L, Ayrault O, Zindy F, Gan L, Roussel MF, Zuo J (2012) Age-dependent in vivo conversion of mouse cochlear pillar and deiters’ cells to immature hair cells by atoh1 ectopic expression. J Neurosci 32(19):6600–6610
Pan N, Jahan I, Kersigo J, Duncan JS, Kopecky B, Fritzsch B (2012) A novel Atoh1 “self-terminating” mouse model reveals the necessity of proper Atoh1 level and duration for hair cell differentiation and viability. PLoS One 7(1):e30358
Lanford PJ, Shailam R, Norton CR, Gridley T, Kelley MW (2000) Expression of Math1 and HES5 in the cochleae of wildtype and Jag2 mutant mice. J Assoc Res Otolaryngol 1(2):161–171
Yang H, Xie X, Deng M, Chen X, Gan L (2010) Generation and characterization of Atoh1-Cre knock-in mouse line. Genesis 48(6):407–413
Driver EC, Sillers L, Coate TM, Rose MF, Kelley MW (2013) The Atoh1-lineage gives rise to hair cells and supporting cells within the mammalian cochlea. Dev Biol 376(1):86–98
Cai T, Seymour ML, Zhang H, Pereira FA, Groves AK (2013) Conditional deletion of Atoh1 reveals distinct critical periods for survival and function of hair cells in the organ of Corti. J Neurosci 33(24):10110–10122
Millimaki BB, Sweet EM, Riley BB (2010) Sox2 is required for maintenance and regeneration, but not initial development, of hair cells in the zebrafish inner ear. Dev Biol 338(2):262–269
Kiernan AE, Pelling AL, Leung KK, Tang AS, Bell DM, Tease C, Lovell-Badge R, Steel KP, Cheah KS (2005) Sox2 is required for sensory organ development in the mammalian inner ear. Nature 434(7036):1031–1035
Neves J, Parada C, Chamizo M, Giraldez F (2011) Jagged 1 regulates the restriction of Sox2 expression in the developing chicken inner ear: a mechanism for sensory organ specification. Development 138(4):735–744
Neves J, Uchikawa M, Bigas A, Giraldez F (2012) The prosensory function of Sox2 in the chicken inner ear relies on the direct regulation of Atoh1. PLoS One 7(1):e30871
Neves J, Vachkov I, Giraldez F (2013) Sox2 regulation of hair cell development: incoherence makes sense. Hear Res 297:20–29
Pan W, Jin Y, Stanger B, Kiernan AE (2010) Notch signaling is required for the generation of hair cells and supporting cells in the mammalian inner ear. Proc Natl Acad Sci U S A 107(36):15798–15803
Pan W, Jin Y, Chen J, Rottier RJ, Steel KP, Kiernan AE (2013) Ectopic expression of activated notch or SOX2 reveals similar and unique roles in the development of the sensory cell progenitors in the mammalian inner ear. J Neurosci 33(41):16146–16157
Raft S, Groves AK (2014) Segregating neural and mechanosensory fates in the developing ear: patterning, signaling, and transcriptional control. Cell Tissue Res
Van Der Heide LP, Hoekman MF, Smidt MP (2004) The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J 380(Pt 2):297–309
Reich NC, Liu L (2006) Tracking STAT nuclear traffic. Nat Rev Immunol 6(8):602–612
MacDonald BT, Tamai K, He X (2009) Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 17(1):9–26
Yang SM, Chen W, Guo WW, Jia S, Sun JH, Liu HZ, Young WY, He DZ (2012) Regeneration of stereocilia of hair cells by forced Atoh1 expression in the adult mammalian cochlea. PLoS One 7(9):e46355
Klisch TJ, Xi Y, Flora A, Wang L, Li W, Zoghbi HY (2011) In vivo Atoh1 targetome reveals how a proneural transcription factor regulates cerebellar development. Proc Natl Acad Sci U S A 108(8):3288–3293
Cruz RM, Lambert PR, Rubel EW (1987) Light microscopic evidence of hair cell regeneration after gentamicin toxicity in chick cochlea. Arch Otolaryngol Head Neck Surg 113(10):1058–1062
Corwin JT, Cotanche DA (1988) Regeneration of sensory hair cells after acoustic trauma. Science 240(4860):1772–1774
Ma EY, Raible DW (2009) Signaling pathways regulating zebrafish lateral line development. Curr Biol 19(9):R381–386
Roberson DF, Weisleder P, Bohrer PS, Rubel EW (1992) Ongoing production of sensory cells in the vestibular epithelium of the chick. Hear Res 57(2):166–174
Corwin JT (1981) Postembryonic production and aging in inner ear hair cells in sharks. J Comp Neurol 201(4):541–553
Stone JS, Cotanche DA (2007) Hair cell regeneration in the avian auditory epithelium. Int J Dev Biol 51(6–7):633–647
Ma EY, Rubel EW, Raible DW (2008) Notch signaling regulates the extent of hair cell regeneration in the zebrafish lateral line. J Neurosci 28(9):2261–2273
Roberson DW, Alosi JA, Cotanche DA (2004) Direct transdifferentiation gives rise to the earliest new hair cells in regenerating avian auditory epithelium. J Neurosci Res 78(4):461–471
Cafaro J, Lee GS, Stone JS (2007) Atoh1 expression defines activated progenitors and differentiating hair cells during avian hair cell regeneration. Dev Dyn 236(1):156–170
Cox BC, Chai R, Lenoir A, Liu Z, Zhang L, Nguyen DH, Chalasani K, Steigelman KA, Fang J, Cheng AG, Zuo J (2014) Spontaneous hair cell regeneration in the neonatal mouse cochlea in vivo. Development 141(4):816–829
Bramhall NF, Shi F, Arnold K, Hochedlinger K, Edge AS (2014) Lgr5-positive supporting cells generate new hair cells in the postnatal cochlea. Stem Cell Reports 2(3):311–322
Kelley MW, Talreja DR, Corwin JT (1995) Replacement of hair cells after laser microbeam irradiation in cultured organs of corti from embryonic and neonatal mice. J Neurosci 15(4):3013–3026
Roberson DW, Rubel EW (1994) Cell division in the gerbil cochlea after acoustic trauma. Am J Otol 15(1):28–34
Liu Z, Fang J, Dearman J, Zhang L, Zuo J (2014) In vivo generation of immature inner hair cells in neonatal mouse cochleae by ectopic atoh1 expression. PLoS One 9(2):e89377
Yang J, Bouvron S, Lv P, Chi F, Yamoah EN (2012) Functional features of trans-differentiated hair cells mediated by Atoh1 reveals a primordial mechanism. J Neurosci 32(11):3712–3725
Husseman J, Raphael Y (2009) Gene therapy in the inner ear using adenovirus vectors. Adv Otorhinolaryngol 66:37–51
Izumikawa M, Minoda R, Kawamoto K, Abrashkin KA, Swiderski DL, Dolan DF, Brough DE, Raphael Y (2005) Auditory hair cell replacement and hearing improvement by Atoh1 gene therapy in deaf mammals. Nat Med 11(3):271–276
Izumikawa M, Batts SA, Miyazawa T, Swiderski DL, Raphael Y (2008) Response of the flat cochlear epithelium to forced expression of Atoh1. Hear Res 240(1–2):52–56
Smeti I, Savary E, Capelle V, Hugnot JP, Uziel A, Zine A (2011) Expression of candidate markers for stem/progenitor cells in the inner ears of developing and adult GFAP and nestin promoter-GFP transgenic mice. Gene Expr Patterns 11(1–2):22–32
Waldhaus J, Cimerman J, Gohlke H, Ehrich M, Muller M, Lowenheim H (2012) Stemness of the organ of Corti relates to the epigenetic status of Sox2 enhancers. PLoS One 7(5):e36066
Acar M, Jafar-Nejad H, Giagtzoglou N, Yallampalli S, David G, He Y, Delidakis C, Bellen HJ (2006) Senseless physically interacts with proneural proteins and functions as a transcriptional co-activator. Development 133(10):1979–1989
Flora A, Garcia JJ, Thaller C, Zoghbi HY (2007) The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc Natl Acad Sci U S A 104(39):15382–15387
Masuda M, Pak K, Chavez E, Ryan AF (2012) TFE2 and GATA3 enhance induction of POU4F3 and myosin VIIa positive cells in nonsensory cochlear epithelium by ATOH1. Dev Biol 372(1):68–80
Powell LM, Chen A, Huang YC, Wang PY, Kemp SE, Jarman AP (2012) The SUMO pathway promotes basic helix-loop-helix proneural factor activity via a direct effect on the Zn finger protein senseless. Mol Cell Biol 32(14):2849–2860
Powell LM, Deaton AM, Wear MA, Jarman AP (2008) The specificity of atonal and Scute bHLH factors: analysis of cognate E box binding sites and the influence of Senseless. Genes to Cells 13(9):915–929
Kiernan AE (2013) Notch signaling during cell fate determination in the inner ear. Semin Cell Dev Biol 24(5):470–479
Bok J, Zenczak C, Hwang CH, Wu DK (2013) Auditory ganglion source of Sonic hedgehog regulates timing of cell cycle exit and differentiation of mammalian cochlear hair cells. Proc Natl Acad Sci U S A 110(34):13869–13874
Tateya T, Imayoshi I, Tateya I, Hamaguchi K, Torii H, Ito J, Kageyama R (2013) Hedgehog signaling regulates prosensory cell properties during the basal-to-apical wave of hair cell differentiation in the mammalian cochlea. Development 140(18):3848–3857
Shi F, Cheng YF, Wang XL, Edge AS (2010) Beta-catenin up-regulates Atoh1 expression in neural progenitor cells by interaction with an Atoh1 3′ enhancer. J Biol Chem 285(1):392–400
Tsuchiya K, Nakamura T, Okamoto R, Kanai T, Watanabe M (2007) Reciprocal targeting of Hath1 and beta-catenin by Wnt glycogen synthase kinase 3beta in human colon cancer. Gastroenterology 132(1):208–220
Mutoh H, Sakamoto H, Hayakawa H, Arao Y, Satoh K, Nokubi M, Sugano K (2006) The intestine-specific homeobox gene Cdx2 induces expression of the basic helix-loop-helix transcription factor Math1. Differentiation 74(6):313–321
Ebert PJ, Timmer JR, Nakada Y, Helms AW, Parab PB, Liu Y, Hunsaker TL, Johnson JE (2003) Zic1 represses Math1 expression via interactions with the Math1 enhancer and modulation of Math1 autoregulation. Development 130(9):1949–1959
Zhao H, Ayrault O, Zindy F, Kim JH, Roussel MF (2008) Post-transcriptional down-regulation of Atoh1/Math1 by bone morphogenic proteins suppresses medulloblastoma development. Genes Dev 22(6):722–727
Kamaid A, Neves J, Giraldez F (2010) Id gene regulation and function in the prosensory domains of the chicken inner ear: a link between Bmp signaling and Atoh1. J Neurosci 30(34):11426–11434
Lanford PJ, Lan Y, Jiang R, Lindsell C, Weinmaster G, Gridley T, Kelley MW (1999) Notch signalling pathway mediates hair cell development in mammalian cochlea. Nat Genet 21(3):289–292
Zine A, Aubert A, Qiu J, Therianos S, Guillemot F, Kageyama R, de Ribaupierre F (2001) Hes1 and Hes5 activities are required for the normal development of the hair cells in the mammalian inner ear. J Neurosci 21(13):4712–4720
Zheng JL, Shou J, Guillemot F, Kageyama R, Gao WQ (2000) Hes1 is a negative regulator of inner ear hair cell differentiation. Development 127(21):4551–4560
Kiernan AE, Cordes R, Kopan R, Gossler A, Gridley T (2005) The Notch ligands DLL1 and JAG2 act synergistically to regulate hair cell development in the mammalian inner ear. Development 132(19):4353–4362
Yamamoto N, Tanigaki K, Tsuji M, Yabe D, Ito J, Honjo T (2006) Inhibition of Notch/RBP-J signaling induces hair cell formation in neonate mouse cochleas. J Mol Med (Berl) 84(1):37–45
Tateya T, Imayoshi I, Tateya I, Ito J, Kageyama R (2011) Cooperative functions of Hes/Hey genes in auditory hair cell and supporting cell development. Dev Biol 352(2):329–340
Doetzlhofer A, Basch ML, Ohyama T, Gessler M, Groves AK, Segil N (2009) Hey2 regulation by FGF provides a Notch-independent mechanism for maintaining pillar cell fate in the organ of Corti. Dev Cell 16(1):58–69
Hayashi T, Kokubo H, Hartman BH, Ray CA, Reh TA, Bermingham-McDonogh O (2008) Hesr1 and Hesr2 may act as early effectors of Notch signaling in the developing cochlea. Dev Biol 316(1):87–99
Slowik AD, Bermingham-McDonogh O (2013) Hair cell generation by notch inhibition in the adult mammalian cristae. J Assoc Res Otolaryngol 14(6):813–828
Takebayashi S, Yamamoto N, Yabe D, Fukuda H, Kojima K, Ito J, Honjo T (2007) Multiple roles of Notch signaling in cochlear development. Dev Biol 307(1):165–178
Liu Z, Owen T, Zhang L, Zuo J (2010) Dynamic expression pattern of Sonic hedgehog in developing cochlear spiral ganglion neurons. Dev Dyn 239(6):1674–1683
D’Angelo A, Bluteau O, Garcia-Gonzalez MA, Gresh L, Doyen A, Garbay S, Robine S, Pontoglio M (2010) Hepatocyte nuclear factor 1alpha and beta control terminal differentiation and cell fate commitment in the gut epithelium. Development 137(9):1573–1582
Briggs KJ, Eberhart CG, Watkins DN (2008) Just say no to ATOH: how HIC1 methylation might predispose medulloblastoma to lineage addiction. Cancer Res 68(21):8654–8656
Jahan I, Pan N, Kersigo J, Fritzsch B (2010) Neurod1 suppresses hair cell differentiation in ear ganglia and regulates hair cell subtype development in the cochlea. PLoS One 5(7):e11661
Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H (1990) The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell 61(1):49–59
Jones JM, Montcouquiol M, Dabdoub A, Woods C, Kelley MW (2006) Inhibitors of differentiation and DNA binding (Ids) regulate Math1 and hair cell formation during the development of the organ of Corti. J Neurosci 26(2):550–558
Ahmed M, Wong EY, Sun J, Xu J, Wang F, Xu PX (2012) Eya1-Six1 interaction is sufficient to induce hair cell fate in the cochlea by activating Atoh1 expression in cooperation with Sox2. Dev Cell 22(2):377–390
Jeon SJ, Fujioka M, Kim SC, Edge AS (2011) Notch signaling alters sensory or neuronal cell fate specification of inner ear stem cells. J Neurosci 31(23):8351–8358
Dabdoub A, Puligilla C, Jones JM, Fritzsch B, Cheah KS, Pevny LH, Kelley MW (2008) Sox2 signaling in prosensory domain specification and subsequent hair cell differentiation in the developing cochlea. Proc Natl Acad Sci U S A 105(47):18396–18401
Bardot ES, Valdes VJ, Zhang J, Perdigoto CN, Nicolis S, Hearn SA, Silva JM, Ezhkova E (2013) Polycomb subunits Ezh1 and Ezh2 regulate the Merkel cell differentiation program in skin stem cells. Embo J 32(14):1990–2000
Chellappa R, Li S, Pauley S, Jahan I, Jin K, Xiang M (2008) Barhl1 regulatory sequences required for cell-specific gene expression and autoregulation in the inner ear and central nervous system. Mol Cell Biol 28(6):1905–1914
Hu X, Huang J, Feng L, Fukudome S, Hamajima Y, Lin J (2010) Sonic hedgehog (SHH) promotes the differentiation of mouse cochlear neural progenitors via the Math1-Brn3.1 signaling pathway in vitro. J Neurosci Res 88(5):927–935
Scheffer D, Sage C, Corey DP, Pingault V (2007) Gene expression profiling identifies Hes6 as a transcriptional target of ATOH1 in cochlear hair cells. FEBS Lett 581(24):4651–4656
Scheffer D, Sage C, Plazas PV, Huang M, Wedemeyer C, Zhang DS, Chen ZY, Elgoyhen AB, Corey DP, Pingault V (2007) The alpha1 subunit of nicotinic acetylcholine receptors in the inner ear: transcriptional regulation by ATOH1 and co-expression with the gamma subunit in hair cells. J Neurochem 103(6):2651–2664
Krizhanovsky V, Soreq L, Kliminski V, Ben-Arie N (2006) Math1 target genes are enriched with evolutionarily conserved clustered E-box binding sites. J Mol Neurosci 28(2):211–229
Bertrand N, Castro DS, Guillemot F (2002) Proneural genes and the specification of neural cell types. Nat Rev Neurosci 3(7):517–530
Jahan I, Pan N, Kersigo J, Calisto LE, Morris KA, Kopecky B, Duncan JS, Beisel KW, Fritzsch B (2012) Expression of Neurog1 instead of Atoh1 can partially rescue organ of Corti cell survival. PLoS One 7(1):e30853
Lai HC, Klisch TJ, Roberts R, Zoghbi HY, Johnson JE (2011) In vivo neuronal subtype-specific targets of Atoh1 (Math1) in dorsal spinal cord. J Neurosci 31(30):10859–10871
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cai, T., Groves, A.K. The Role of Atonal Factors in Mechanosensory Cell Specification and Function. Mol Neurobiol 52, 1315–1329 (2015). https://doi.org/10.1007/s12035-014-8925-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8925-0