
Abstract
The endoplasmic reticulum (ER) serves many crucial cellular functions. However, when misfolded or unfolded proteins accumulated in the ER, the stress of ER will be induced. Meanwhile, the intracellular signaling network, which is called unfolded protein response, will also be activated to cope with. Those unfolded proteins can be recognized by three kinds of stress sensors which are IRE1, PERK, and ATF6. Based on lots of medical reports, ER stress in postmortem brains from Alzheimer’s disease (AD) patients, animals, and vitro models have indicated that ER dysfunction might work as an important part in causing AD. In this review, we demonstrated that the effect of ER stress contributed to the pathogenesis of AD. ER stress associates almost the whole brain pathology processes which can be observed in AD, such as gene mutation of presenilin1, the abnormal clipped mRNA of presenilin2, β-amyloid production, tau phosphorylation, and cell death. The status of ER stress and unfolded protein response in the pathogenesis of AD also suggests they can be used as potential therapeutic agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease is a neurodegenerative disease which is characterized by progressive cognitive impairment and behavioral disorder. According to a recent report from Alzheimer’s Association, AD has become the sixth leading cause of all deaths in the USA and the fifth leading cause of death in Americans aged ≥65 years and deaths depend on it have been rising dramatically [1, 2]. Although the causes of AD are largely not yet known, most experts agree that AD, like other common chronic diseases, probably develops as a result of multiple factors rather than a single cause. Neuropathologically, AD has been mainly characterized by the accumulation of extracellular plaques of the Aβ and intracellular neurofibrillary tangles [3]. In addition, oxidative stress, synaptic loss, and neuronal degeneration are also being taken into consideration. However, although so many hypotheses there are, the exact pathogenesis of AD still remains unclear.
Several lines of evidence have indicated the importance of stress in subcellular organelles as a pathophysiological mechanism. Perturbations in the function of ER are emerging as relevant factors of AD. ER is a vital organelle which plays an important role in maintaining the balance of cellular Ca2+ and modifying the proteins after translation. Much pathological and physiological stimulation, such as ischemia, hypoxia, and poison, induces ER stress, which is characterized as overexpression of ER molecular chaperones and UPR in order to promote normal protein folding and degrade the abnormal. However, when activation of the UPR is severe or prolonged enough, the final cellular outcome is pathologic apoptotic cell death. The accumulation of unfolded protein in the ER lumen could be sensed and then induce a series of reactions. The mechanisms associated with ER stress in AD are diverse and complex, from gene to protein folding. Twenty years ago, Philip J. Thomas demonstrated the pathobiology and processes of the effect of altering protein folding into many disease-causing mutations and modifications [4]. Following by more and more UPR activation markers being detected in postmortem brain for AD patients and animal or vitro models, the relationship between ER stress and AD attracts increasing attention. Here, we will review the recent progress in the studies on the ER dysfunction, including its classical signaling pathways, and its association with onset of Alzheimer’s disease in various aspects in order to develop new treatments toward AD.
The Physiological Functions of ER
ER is a type of organelle in the cells of eukaryotic organisms whose membranes are continuous with the outer membrane of the nuclear envelope. ER serves as many general functions such as the folding of protein molecules in cisternae and the transport of synthesized proteins in vesicles to the Golgi apparatus.
Only properly folded proteins are transported from the rough ER to the Golgi apparatus. The so-called unfolded proteins, which are not properly folded into their functionally active structures, are retained in the lumen of ER until they attain their proper conformations. If this final tertiary structure cannot be achieved, unfolded proteins are then transported back to the cytosol and subjected to ubiquitination and proteasome-dependent degradation, a process referred to as ER-associated degradation (ERAD) [5, 6]. In the ubiquitin-proteasome pathway, energy from ATP is used to tag an unwanted protein with a chain of ubiquitin marking it for destruction. The protein is then hydrolyzed into small peptide fragments by the proteasome (Fig. 1).

Normal state. Proteins that enter the ER are folded with the help of chaperones and transported to the Golgi apparatus. GRP78 is bound to the luminal domains of PERK, IRE1, and ATF6 so that to remain them inactive. Misfolded proteins are transported back to the cytosol and subjected to ubiquitination and proteasome-dependent degradation
The ER Stress Signaling Pathways
Unfolded Protein Response
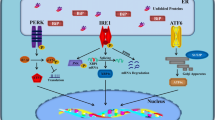
UPR is a kind of self-protection system triggered by ER stress in case of being damaged from some physical or biological factors. These responses include molecular chaperone increase, translational attenuation, and Endoplasmic reticulum-associated protein degradation (ERAD) activation in eukaryotic cells. One of the most representative examples of ER-resident chaperones is the immunoglobulin binding protein binding immunoglobulin protein (BiP), which supposed to prevent the secretion of incompletely assembled immunoglobulin [7]. Three transmembrane signal transducers work as sensors of the UPR including PERK, IRE1, and ATF6 (Fig. 2).

Three UPR arms in eukaryotes. During ER stress, GRP78 dissociates from PERK, IRE1, and ATF6 and lead them active. PERK phosphorylate eIF2α which transient attenuate protein translation in order to decrease in protein loading into ER. However, eIF2α phosphorylation selectively translates ATF4 and induces Nrf2 nuclear import. IRE1 arm splices XBP-1 mRNA which lead a frameshift of this protein and its nuclear import regulate UPR target genes. Activated ATF6 translocate to the Golgi by COPII vesicle where it is cleaved by S1P and S2P, and the cytoplasmic tail of ATF6 acts as a transcription factor to regulate UPR target genes
PKR-Like Endoplasmic Reticulum eIF2α Kinase (PERK)
PERK is a type I transmembrane kinase resident in the ER which plays a central role in the regulation of the UPR. Normally, PERK is held in an inactive state by binding of the GRP78 as supported by reports that GRP78 and PERK form a complex under normal conditions [8]. However, in ER stressed cells, as an increase of unfolded proteins in the lumen of ER, GRP78 is titrated away from PERK and this dissociation leads PERK to oligomerization and self-phosphorylation.
As PERK being activated, it could phosphorylate the α-subunit of eukaryotic translation initiation factor-2 (eIF2α) at Ser51 [9]. In this way, PERK helps reduce the flux of protein which is destined to enter the already stressed ER lumen so that to alleviate ER stress [5]. As the proteins being inhibited ubiquitously, an activating transcription factor 4 (ATF4) escaped. The target genes of ATF4 are transcription factor C/EBP homologous protein (CHOP), growth arrest, and DNA damage-inducible 34 (GADD34) and activating transcription factor 3 (ATF3).
CHOP is widely regarded as a participator in the initiation of apoptotic or organ regeneration which activates the transcription of several genes that may potentiate apoptosis. However, in a model of severe ER stress, the upregulation of CHOP increase the intensity of stress and thus induce cell death. Nevertheless, another model of milder one shows the opposite. This indicates that the intensity and duration of the stress should be emphasized; sine the beneficial or deleterious outcome is based on them [10]. GADD34 protein binds the catalytic subunit of protein phosphatase 1 (PP1c) and serves as a negative feedback loop to deactivate PERK action by dephosphorylating eIF2α, and so brings about the recovery of protein translation after its initial inhibition [11]. ATF3 is a member of the ATF/CREB subfamily of basic-region leucine zipper (bZIP) proteins, which contributes to expression of CHOP and GADD34.
Another effector protein that is phosphorylated by PERK is nuclear factor erythroid2-related factor 2 (Nrf2), which is a component of Nrf2/Keap1 complex. PERK-dependent phosphorylation triggers the dissociation of Nrf2/Keap1 complexes and thereby allows Nrf2 nuclear import. Moreover, Nrf2 nuclear translocation is independent of eIF2α phosphorylation. Finally, targeted deletion of Nrf2 reduces cell survival following ER stress. These findings suggest that Nrf2 functions as an effector of PERK cell survival signaling [12].
Inositol-Requiring Enzyme 1 (IRE1)
Like PERK, IRE1 is also a type I transmembrane kinase. There are two subtypes of IRE1 in mammalian cells, IRE1α and IRE1β. IRE1α is expressed ubiquitously, while the expression of IRE1β is limited to cells of the gut. Besides, mice knockouting IRE1α exhibit early embryonic lethality, while mice knockouting IRE1β express phenotypic normally [13, 14]. But both are essential for activation of the UPR. During ER stress, IRE1-bound GRP78 is released from IRE1. Once this release occurs, the structure of IRE1 allows it to bind to unfolded proteins, IRE1 active and protect cells from further damage. On the other hand, the splicing of X-box binding protein 1 (XBP1) mRNA can be induced by IRE1α when ER stress occurs which lead to the removal of a 26-nucleotideintron from XBP1 mRNA and a protein-frameshift that generates the active form of this protein [15]. It used to be widely accepted that IRE1α signaling was suspended during irremediable ER stress to initiate apoptosis [16]. However, more and more evidences show that IRE1α persistently adjusts the capacity of protein folding and directs UPR signaling [17, 18]. By the way, a recent research shows that the action of IRE1α on miRNA biogenesis is direct and that it antagonizes classical processing by DICER to derepress a translational block to entry into apoptosis [17].
Activating Transcription Factor 6 (ATF6)
Similar with the former two sensors, ATF6 is also a transmembrane protein which remains inactive in the unstressed cells because of binding to BiP. However, in response to ER stress, BiP dissociation permits Golgi localization sequence sites of ATF6 explosion and ATF6 transforming to the Golgi complex via COP II vesicles, where ATF6 is sequentially cleaved by two proteases (site-1 protease and site-2 protease) to yield a N-terminal cytosolic fragment, ATF6(N) that migrates to the nucleus to further activate transcription of UPR-responsive genes [19]. However, it is unclear if BiP dissociation is the sole driving force for ATF6 translocation. The serine protease site-1 protease (S1P) cleaves ATF6 in the luminal domain and the N-terminal portion is subsequently cleaved by the metalloprotease site-2 protease (S2P) [20]. ATF6 cleavage does not, however, occur when cells are deprived of sterols, nor is cleavage dependent on the sterol-responsive element binding protein (SREBP) cleavage activating protein (SCAP), which escorts SREBP to the Golgi for proteolysis [21].
Evidence of ER Dysfunction in AD
A number of reports have described manifestations of ER stress in postmortem brain samples from AD patients; continuously, there are also papers that report the presence of ER stress in animal and vitro models. To human samples, for instance, BiP increased in neurons which associated with amyloid deposits in AD brains [22]. Besides, PERK and eIF2α phosphorylation is also increased in AD brains [23, 24], and CHOP is upregulated in the temporal cortex of AD brains [25]. There’s also occurrence of XBP1 mRNA splicing in AD temporal cortex and hippocampal tissue [25]. In addition, heat shock protein (HSP72) is increased surrounding neuritic plaques and neurofibrillary tangles from AD brains [26]. In animal and vitro models, using drugs or gene knockout and gene silence technology further confirm the association between ER dysfunctions and AD.
Mechanisms Linking ER Stress to AD
ER Stress and Presenilin
PS1 and PS2 are highly homologous; they both are important components of the γ-secretase complex. γ-secretase containing mutation-altered presenilin could still catalyze the cleavage of β-amyloid precursor protein (APP), but the proteolytic site is altered. General γ-secretase yields mostly Aβ40 with smaller amounts of Aβ42, but mutant γ-secretase produces more Aβ42. However, Aβ42 is more amyloid genic and more prone to aggregate than Aβ40. In addition, calcium dysregulation has been presumed as another pathway by which the presenilins contribute to the pathogenesis of AD. That’s why presenilin could cause AD. On the other hand, presenilin has a close association with ER stress. We will review the association between ER stress to PS1 and PS2 separately in the following article.
ER Stress and PS1
PS1 is a component of the γ-secretase which is taking part in the cleavage process of APP. A research of a family with Alzheimer’s disease shows that mutation of PS1 gene was discovered among patients and their progeniture are also carriers of this mutation. Mutated presenilin transfected into cultures of murine neuroblastoma and human kidneys provoked production of β-amyloid with increased Aβ42/40 ratio [27]. Mutant PS1 may selectively increase Aβ42 secretion when N-glycosylation is impaired. As PS1 is an integral membrane protein which is mainly located in the ER, it leads us to study the relationship between PS1 mutations and the ER stress.
PS1mutations connect with ER stress by interfering with the signaling pathways of the UPR. And the decrease of GRP78 mRNA in PS1 mutation brain indicates that this mutation could perturb the stress response by inhibiting the unfold protein response and therefore increase cells susceptibility to ER stress. However, another research shows contrarily: neither the activation of IRE1α and PERK, nor the coordinate induction of BiP and CHOP mRNA and protein is impaired in cells lacking PS1 function. In addition, in contrast to previous work that reported diminished levels of BiP in brains of patients with PS1-linked FAD, they failed to find significant decreases in the levels of BiP in the brains of individuals with sporadic AD, or patients with FAD carrying PS1 mutations [28]. These conflicts are possibly because the effects of PS1 mutations could be masked by treatment with excessive doses of ER stress inducers or by prolonged stimulation. Another possibility might be the cells latter used are less sensitive to ER stress. What’s more, to detect those subtle defects of BiP/GRP78 mRNA in PS1 mutation-expressing cells, the cells need to be carefully handled under the same experimental conditions. At present, this area is still controversial. It also indicates that the expression of BiP alone is inadequate to assess the activation of unfolded-protein-response signaling. The phosphorylation of eIF2α is proven to be increased in PS1 mutant knock-in mice, in other words, PS1 could inhibit eIF2α phosphorylation [29]. In fact, FAD- mutant PS1 disturbs the UPR by attenuating both the activation of PERK and the phosphorylation of eIF2α. From the researches upon, FAD- mutant PS1 influences the UPR by various aspects.
Damaged ER organization and loss of general ER functions were also found in connection with presenilin-1 mutation. Furthermore, disordered ER homeostasis, not reversed by the mechanisms of UPR, could result in autophagy [24, 30]. There is much evidence that the homeostasis of autophagocytosis is disturbed in AD which displays protein aggregation [31]. Aging, a major risk factor of AD, is also associated with defects in autophagocytosis [32]. What’s more, recent studies have revealed that the stimulation of autophagy can reduce amyloid-β accumulation and alleviate memory deficits in the transgenic AD mice [33].
In addition, some cell culture studies show that the PS1 mutation perturbs subcellular calcium homeostasis and increases production of free radicals in affected cells. PS1 mutations sensitize neurons to DNA damage-induced death by promoting ER-mediated apoptotic proteolytic cascades such as activation of calpains and caspase-12 [34]. Moreover, mutations in PS1 increase cellular susceptibility to apoptosis induced by various insults, including withdrawal of trophic factors and exposure to Aβ.
ER Stress and PS2
PS2 is another component of the γ-secretase except PS1. Recently, a study shows that PS2 mutation has a close relationship with the onset of cognitive impairment in associative trace eyeblink conditioning in APP transgenic mice. This is the first study to elucidate the effect of PS2 mutation on mouse eyeblink conditioning [35]. An alternative spliced form of the PS2 genes (PS2V) lacking exon 5 has previously been reported to be expressed in human brains in sporadic Alzheimer’s disease (AD). Gene lack of exon 5 expresses frameshift mutations in 6 exon and produces terminator codon leading to premature translation termination. PS2V-encoding protein expresses mainly in the hippocampal CA1 region and temporal cortex in AD patients.
Under the condition of the ER stress, genes such as GRP78/BiP and GRP94 are widely known to be increased right away to refold the unfolded proteins and protect cells from injury. However, the process seems to be attenuated in PS2 expressed cells. The decrease of GRP78 mRNA is because of the impaired phosphorylation of IRE1, which supposed to be phosphorylated dealing with the unfolded protein in the ER [36]. What’s more, PS2V protein could significantly stimulate the production of both amyloidβ40 and β42 and changes the structure of tau protein, which is a major component of neurofibrillary tangles [37]. In order to explain the impaired phosphorylation, researchers do some experiments showing that PS2V directly binds to IRE1 on the membrane of the ER. Nevertheless, the cause of the decrease in phosphorylated IRE1 by PS2V is yet to be studied [36]. Researchers purify high mobility group A protein 1α which could bind to DNA as a transcription factor and adjacent to the 5′splice site of the PS2 pre-mRNA. The expression of HMGA1 proteins is detected to be increased though researches of the hippocampus of sporadic AD patients [38]. Therefore, hippocampus of sporadic AD patients exist abnormal mRNA clip which induces the impairment of ER stress response mechanism as an important factor of neurodegeneration.
ER Stress and Aβ
Aβ is the main component of extracellular senile plaques which produced from sequential proteolytic cleavages of the type 1 transmembrane APP by β- and γ-secretase whose neurotoxicity plays a significant role in the development of AD. It induces neuronal degeneration and apoptosis which relate closely to the cognitive dysfunction of AD patients. There’s a new hypothesis that AD memory failure is caused by small soluble oligomers of the Aβ peptide, toxins that target and disrupt particular synapses. It is now supported by more than a decade of further investigation, with over 1,000 papers addressing the oligomer hypothesis [39].
Under normal circumstances, GRP78/BiP could bind the APP in order to inhibit the production of Aβ. However, when ER stress responses abnormally, such as PS1 mutation and the aberrant splicing of PS2, the expression of molecular chaperone would be inhibited leading to the increase of the production of Aβ and the vulnerability to ER stress [36]. APP supposed to be glycosylated at its amino terminal and then transports to Golgi complex to process c-terminus glycosylation. But in cells lack of ΔIREl, this process will be inhibited leading unfolded and abnormal hydrolysis of APP in ER to the increase of Aβ. On the other hand, it has been proposed that Aβ can directly mediate ER stress responses and apoptosis. In addition, ER Ca2+ release which involved in oligomer-induced GSK-3β activation and tau phosphorylation could cause AD [40]. What’s more, Aβ-induced sustained activation of the ER stress which could be detected by the significant increases of ATF4, unspliced XBP1, spliced XBP1, active ATF6α, and the ER chaperone GRP78/BiP in cultured cells could cause brain endothelial cell death which deposits in the cerebral vessels in many AD patients and transgenic mice. To elaborate, incubation of rat brain endothelial cells treated with Aβ show significant decrease in cell lifespan and cell viability but increase in the number of apoptotic cells. Additionally, as the result of treatment with Aβ, the calcium homeostasis in or outside ER may be impaired. More Ca2+ release from ER to cytosolic, though before the activation of apoptotic cell death, also contribute significantly to the pathological process of AD [41]. As to apoptosis, Aβ1–42 activates caspase-12 in primary neurons through calpain activation and caspase-12 knockout neurons are partially resistant to Aβ-induced cell death [34]. The presence of functional mitochondria is required for ER stress-mediated apoptotic cell death and there is a crosstalk between ER and mitochondria in Aβ-treated cells [42]. On the contrary, mitochondrial dysfunction affects the ER stress response by enhancing the neuronal susceptibility to Aβ-induced ER stress [43].
In addition, Aβ could disrupt the anchoring between ER and microtubules (MT) in order to affect the architecture of the ER and induce collapse of ER which may play an important role in Aβ peptide-triggered neurodegenerative processes. Remarkably, this process is independent on UPR. After ER architecture being altered, autophagy is triggered and lysosomal degradation enhanced, as shown by electron microscopy and live-cell imaging [44]. Using drugs to stabilize MT could partially inhibit collapse of the ER and induction of autophagy which could be a therapy approach of AD.
What is the effect of Aβ on the unfold protein response? Increased BiP expression detected on application of exogenous amyloid β to primary cortical neurons suggests that accumulation of the peptide could activate ER stress signaling ways [40, 45]. Actually, phosphorylation of PERK, p-eIF2a, and cleavage of ATF6 also could be markers of the evidence of activation of UPR in Aβ treated neuronal cells. However, short-term treatment of Aβ (within 6 h) preferential augmented activation of the PERK pathway in neurons [45]. In amyloid β- induced ER stress, PERK and eIF2α were phosphorylated which could promote the induction of ER chaperones and confers resistant to aggregated protein toxicity in neuronal cells. On the contrary, PERK silenced limited the eIF2α phosphorylation and enhanced cell death [40, 45]. In addition, amyloid-β42 treatment induces CHOP (also termed GADD153) expression both in cultured cells and rabbit hippocampus and pretreatment with CHOP antisense RNA improves survival after exposure to amyloid β, which suggests a role for CHOP in amyloid-β-mediated cell death. That’s because CHOP acts to transcriptionally inhibit protective cellular molecules such as Bcl-2 and glutathione. Moreover, IP3-mediated calcium release from ER, stimulated by Aβ exposure, mediated CHOP expression [46]. Moreover, Aβ could activate the ER stress response factor XBP1 in transgenic flies and in mammalian cultured neurons, and its active form XBP1s shows neuroprotective effect on the two different AD models (prevents the accumulation of free calcium in the cytosol), whereas knockdown of XBP1 exacerbates amyloid-β toxicity [47]. Intraneuronal accumulation of Aβ may be associated with ER stress in the brains of AD patients at an early stage.
ER Stress and Tau
In addition to senile plaques, intracellular neurofibrillary tangling of hyperphosphorylated aggregates of microtubule-associated protein tau leading to the formation of NFTs is another pathological hallmark of AD. As mentioned above, activation of UPR can be found in postmortem brains of AD patients, in turn, markers for activation of UPR are abundant in neurons with phosphorylated tau. Those observations suggest a close linkage between ER-stress and tau pathology.
UPR activation is observed in postmortem AD brains and is associated with the early stage of neurofibrillary degeneration and tau phosphorylation [48]. The immunoreactivity of p-PERK which was found to be co-localized with tau phosphorylation was markedly increased in the hippocampus of TgTauP301L mice compared to age-matched controls indicating that ER-stress was increased in aged TgTauP301Lmice [49]. OA which could reduce protein phosphatase 2A activity thereby induces hyperphosphorylation of tau [50] could trigger UPR [49]. The evidence could be the increase of the immunoreactivity of p-PERK, p-eIF2α, splicing of mRNA for xbp-1, and elevated levels of mRNA for GADD153. UPR activation (pPERK and pIRE1 phosphorylation) is prominently presenting cases that can be neuropathologically classified as FTLD-tau (frontotemporal lobar degeneration with taupathology), but is undetectable in other FTLD cases [51]. These data indicated that UPR activation is intimately connected with the accumulation and aggregation of tau. Brains from AD patients and rTg4510 tau transgenic mice all show abnormal accumulation of CD3∂ which is an ERAD substrate in ER and activation of PERK. The mechanism that how tau accumulation facilitating its deleterious is interacting with ER membrane and associated proteins that are essential for ERAD, including VCP and Hrd1. Interestingly, this process could be reversed if soluble tau was depleted, suggesting that strategies aimed at reducing soluble tau could be beneficial for tauopathies including AD [52]. Thapsigargin could inhibit intracellular Ca2+-transport ATPase and induce perturbation of intracellular calcium homeostasis in order to induce ER stress. The band intensity of phosphorylated tau at Thr231, Ser262, and Ser396 was found to increase by treatment with Thapsigargin. Those all indicate that ER stress could induce tau phosphorylated. These findings suggested that ER-stress and phosphorylation of tau could be induced by each other to form a vicious cycle in AD [49].
Another vitro studies suggest that Aβ1–42 oligomers could not only induce ER stress but also increase tau phosphorylation and compromise cell survival through a mechanism mediated by GSK-3β activation [40]. Moreover, GSK-3β is found to be increased co-localizing with pPERK in neurons form AD brain [48]. These findings suggest that UPR activation induced by Aβ is an early event during tau pathology and point to a functional crosstalk between these molecular mechanisms in tauopathies [53] (Fig. 3).

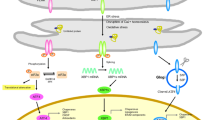
ER stress and neuronal cell death. IRE1α induces the activation of the ASK1, and then JNK activation followed, ending with apoptosis. Besides, Aβ could activate the ASK1 cascade which induces apoptosis in cells. In addition, PERK pathway induces CHOP which activated GADD34 to promote protein dephosphorylation of elF2α and enhances ROS production as a result of cell death. However, CHOP inhibited BCL-2 family expression. Activated IRE1 on the ER membrane recruitsTRAF2 and then activates JNK which could induce autophagy during ER stress. There is a crosstalk between ER and mitochondria in Aβ-treated cells. Under ER stress, a caspase-mediated apoptotic cell death pathway that involves caspase-9 and caspase-3 activation is triggered, leading to cell death
ER Stress and Neuronal Cell Death
Neuronal death is the most common and critical feature of neurodegenerative disorders such as Alzheimer’s disease. As autophagy plays protective roles in the pathogenesis of AD, apoptotic plays the opposite. In spite of many studies used to focus on mitochondria as a key mediator of cell death in AD, several recent reports suggested that neuronal death in AD has its origin in ER (Fig. 4).

ER stress and tau. The phosphorylation of PERK and Aβ1–42 oligomers both could activate GSK-3β to induce tau phosphorylation. On the other hand, p-PERK, p-eIF2α, splicing of mRNA for xbp-1, and elevated levels of mRNA for GADD153 could also be detected under ER stress. Microtubule bound with tau remains stable but dissociation when loss of tau binding and tau sequestered in NTFs
ER Stress and Apoptotic
ER stress triggers an adaptive program called the UPR, which aims to clear unfolded proteins and restore ER homeostasis. However, when the insult is too great, ER stress cannot be reversed; apoptotic cell death will often follows [10].
Among the UPR signaling pathways, IRE1α is the most important protein that plays as a regulator of cell fate in apoptosis activation [17]. IRE1α could induce the activation of the apoptotic-signaling kinase-1 (ASK1), which subsequently causes activation downstream of stress kinases Jun-N-terminal kinase (JNK) and p38 MAPK that promote apoptosis as a component of the IRE1-TRAF2-ASK1 cascade [54]. The substrates of JNK are Bcl-2 and Bim, which are, respectively, inhibited and activated by JNK phosphorylation. Besides, ASK1-mediated JNK activation has the potential to incite AD pathogenesis through increasing Aβ production, enhancing inflammation response, or even inducing NFTs aggregation. In addition, p38 MAPK phosphorylates and activates CHOP, which causes changes in gene expression that favor apoptosis, including increasing expression of Bim and DR5, while decreasing expression of Bcl-2 [55]. By the way, Aβ could activate the ASK1 cascade which induces apoptosis in SH-SY5Y cells [56]. Those evidences prove that ASK1 might be a new approach of therapeutic intervention to prevent or treat AD. In addition, PERK/eIF2α-dependent pathway induces the pro-apoptotic transcriptional factor CHOP. CHOP-mediated activation of GADD34 promotes protein dephosphorylation of elF2α reversing translational inhibition which contributes to permit translation of mRNAs encoding pro-apoptotic proteins [55]. GADD34 expression enhances reactive oxygen species (ROS) production possibly by promoting protein synthesis, which may trigger proteotoxicity and induces cell death. Most attention has been paid to BCL-2 family members in cell death induced by CHOP. BCL-2 is a superfamily of proteins that includes both pro-apoptotic and anti-apoptotic proteins. The expression of BCL-2 is reduced by CHOP under ER stress which induces cell apoptosis. In addition, some components of the BCL-2 family such as BH3-only proteins, BIM, PUMA, and NOXA also exhibited an increase under ER stress conditions. Remarkable, the hypersensitivity of PERK-deficient cells to ER stress is reversed by silencing of NOXA, suggesting an important role of the BCL-2 family of proteins in the cell death process [57].
As mentioned above, we have demonstrated that the PS1 mutants and the aberrant splicing of PS2V increase the vulnerability of cells to ER stress and induce apoptosis in FAD and SAD, respectively. However, what are the specific molecules which are taking part in cell death in the case of AD still remains unclear [58]. It has been reported that caspase-12 knockout mice show resistance to death caused by apoptosis induced β-amyloid protein, which is approved to play the most important role in the occurrence of neuronal apoptosis in AD [59]. Caspase-12 is specifically localized on the outer membrane of ER which needs cleavage for activation and calpain is in charge of this cleavage at N-terminal region. It has been reported that GRP78 binds to caspase-7 and caspase-12, and prevents the release of caspase-12 from ER. Under ER stress conditions, caspase-7 associates with caspase-12 and cleaves it into active form [60]. However, unfortunately it was later found that caspase-12 is only expressed in mice and rats. Later on, researchers using the sequence of mouse caspase-12 as a probe, obtain caspase-4 with a high similarity to caspase-12 which may be considered as human caspase-12 [58]. Since this enzyme is known to only exist in human cells, so it is impossible to conduct the caspase-4 knockout experiment in mice to insure its association with cell death. However, immunostaining on the ER membrane was decreased by using the RNAi-based knockdown technique, and cell death due to Tg stimulation was reduced. In contrast, cell death due to non-ER stress (Etop) was not affected. In SK-N-SH cell immunostaining of caspase-4 shows increase in the cytoplasm of pyramidal cells from the hippocampal region of the brains of AD patients [58]. These findings suggest that caspase-4 may be a particular caspase in human and has a close connection with cell death in AD patients.
In addition, the perturbation of ER Ca2+ homeostasis may be another important link in ER stress and apoptosis. Acute and large release of Ca2+ from the ER can trigger a variety of signaling mechanisms that induce cell death while the depletion of Ca2+ in ER can induce ER stress. So it is an interactional process. As mentioned above, ER stress response could lead to the increase of the production of Aβ which could exactly activate the mitochondrial-mediated apoptotic pathway by a mechanism involving the release of Ca2+ from ER through channels associated with ryanodine receptors (RyR) and inositol 1,4,5-trisphosphate receptors (IP3R) [61]. When large numbers of Ca2+ are released to cytoplasm, mitochondria can capture them and collapse mitochondrial membrane resulting in the release of apoptosis factor, including cytochrome c, Smac/Diablo, HtrA2/Omi, and others [62, 61]. Due to the researches upon, apoptosis do have a great relationship with ER stress and increase the vulnerability to AD.
ER Stress and Autophagy
Autophagy is a self-sacrificing mechanism, which has important role in cell fate as well as maintaining cell metabolic balance [63]. Neurons, because of their extreme polarization, size and post-mitotic nature, they may be particularly susceptible to the accumulation of aggregated or damaged cytosolic compounds, or membranes, and depend on autophagy for survival [64]. Thus, the significant roles of autophagy in nervous system are associated with maintaining the balance between the formation and degradation of cellular proteins as defects in autophagy pathway have been related to neurodegenerative diseases, such as AD. Alterations of autophagy have also been observed in Alzheimer’s disease, for instance, some autophagosome-like structures were found in dystrophic neurites of Alzheimer’s disease patients and model mice. This probably owes to the impairment of autophagosome maturation into autolysosomes. There’s also a hypothesis that impaired autophagic flux provides a novel site for Aβ peptide production [65]. As mentioned above, ERAD is recognized as the predominant cellular mechanism for removal of unfolded proteins in the context of ER stress; however, recent studies have shown that autophagosome formation is increased during ER stress [66].
It has been shown that activated IRE1 on the ER membrane recruits tumor necrosis factor receptor-associated factor 2 (TRAF2) and then activates JNK. In IRE1-deficient cells or cells treated with JNK inhibitor, the autophagy induced by ER stress was inhibited. Those data indicate that activation of JNK through the IRE1-TRAF2 pathway is required and plays a role specifically in autophagy activation during ER stress [63]. However, in other cases of IRE1-deficient cells, the autophagy could be induced, indicating that a signaling pathway other than the IRE1-JNK pathway may also play important roles in the activation of autophagy signaling under ER stress. The previous findings proposed that the PERK-eIF2α pathway may associate with activation of autophagy under ER stress. However, recent data showed that the PERK-eIF2α pathway is not essential for ER stress-induced autophagy. PERK-deficient cells and ATF6 knockdown cells showed that autophagy was induced after ER stress in a manner similar to the wild-type cells [63]. Disturbance of autophagy rendered cells vulnerable to ER stress, suggesting that autophagy plays roles in protecting against the cell death induced by ER stress. So it is reasonable to assume that autophagy could be a therapeutic target for the treatment of AD. For example, upregulation of autophagy by the regulatory of rapamycin or its analog CCI-779 which are target of rapamycin (TOR) inhibitors, to protect against neurodegeneration seen in models in Drosophila and mice [67]. What’s more, ER stress induced by thapsigargin could be alleviated by 4-phenylbutyric acid (4-PBA) through the unfolded protein response related proteins including GRP78, GRP94, C/EBP homologous protein, phospho-eIF-2α, eIF-2α, phospho-JNK1, and phosphor-JNK2/3, JNK1, IRE-1α, PERK, and sXBP-1. This suggests that 4-PBA may also be a potential therapeutic agent against ER stress-associated pathologic situations [68].
Regulating the Machinery of ER Stress as a Target in the Treatment of AD
Various approaches can be optional for the therapy pathways. First of all, it is required that strategies should be made to enhance the resistance of cells to conditions related to ER dysfunction. Secondly, to block the pathological process which is the main cause to the functional ER impairment should be considered into therapeutic intervention design. Finally, action should be taken to stimulate the refolding of unfolded proteins and to protect cells from irreversible damage which leading by the accumulation of unfolded protein in ER stress conditions. Now, based on lab data, all three approaches are proved to have therapeutic value in suppressing or blocking the pathological process which cause ER stress [69].
The first is therapy approach enhance cells resistance. As represented above, the onset of Alzheimer’s disease has a close association with ER stress, so we hypothesize that enhancing cells resistance could confer protection under ER stress circumstances. Study shows HCT116 cells with a DICER hypomorphic mutation (Exn5/Exn5) or where DICER or DROSHA were knocked down were resistant to ER stress-induced cell death, showing that loss of miRNA biogenesis increased resistance to ER stress-induced cell death [70]. This suggests that disrupted miRNA biogenesis may be a target for future therapeutic treatments.
The secondly approach is to block the pathological process. A major pathological process resulting in ER dysfunction in various acute disorders and degenerative diseases of the brain is oxidative stress caused by a rise in reactive oxygen species to levels exceeding antioxidant activity. This allows us to predict that drugs with antioxidant activity are prominent candidates for AD prevention and therapy [69]. The current clinical commonly used drugs are Selegiline, Vitamin E, melatonin, gingko biloba, and so on. However, there is still a long way to go in the development of the antioxidant drugs.
Finally, its turn for the last available approach help in stimulating refolding of proteins. Chemical chaperones can be the chief element to handle this problem. Chemical chaperones are a group of low-molecular mass compounds that stabilize the folding of proteins and buffer abnormal protein aggregation. Chemical chaperones like DMSO and TMAO have been studied in vitro, and showed reduced cytotoxicity and cell death, which has been reported to be a good therapeutic strategy [71]. Drugs such as geldanamycin could modulate and enhance chaperone levels [72–74]. Once a study shows Geldanamycin, which specifically binds hsp90 and GRP94, is a potent inducer of the cellular response to stress in the ER, resulting in the transcriptional upregulation of ER chaperones and expression of the gadd153/CHOP transcription factor. Besides, a lot of data strongly suggest that the increase in mRNA levels is due to geldanamycin effects on GRP94 but not hsp90. But then again, Hsp90 is a major cellular chaperone whose function is to maintain protein quality control and assist in protein degradation. However, much evidence indicates that as the development of the age and the enhanced oxidative stress, this system becomes less efficient which leads to oxidation and nitration of proteins, as well as the chaperones themselves, allowing for the accumulation of more misfolded proteins [75–77]. This indicates that Hsp90 inhibitors could provide therapeutic benefits in AD. For instance, phenylbutyrate, blocking the binding of ATP to Hsp90 protein [78], delays cognitive deficits and reduces tau pathology in AD mouse model [79]. A study conducted by Terracciano, S. et al finds that dimeric and trimeric triazole-based molecules seem to be a new class of Hsp90 molecular chaperone inhibitors which may provide a new approach for the treatment of AD.
Conclusions and Future Perspectives
Thanks to neuroscientists’ hard work, the processes induced by ER dysfunction and their definitive associations with AD have been demonstrated in considerable detail in the last decade. And modulation of these machineries allows the development of therapeutic strategies for neurological diseases that are caused by ER dysfunction. However, as we continue to explore, there are many terra incognita waiting.
Future studies are much required to understand the physiological significance of ER stress and UPR signaling in disease pathogenesis. For example, Salubrinal could prolong the phosphorylation of eIF2α and has been shown to be protective in cells under ER stress [80]. However, there is conflicting evidence that whether inhibiting or activating eIF2α-P is the prudent approach to be take when modulating the UPR. The answer may lie in the nature of the ER stress, and more importantly, in its duration. Future studies should be conducted to detect what level of UPR signaling is ideal for enabling cellular adaptation and survival? And which process of UPR could be the most effective one for drugs to target? How can we selectively modulate the ER stress responses in sick but not healthy cells? As ER stress pathways have been found to promote antitumor immunity by enhancing immunogenicity of dying cancer cells, there’s an emerging link between ER stress, cell fate decisions, and immunomodulation and the potential therapeutic benefit of targeting this multifaceted signaling pathway in anticancer therapy [81]. Does ER stress in the onset of other diseases like cancer and diabetes have the same mechanism to neurological diseases? Whether we can draw on the experience of treatment of other diseases? What’s more, it remains to be determined whether or not the UPR participates in cognitive functions of the nervous system. This could also contribute a lot for treating nervous system diseases. The mechanisms leading to accelerated neuronal cell death in AD are still largely unknown, we wonder if ER interconnects with other cellular organelles to modulate cell death. All in all, we sincerely hope researchers could put more efforts on the precise mechanism of UPR and ER stress for new treatments which would surely benefit a lot to protect AD patients.
References
2011 Alzheimer’s disease facts and figures (2011) Alzheimer’s & dementia 7 (2):208-244 doi:10.1016/j.jalz.2011.02.004
Jiang T, Yu JT, Tian Y, Tan L (2013) Epidemiology and etiology of Alzheimer’s disease: from genetic to non-genetic factors. Curr Alzheimer’s Res 10(8):852–867
Jiang T, Yu JT, Tan L (2012) Novel disease-modifying therapies for Alzheimer’s disease. J Alzheimer’s Dis JAD 31(3):475–492. doi:10.3233/JAD-2012-120640
Thomas PJ, Qu B-H, Pedersen PL (1995) Defective protein folding as a basis of human disease. Trends Biochem Sci 20(11):456–459. doi:10.1016/S0968-0004(00)89100-8
Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334(6059):1081–1086. doi:10.1126/science.1209038
Smith MH, Ploegh HL, Weissman JS (2011) Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334(6059):1086–1090. doi:10.1126/science.1209235
Haas IG, Wabl M (1983) Immunoglobulin heavy chain binding protein. Nature 306(5941):387–389
Sanderson TH, Deogracias MP, Nangia KK, Wang J, Krause GS, Kumar R (2010) PKR-like endoplasmic reticulum kinase (PERK) activation following brain ischemia is independent of unfolded nascent proteins. Neuroscience 169(3):1307–1314. doi:10.1016/j.neuroscience.2010.05.076
Marciniak SJ, Garcia-Bonilla L, Hu J, Harding HP, Ron D (2006) Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J Cell Biol 172(2):201–209. doi:10.1083/jcb.200508099
Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ (2013) Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol 12(1):105–118. doi:10.1016/S1474-4422(12)70238-7
Park SW, Ozcan U (2013) Potential for therapeutic manipulation of the UPR in disease. Semin Immunopathol 35(3):351–373. doi:10.1007/s00281-013-0370-z
Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23(20):7198–7209
Iwawaki T, Akai R, Yamanaka S, Kohno K (2009) Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci U S A 106(39):16657–16662. doi:10.1073/pnas.0903775106
Tsuru A, Fujimoto N, Takahashi S, Saito M, Nakamura D, Iwano M, Iwawaki T, Kadokura H, Ron D, Kohno K (2013) Negative feedback by IRE1beta optimizes mucin production in goblet cells. Proc Natl Acad Sci U S A 110(8):2864–2869. doi:10.1073/pnas.1212484110
Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415(6867):92–96. doi:10.1038/415092a
Chen Y, Brandizzi F (2013) IRE1: ER stress sensor and cell fate executor. Trends Cell Biol 23(11):547–555. doi:10.1016/j.tcb.2013.06.005
Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, Papa FR, Oakes SA (2012) IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 338(6108):818–822. doi:10.1126/science.1226191
Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR (2009) IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138(3):562–575. doi:10.1016/j.cell.2009.07.017
Malhotra JD, Kaufman RJ (2007) The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 18(6):716–731. doi:10.1016/j.semcdb.2007.09.003
Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6(6):1355–1364
Patil C, Walter P (2001) Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 13(3):349–355. doi:10.1016/S0955-0674(00)00219-2
Hoozemans JJ, Stieler J, van Haastert ES, Veerhuis R, Rozemuller AJ, Baas F, Eikelenboom P, Arendt T, Scheper W (2006) The unfolded protein response affects neuronal cell cycle protein expression: implications for Alzheimer’s disease pathogenesis. Exp Gerontol 41(4):380–386. doi:10.1016/j.exger.2006.01.013
O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hebert SS, De Strooper B, Haass C, Bennett DA, Vassar R (2008) Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 60(6):988–1009. doi:10.1016/j.neuron.2008.10.047
Nijholt DA, de Graaf TR, van Haastert ES, Oliveira AO, Berkers CR, Zwart R, Ovaa H, Baas F, Hoozemans JJ, Scheper W (2011) Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: implications for Alzheimer’s disease. Cell Death Differ 18(6):1071–1081. doi:10.1038/cdd.2010.176
Lee JH, Won SM, Suh J, Son SJ, Moon GJ, Park UJ, Gwag BJ (2010) Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp Mol Med 42(5):386–394. doi:10.3858/emm.2010.42.5.040
Hamos JE, Oblas B, Pulaski-Salo D, Welch WJ, Bole DG, Drachman DA (1991) Expression of heat shock proteins in Alzheimer’s disease. Neurology 41(3):345–350
Kulczycki J, Bertrand E, Lojkowska W, Dowjat W, Wisniewski T, Lyczywek-Zwierz M (2001) Familial Alzheimer’s disease connected with mutation in presenilin gene 1 (P117L). Neurol Neurochir Pol 35(2):213–224
Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M (1999) Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol 1(8):479–485. doi:10.1038/70265
Milhavet O, Martindale JL, Camandola S, Chan SL, Gary DS, Cheng A, Holbrook NJ, Mattson MP (2002) Involvement of Gadd153 in the pathogenic action of presenilin-1 mutations. J Neurochem 83(3):673–681
Hoyer-Hansen M, Jaattela M (2007) Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 14(9):1576–1582. doi:10.1038/sj.cdd.4402200
Nixon RA, Yang DS (2011) Autophagy failure in Alzheimer’s disease—locating the primary defect. Neurobiol Dis 43(1):38–45. doi:10.1016/j.nbd.2011.01.021
Salminen A, Kaarniranta K (2009) Regulation of the aging process by autophagy. Trends Mol Med 15(5):217–224. doi:10.1016/j.molmed.2009.03.004
Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA (2011) Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain J Neurol 134(Pt 1):258–277. doi:10.1093/brain/awq341
Takuma K, Yan SS, Stern DM, Yamada K (2005) Mitochondrial dysfunction, endoplasmic reticulum stress, and apoptosis in Alzheimer’s disease. J Pharmacol Sci 97(3):312–316
Kishimoto Y, Kirino Y (2013) Presenilin 2 mutation accelerates the onset of impairment in trace eyeblink conditioning in a mouse model of Alzheimer’s disease overexpressing human mutant amyloid precursor protein. Neurosci Lett 538:15–19. doi:10.1016/j.neulet.2013.01.025
Sato N, Imaizumi K, Manabe T, Taniguchi M, Hitomi J, Katayama T, Yoneda T, Morihara T, Yasuda Y, Takagi T, Kudo T, Tsuda T, Itoyama Y, Makifuchi T, Fraser PE, St George-Hyslop P, Tohyama M (2001) Increased production of beta-amyloid and vulnerability to endoplasmic reticulum stress by an aberrant spliced form of presenilin 2. J Biol Chem 276(3):2108–2114. doi:10.1074/jbc.M006886200
Manabe T, Ohe K, Katayama T, Matsuzaki S, Yanagita T, Okuda H, Bando Y, Imaizumi K, Reeves R, Tohyama M, Mayeda A (2007) HMGA1a: sequence-specific RNA-binding factor causing sporadic Alzheimer’s disease-linked exon skipping of presenilin-2 pre-mRNA. Genes Cells Devoted Mol Cell Mech 12(10):1179–1191. doi:10.1111/j.1365-2443.2007.01123.x
Manabe T, Katayama T, Sato N, Gomi F, Hitomi J, Yanagita T, Kudo T, Honda A, Mori Y, Matsuzaki S, Imaizumi K, Mayeda A, Tohyama M (2003) Induced HMGA1a expression causes aberrant splicing of Presenilin-2 pre-mRNA in sporadic Alzheimer’s disease. Cell Death Differ 10(6):698–708. doi:10.1038/sj.cdd.4401221
Ferreira ST, Klein WL (2011) The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem 96(4):529–543. doi:10.1016/j.nlm.2011.08.003
Resende R, Ferreiro E, Pereira C, Oliveira CR (2008) ER stress is involved in Abeta-induced GSK-3beta activation and tau phosphorylation. J Neurosci Res 86(9):2091–2099. doi:10.1002/jnr.21648
Fonseca AC, Ferreiro E, Oliveira CR, Cardoso SM, Pereira CF (2013) Activation of the endoplasmic reticulum stress response by the amyloid-beta 1-40 peptide in brain endothelial cells. Biochim Biophys Acta 1832(12):2191–2203. doi:10.1016/j.bbadis.2013.08.007
Costa RO, Ferreiro E, Cardoso SM, Oliveira CR, Pereira CM (2010) ER stress-mediated apoptotic pathway induced by Abeta peptide requires the presence of functional mitochondria. J Alzheimer’s Dis JAD 20(2):625–636. doi:10.3233/JAD-2010-091369
Costa RO, Ferreiro E, Oliveira CR, Pereira CMF (2013) Inhibition of mitochondrial cytochrome c oxidase potentiates Aβ-induced ER stress and cell death in cortical neurons. Mol Cell Neurosci 52:1–8. doi:10.1016/j.mcn.2012.09.005
Lai CS, Preisler J, Baum L, Lee DH, Ng HK, Hugon J, So KF, Chang RC (2009) Low molecular weight Abeta induces collapse of endoplasmic reticulum. Mol Cell Neurosci 41(1):32–43. doi:10.1016/j.mcn.2009.01.006
Lee do Y, Lee KS, Lee HJ, Kim do H, Noh YH, Yu K, Jung HY, Lee SH, Lee JY, Youn YC, Jeong Y, Kim DK, Lee WB, Kim SS (2010) Activation of PERK signaling attenuates Abeta-mediated ER stress. PloS One 5(5):e10489. doi:10.1371/journal.pone.0010489
Schapansky J, Olson K, Van Der Ploeg R, Glazner G (2007) NF-kappaB activated by ER calcium release inhibits Abeta-mediated expression of CHOP protein: enhancement by AD-linked mutant presenilin 1. Exp Neurol 208(2):169–176. doi:10.1016/j.expneurol.2007.04.009
Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez-Velazquez M, Rincon-Limas DE, Fernandez-Funez P (2011) The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum Mol Genet 20(11):2144–2160. doi:10.1093/hmg/ddr100
Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W (2009) The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol 174(4):1241–1251. doi:10.2353/ajpath.2009.080814
Ho YS, Yang X, Lau JC, Hung CH, Wuwongse S, Zhang Q, Wang J, Baum L, So KF, Chang RC (2012) Endoplasmic reticulum stress induces tau pathology and forms a vicious cycle: implication in Alzheimer’s disease pathogenesis. J Alzheimer’s Dis JAD 28(4):839–854. doi:10.3233/JAD-2011-111037
Kins S, Crameri A, Evans DR, Hemmings BA, Nitsch RM, Gotz J (2001) Reduced protein phosphatase 2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J Biol Chem 276(41):38193–38200. doi:10.1074/jbc.M102621200
Nijholt DA, van Haastert ES, Rozemuller AJ, Scheper W, Hoozemans JJ (2012) The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol 226(5):693–702. doi:10.1002/path.3969
Abisambra JF, Jinwal UK, Blair LJ, O’Leary JC 3rd, Li Q, Brady S, Wang L, Guidi CE, Zhang B, Nordhues BA, Cockman M, Suntharalingham A, Li P, Jin Y, Atkins CA, Dickey CA (2013) Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci Off J Soc Neurosci 33(22):9498–9507. doi:10.1523/JNEUROSCI.5397-12.2013
Ferreiro E, Pereira CM (2012) Endoplasmic reticulum stress: a new playER in tauopathies. J Pathol 226(5):687–692. doi:10.1002/path.3977
Song J, Park KA, Lee WT, Lee JE (2014) Apoptosis signal regulating kinase 1 (ASK1): potential as a therapeutic target for Alzheimer’s disease. Int J Mol Sci 15(2):2119–2129. doi:10.3390/ijms15022119
Sano R, Reed JC (2013) ER stress-induced cell death mechanisms. Biochim Biophys Acta (BBA) Mol Cell Res 1833(12):3460–3470. doi:10.1016/j.bbamcr.2013.06.028
Akterin S, Cowburn RF, Miranda-Vizuete A, Jimenez A, Bogdanovic N, Winblad B, Cedazo-Minguez A (2006) Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer’s disease. Cell Death Differ 13(9):1454–1465. doi:10.1038/sj.cdd.4401818
Gupta S, Giricz Z, Natoni A, Donnelly N, Deegan S, Szegezdi E, Samali A (2012) NOXA contributes to the sensitivity of PERK-deficient cells to ER stress. FEBS Lett 586(22):4023–4030. doi:10.1016/j.febslet.2012.10.002
Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M (2004) Induction of neuronal death by ER stress in Alzheimer’s disease. J Chem Neuroanat 28(1–2):67–78. doi:10.1016/j.jchemneu.2003.12.004
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403(6765):98–103. doi:10.1038/47513
Martinez JA, Zhang Z, Svetlov SI, Hayes RL, Wang KK, Larner SF (2010) Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis Int J Program Cell Death 15(12):1480–1493. doi:10.1007/s10495-010-0526-4
Ferreiro E, Oliveira CR, Pereira CM (2008) The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol Dis 30(3):331–342. doi:10.1016/j.nbd.2008.02.003
Vaux DL (2011) Apoptogenic factors released from mitochondria. Biochim Biophys Acta 1813(4):546–550. doi:10.1016/j.bbamcr.2010.08.002
Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A (2009) Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ 16(7):966–975. doi:10.1038/cdd.2009.33
Tooze SA, Schiavo G (2008) Liaisons dangereuses: autophagy, neuronal survival and neurodegeneration. Curr Opin Neurobiol 18(5):504–515. doi:10.1016/j.conb.2008.09.015
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA (2005) Macroautophagy—a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol 171(1):87–98. doi:10.1083/jcb.200505082
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K (2006) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26(24):9220–9231. doi:10.1128/MCB.01453-06
Jiang T, Yu JT, Zhu XC, Tan MS, Wang HF, Cao L, Zhang QQ, Shi JQ, Gao L, Qin H, Zhang YD, Tan L (2014) Temsirolimus promotes autophagic clearance of amyloid-beta and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol Res Off J Ital Pharmacol Soc. doi:10.1016/j.phrs.2014.02.008
Kim DS, Li B, Rhew KY, Oh HW, Lim HD, Lee W, Chae HJ, Kim HR (2012) The regulatory mechanism of 4-phenylbutyric acid against ER stress-induced autophagy in human gingival fibroblasts. Arch Pharm Res 35(7):1269–1278. doi:10.1007/s12272-012-0718-2
Paschen W, Mengesdorf T (2005) Cellular abnormalities linked to endoplasmic reticulum dysfunction in cerebrovascular disease–therapeutic potential. Pharmacol Ther 108(3):362–375. doi:10.1016/j.pharmthera.2005.05.008
Cawley K, Logue SE, Gorman AM, Zeng Q, Patterson J, Gupta S, Samali A (2013) Disruption of microRNA biogenesis confers resistance to ER stress-induced cell death upstream of the mitochondrion. PloS One 8(8):e73870. doi:10.1371/journal.pone.0073870
Yoshida H, Yoshizawa T, Shibasaki F, Shoji S, Kanazawa I (2002) Chemical chaperones reduce aggregate formation and cell death caused by the truncated Machado-Joseph disease gene product with an expanded polyglutamine stretch. Neurobiol Dis 10(2):88–99
Verhoef LG, Lindsten K, Masucci MG, Dantuma NP (2002) Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum Mol Genet 11(22):2689–2700
Winklhofer KF, Reintjes A, Hoener MC, Voellmy R, Tatzelt J (2001) Geldanamycin restores a defective heat shock response in vivo. J Biol Chem 276(48):45160–45167. doi:10.1074/jbc.M104873200
Piper PW (2001) The Hsp90 chaperone as a promising drug target. Curr Opin Investig Drugs 2(11):1606–1610
Cuervo AM, Dice JF (2000) Age-related decline in chaperone-mediated autophagy. J Biol Chem 275(40):31505–31513. doi:10.1074/jbc.M002102200
Lund J, Tedesco P, Duke K, Wang J, Kim SK, Johnson TE (2002) Transcriptional profile of aging in C. elegans. Curr Biol CB 12(18):1566–1573
Tonoki A, Kuranaga E, Tomioka T, Hamazaki J, Murata S, Tanaka K, Miura M (2009) Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol Cell Biol 29(4):1095–1106. doi:10.1128/MCB.01227-08
Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K (2005) Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 280(29):26729–26734. doi:10.1074/jbc.C500186200
Ricobaraza A, Cuadrado-Tejedor M, Perez-Mediavilla A, Frechilla D, Del Rio J, Garcia-Osta A (2009) Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 34(7):1721–1732. doi:10.1038/npp.2008.229
Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J (2005) A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307(5711):935–939. doi:10.1126/science.1101902
Verfaillie T, Garg AD, Agostinis P (2013) Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett 332(2):249–264. doi:10.1016/j.canlet.2010.07.016
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China to L.T. (81171209, 81371406) and J.T.Y. (81000544), the grants from the Shandong Provincial Natural Science Foundation to L.T. (ZR2011HZ001) and J.T.Y. (ZR2010HQ004), the Medicine and Health Science Technology Development Project of Shandong Province to L.T. (2011WSA02018) and J.T.Y. (2011WSA02020), and the Innovation Project for Postgraduates of Jiangsu Province to T.J. (CXLX13_561).
Conflicts of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Li, JQ., Yu, JT., Jiang, T. et al. Endoplasmic Reticulum Dysfunction in Alzheimer’s Disease. Mol Neurobiol 51, 383–395 (2015). https://doi.org/10.1007/s12035-014-8695-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8695-8