
Abstract
Remote neurodegeneration significantly influences the clinical outcome in many central nervous system (CNS) pathologies, such as stroke, multiple sclerosis, and traumatic brain and spinal cord injuries. Because these processes develop days or months after injury, they are accompanied by a therapeutic window of opportunity. The complexity and clinical significance of remote damage is prompting many groups to examine the factors of remote degeneration. This research is providing insights into key unanswered questions, opening new avenues for innovative neuroprotective therapies. In this review, we evaluate data from various remote degeneration models to describe the complexity of the systems that are involved and the importance of their interactions in reducing damage and promoting recovery after brain lesions. Specifically, we recapitulate the current data on remote neuronal degeneration, focusing on molecular and cellular events, as studied in stroke and brain and spinal cord injury models. Remote damage is a multifactorial phenomenon in which many components become active in specific time frames. Days, weeks, or months after injury onset, the interplay between key effectors differentially affects neuronal survival and functional outcomes. In particular, we discuss apoptosis, inflammation, oxidative damage, and autophagy—all of which mediate remote degeneration at specific times. We also review current findings on the pharmacological manipulation of remote degeneration mechanisms in reducing damage and sustaining outcomes. These novel treatments differ from those that have been proposed to limit primary lesion site damage, representing new perspectives on neuroprotection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since Broca’s assumption was first proposed, lesion mapping and functional localization have become keystones in interpreting brain function. Subsequently, localization theories have been challenged by network approaches that have shifted the study of structure-function correlations from strict cortical localization—sometimes called a topological perspective—to a more hodological, or network-based, approach [1]. According to the latter model, brain functions emerge from the flow of information across networks rather than from the activity of single cortical areas. The network-based approach emphasizes that deficits are related to the local effects of damaged regions and to the dysfunction of anatomically intact brain regions that are connected functionally to the lesioned areas [2].
There is much debate over the nature of these remote dysfunctions and how “anatomically intact” they are. Furthermore, the network model of brain injury highlights the potential of remote damage to act as a decisional node in functional outcomes. The growing interest in remote changes has been prompted primarily by neuroimaging evidence [3], but little neurobiological data on the mechanisms of such remote changes exist.
The development of functional neuroimaging techniques is providing greater insight into the functional interactions between brain areas but little information on the underlying cellular mechanisms. To examine the mechanisms that are associated with remote changes, data from experimental models must be considered [4]. In this review, we evaluate the findings from several remote degeneration models to describe the complexity of the systems that are involved and the significance of their interactions in mitigating damage and promoting recovery after development of a brain lesion.
Specifically, in the present review, we address the following aspects of remote damage:
-
Remote damage and recovery;
-
Animal models for remote central nervous system (CNS) degeneration;
-
Structural and functional changes;
-
Inflammation;
-
Apoptosis;
-
Autophagy;
-
Oxidative and nitrosative stress;
-
Retrograde signaling;
-
Purinergic system;
-
Nitrergic system;
-
Endocannabinoid system;
-
Systems interactions in the remote damage;
-
Therapeutic approaches: glucocorticoids, minocycline, rapamycin, and cannabinoid-based drugs.
Remote Damage
Remote Cell Death Mechanisms
Remote cell death is a complex phenomenon, many aspects of which are unknown. In contrast to chronic neurodegenerative disorders, for which much effort has been made to characterize the molecular components of neuronal death, the study of the effects and mechanisms of remote cell death damage is in its infancy. A more expansive understanding of the mechanisms and factors of remote cell death will help identify new effective treatments for this deceitful aspect of brain damage.
Following damage, two events characterize a brain injury: (1) immediate, or primary, damage that induces degeneration and cell death directly and (2) delayed, or secondary, damage that effects delayed degeneration and cell death through active, independent mechanisms. Secondary neuronal degeneration entails destructive downstream events that can affect cells that were unaffected or only marginally affected by the initial damage [5]. Usually, the degree of secondary neuronal damage is proportional to the extent of the initial injury—thus, more extensive and longer-lasting primary insults result in greater release of mediators of secondary degeneration.
The mechanisms of secondary degeneration are not limited to the lesion site and can involve remote areas. Secondary damage can occur next to an area that has experienced irreversible primary damage and distal areas that are functionally related to the primary site of injury. This delayed phenomenon has been termed “remote damage” [6] and can last for days, weeks, or months.
Remote damage might result from an axonal damage or from transneuronal effects. Transneuronal (or transsynaptic) degeneration may spread along anatomical and functional connections, and it can be either anterograde or retrograde, indicating the direction of the degeneration relative to the original site of damage [7].
Retrograde transneuronal degeneration is a form of degeneration involving neurons that are distal to the insult that lose their projection target. It is also termed “dying backward.” Conversely, anterograde transneuronal degeneration is a form of degeneration caused by loss of inputs. It is also termed “dying forward.” The mechanism of transsynaptic degeneration has been described in humans in a number of CNS diseases after focal damage in different brain circuits, including the cerebellar [8, 9], visual [10, 11], and corticospinal [12] systems.
Axotomized neurons progress through an orderly series of morphological changes before eventually dying, creating a window of opportunity during which death can be halted or delayed by the appropriate interventions.
The severity of remote cell death is believed to be related to several factors, including the type and extent of the primary insult, the distance of axonal trauma in relation to the soma, the type of connectivity, and the intrinsic vulnerability of the circuits that are involved [13, 14], whether the soma resides in the peripheral nervous system (PNS) or CNS, the animal species, and age at the time of injury [15, 16].
Heterogeneous neuronal populations differ with regard to their vulnerability, but disparities between apparently similar cell populations are a significant aspect of phenotypic variability. Variability in the extent and progression of atrophy and in intensity of degeneration after axotomy has been observed in seemingly homogeneous neurons of the dorsal lateral geniculate nucleus (dLGN) [17, 18] and inferior olive (IO) and pontine (Pn) precerebellar nuclei [19].
The reasons for this variability remain unknown [20]. Nevertheless, the few findings on the differences in morphological and biochemical profiles and cell vulnerability have been valuable in developing therapeutic interventions for specific neuronal systems and times [21].
Animal Models for Studying Remote Degeneration in the CNS
Neurodegeneration has become an important topic in neuroscience, and several animal models have been developed to gain insight into the pathophysiology of neuronal degeneration in acute insults (e.g., stroke, trauma) and chronic neurodegenerative diseases (e.g., amyotrophic lateral sclerosis, Alzheimer's disease). Less attention in neurodegeneration has been paid to animal models of remote damage. In general, remote damage studies have been based on methods that apply axotomy and target deprivation to examine the morphological, biochemical, and ultrastructural changes that occur days to months after injury in various brain circuits [14, 19, 22–26].
In this review, we will focus on three models—hemicerebellectomy (HCb), spinal cord injury (SCI), and occipital cortical ablation (OCA)—which are extremely useful in studying the mechanisms and pharmacology of remote cell death [4, 27–33].
The HCb Paradigm
The HCb paradigm has been used for over 50 years to study the mechanisms of remote neuronal degeneration and their significance in recovery after CNS injuries. HCb is the ablation of half of the cerebellum and has been used widely by many groups in behavioral, neurophysiological, and morphological studies [34]. The aim of HCb is to remove half of the vermis with one cerebellar hemisphere, including the deep cerebellar nuclei, and spare the vestibular nuclei and all surrounding structures. This approach is simple, effects low mortality, and has a high degree of reproducibility.
Because of the crossed input-output cerebellar organization, HCb damages the axons of all neurons of the contralateral IO and Pn and deprives the contralateral cerebral cortex of nearly all cerebellar input. Further, due to the projections of deep cerebellar nuclei (DCN) to the IO, HCb damages the IO due to axotomy and input deprivation. Thus, HCb is considered a mixed model of remote degeneration.
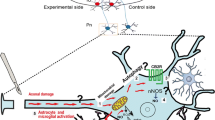
Based on the unilaterality of the lesion and the nearly complete crossover of the cerebellar input-output organization, it is possible to study an intact and a lesioned cerebellar circuit in the same animal. This model allows one to examine a damaged side and a spared, control side in the same anatomical section—a patent advantage when morphological or physiological comparisons are needed (Fig. 1).

Schematic of the hemicerebellectomy (HCb) model. Due to the crossed input-output organization of the cerebellar connections, unilateral lesion of a cerebellar hemisphere induces axotomy and subsequent damage of virtually all neurons of the contralateral (experimental side) IO and Pn, with sparing of the IO and Pn neurons on the ipsilateral side (control side). Thus, in the same section (lower part of the figure), both experimental (damaged) and control (spared) neuronal populations can be observed and compared. Note the easy comparison between neuronal degeneration of the experimental side versus the control one. DCN deep cerebellar nuclei, icp inferior cerebellar peduncle
HCb is performed primarily in rodents [35] and occasionally in cats [36] and monkeys [37, 38]. This unilateral cerebellar lesion causes extensive and persistent (up to 2 months) neuronal death of contralateral precerebellar nuclei [19, 26]. Notably, as in other remote damage models, not all degenerative phenomena develop simultaneously during this time. At any given point, neurons exist in various degenerative states, suggesting differences in neuronal sensitivity and time-specific activation of several reactive/compensative mechanisms. Different cellular and molecular phenomena are relevant in specific time windows. A time window table for the HCb model is reported in Table 1. The characteristics of these time-locked activations are the key element for the successful planning of neuroprotective strategies (see later).
In human pathologies, whereas degenerative cerebellar diseases are usually bilateral, focal cerebellar lesions due to stroke, bleeding, trauma, or surgery are often unilateral. Thus, unilateral HCb is suitable for examining degeneration and recovery clinically after development of a focal cerebellar lesion. A characteristic degenerative phenomenon observed postmortem [39] or antemortem by structural MRI [40, 41] is the so-called hypertrophic olivary degeneration (HOD). HOD occurs in the inferior olive as a consequence of a lesion within the “Guillain and Mollaret triangle” (dento-rubro-olivary pathway). HOD is a transsynaptic and delayed form of degeneration causing hypertrophy and subsequent degeneration of the inferior olivary nucleus. It occurs unilaterally and ipsilateral if the lesion is limited to the central tegmental tract and unilaterally and contralateral if the lesion involves dentate nucleus or superior cerebellar peduncle. If the lesion involves both central tegmental tract and superior cerebellar peduncle, bilateral HOD occurs.
The SCI Paradigm
Several models have been developed to study the mechanisms of injury and recovery after SCI, the most common of which are based on transection, compression, or contusion of the spinal cord with complete or partial injury.
Partial hemisection is generally adopted to study regeneration and supraspinal responses to injury. It typically damages the cervical or thoracic cord. Lesions at the cervical level involve the upper extremities, causing severe impairments, whereas thoracic-level injuries impede only lower limb function.
Hemisection is not debilitating, because it does not cause the bladder, bowel, or respiratory dysfunction that is generally observed after complete cord sectioning. Overall, cord dorsal hemisection is simple and is associated with low mortality and simple postoperative care [42].
Dorsal hemisection injury damages multiple descending spinal tracts including the corticospinal tract, the serotonergic raphespinal tract, and the rubrospinal tract. Although studies on axonal regeneration of these tracts after spinal cord hemisection and different treatments are numerous [43–45], the findings on the responses of the supraspinal remote axotomized neurons are still scarce and the results obtained are often contradictory. In this field, studies have been directed mainly towards the fate of the rubrospinal (see later) and, to a lesser extent, to corticospinal neurons [46–48], while the fate of raphespinal neurons has been completely neglected.
Due to the nature of the present review, we will discuss exclusively of morphological and structural changes occurring in rubrospinal neurons after SCI.
The rat rubrospinal tract emerges from the magnocellular section of the caudal red nucleus (RN), crosses over nearly completely (99 %), and descends in the dorsolateral aspect of the spinal cord [49]. This anatomical position renders severance of the entire rubrospinal tract by partial hemisection easy. When performed at the cervical level, a dorsal hemisection axotomizes nearly the entire contralateral neuronal population of the magnocellular pars. This lesion results in severe shrinkage, atrophy, and eventually death of rubrospinal neurons [50–57].
The response of rubrospinal neurons to spinal hemisection, particularly regarding neuronal death, has been studied extensively, predominantly in rodents [33, 57–60] but also in opossum [61] and primates [62]. Despite the consensus that neuronal atrophy occurs after axotomy, rates of cell loss differ between and within species [63]; thus, whether cells die or merely became atrophic remains unknown [64].
High cervical unilateral hemisection injuries in humans cause Brow-Sequard syndrome, which is characterized by ipsilesional motor weakness or paralysis and loss of proprioception with contralesional deficits in pain and temperature sensation [65, 66]. Because these symptoms also develop after cervical hemisection in rodents, this paradigm is considered a sensitive and reliable method of evaluating forelimb motor functions and supraspinal changes in humans after spinal damage [67].
The OCA Paradigm
Cortical injuries, as occur in stroke and trauma, are common clinical pathologies, and the consequent deficits are caused by impairments to the area that is directly affected by trauma or ischemia and from functional and morphological changes in functionally connected cortical and subcortical areas [3]. One of the most widely used models of cortical damage is on occlusion of the middle cerebral artery (MCAO), which mimics the most common clinical stroke. With regard to remote damage, neuronal death, inflammation, and axonal degeneration in the ipsilateral thalamus—specifically the ventroposterior nucleus—and substantia nigra are observed in the MCAO model [68, 69].
Despite the relevance of how it is affected by remote damage, permanent MCAO is particularly suited for studying reperfusion and cellular mechanisms in the core and penumbra areas—less so for examining mechanisms of remote damage. The chief drawback in using MCAO to study remote damage is the difficulty in providing reproducible lesions. Many factors, such as temperature, physiological variables, age, and sex, impact the reproducibility of cortical damage in the MCAO model [70].
These issues are less pertinent to the OCA paradigm. OCA is a surgical approach that is simple and highly reproducible with regard to size and location, rendering it an appropriate animal model for investigating secondary thalamic damage [27, 71, 72]. OCA entails creating a unilateral, partial aspiration lesion of the occipital cortex without damaging the underlying corpus callosum and hippocampus. Because geniculocortical projection neurons target highly focal regions of the visual cortex with minimal collateralization, target deprivation in this system induces rapid neuronal death in the dLGN [22, 27, 73]. Notably, the OCA model differs from HCb, effecting uniform remote damage with complete degeneration of dLGN neurons in few weeks [27, 71].
Remote thalamic damage is common in stroke and brain trauma, the recovery from which is mediated by the thalamus, based on its function in organizing and integrating sensorimotor information [74]. Thus, thalamic mechanisms of remote damage must be examined, and the OCA paradigm is a simple and reliable method of studying such mechanisms at the cellular and molecular levels.
Remote Responses to Primary Injury
This section is focused on the principal responses to primary injury occurring in the remote axotomized neurons. Notably, many signaling cell pathways are activated after axonal injury, differing significantly between brain structures and within neuronal subsets in the same structure.
Retrograde Structural Changes
After axotomy, due to a distant focal brain injury, CNS neurons undergo a series of cytological changes, such as chromatolysis, reduction in basophilic cytoplasmic substances, nuclear eccentricity, nuclear and nucleolar enlargement, swelling, perikaryal shrinkage, and changes in contour [19, 56, 75]. Morphological changes are accompanied by damage to cellular components and biochemical changes, such as altered RNA content and protein synthesis and DNA condensation and fragmentation [23, 76]. These events are paralleled by many ultrastructural changes: redistribution and subsequent fragmentation of rough endoplasmic reticulum, dilatation and vesiculation of the Golgi, aggregation and lamination of the cytoskeleton, accumulation of mitochondria in the perikaryon, chromatin condensation, and progressive cellular shrinkage [22, 73, 77].
In contrast, little attention has been paid to the subsequent axotomy-dependent structural changes in dendrites. In the early stages after spinal hemisection, few morphological changes develop in rubral dendrites—primarily the formation of varicosities [78, 79]. Conversely, after OCA, gross changes occur in dendritic arborization in dLGN neurons, associated with neuronal loss. The latter finding raises the question of whether the loss of dendritic arborization causes damage to the soma or whether such damage affects alterations in dendritic morphology [17].
Functional Changes
As stated in “Introduction,” deficits after focal lesion highly depend on changes in network functioning [1]. Functional changes, as degenerative ones, are present both pre- and postsynaptically and have been recently the object of increasing attention by neuroscientists and clinicians. Changes in the excitability level of different brain areas are considered a critical point not only to understand postlesional deficits but also for planning rehabilitation strategies and monitor recovery. Interestingly, one of the models here analyzed, namely the HCb one, has provided relevant data on the distant changes induced by focal cerebellar lesion on cortical excitability [80, 81] and electrical modulation of the cerebello-cortical circuits has been proven capable of improving recovery [82]. These relationships among excitability changes, structural modification, and recovery are further intrigued by genetic factors. Different lines of research are converging in suggesting the importance of genetic determinants for neuronal excitability [83] as well as for predicting clinical outcome after brain injury [84]. These aspects are beyond our scope and have been recently the object of interesting reviews [84].
Inflammation
Inflammation has dual functions in the damaged brain, providing neuroprotection and having deleterious effects, depending on the situation [85–88]. Glial cells—specifically microglia and astrocytes—are the key mediators of the inflammatory response, secreting pro-inflammatory cytokines and chemokines.
Inflammation in various acute and chronic pathologies of the CNS is well established [86]. Inflammation has recently been shown to exist distally to the primary site of damage [6]. In such areas of remote inflammation, resident glial cells become activated several days after injury; this response increases for 2–3 weeks and then wanes progressively [68, 88]. As in primary lesion sites, glial activation, other than that secondary to neuronal loss, influences remote degeneration by producing toxic mediators, such as pro-inflammatory cytokines, nitric oxide, glutamate, and free radicals [6]. In this regard, at early times in the OCA paradigm (3–7 days), the decline in dLGN neurons is accompanied by a mild increase in reactive astrogliosis [71].
In SCI, axotomy activates microglial and astrocytic cells in the RN [54, 78]. Notably, the astrocyte response is transient, whereas the microglial reaction remains high and continues for long periods. In this model, the relationships between glial activation and neuronal damage have not been determined.
In the HCb model, remote activation of glial cells is prolonged, lasting for several months after the lesion. Microglial and astrocytic activation is evident by 7 days, plateauing at 3 weeks, and despite decreasing in intensity, it remains until 2 months after the injury [89].
In general, after brain or spinal injury, astrocytic activation at the primary site of lesion, despite limiting axonal regeneration, is protective, reconstituting the BBB, preventing neuronal degeneration, limiting the spread of damage, and favoring the reuptake of excitotoxic glutamate [90–92]. Conversely, under the same conditions, activated microglia are a source of neurotoxic factors, including tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), nitric oxide, and reactive oxygen species (ROS), driving progressive neuronal damage [93–95].
In remote damage, the scenario is quite different of that observed in primary damage. Although microglia and astrocytes are activated in HCb, their functions differ. Whereas modulation of microglial activation does not affect remote degeneration, the inhibition of astroglia impedes remote damage. In this model, astrocytes are the glial cells that release hazardous factors—i.e., IL-1β [89] and inducible nitric oxidase synthase (iNOS)-derived NO [96], which accelerate remote degeneration [96]. There are notable differences in glial reactions in the primary lesion, where pro-inflammatory mediators and hazardous factors are secreted primarily by microglia, not astrocytes. Thus, the two main classes of glial cells differentially regulate brain damage—locally and remotely [97, 98]. These results are consistent with the large body of evidence that the beneficial and harmful effects of glia are regulated, depending on context, through complex inter- and intracellular signaling mechanisms [91, 99].
In conclusion, inflammation is present in all models of remote damage and is central to death/survival choices. Yet, differences in models and between local and remote damage render the generalization of mechanisms in the HCb paradigm difficult. The significance of contextual factors in the regulation of inflammation necessitates the determination of the effects of inflammation on remote damage paradigms.
Apoptosis
Apoptosis is a structurally and biochemically organized form of cell death that occurs through two major pathways: an extrinsic death receptor pathway and an intrinsic mitochondrial pathway [100]. In the death receptor pathway, cell surface receptors transmit apoptotic signals that are initiated by specific ligands, such as caspase-8, activating other caspases to orchestrate apoptosis. In the intrinsic mitochondrial pathway, noxious stimuli target mitochondria directly or through transduction by pro-apoptotic members of the Bcl-2 family, such as Bax and Bak [100].
As the powerhouse of the cell, mitochondria have begun to emerge as active components in cell death due to their association with apoptosis-related proteins [101–103]. Various key events in apoptosis are centered around mitochondria, including the release of apoptogenic factors (such as cytochrome c, apoptosis-inducing factor, endonuclease G, and Smac/DIABLO), changes in electron transport, loss of mitochondrial membrane potential, altered cellular redox states, and participation of pro- and antiapoptotic Bcl-2 family proteins [104–106].
Degeneration in many brain pathologies is primarily an apoptotic process that mitochondria regulate (for a review, see [101]). The significance of apoptotic mechanisms is also being highlighted in remote damage. In the HCb model, axotomy death signals, transported retrogradely to precerebellar neurons, damage mitochondria, inducing massive amounts of cytochrome c (cyt c) to be released into the cytoplasm [107, 108]. In turn, caspase-3-dependent apoptotic signaling affects DNA fragmentation and cell death [108].
Similarly, in the OCA model, the death of LdGN neurons follows the intrinsic mitochondrial pathway [29]. Axotomized dLGN neurons present different signs of mitochondrial damage and apoptotic activation, which manifest as somal mitochondria accumulation with nuclear sequestration and activation of p53, perikaryal accumulation of Bax, and activation of caspase-3 [29].
These findings suggest that identifying, targeting, and manipulating mitochondrial dysfunction will help identify therapeutic targets for the treatment of remote cell death.
Autophagy
Autophagy is an intracellular catabolic process in which eukaryotic cells degrade their cytoplasm and organelles [109, 110]. In addition to being a homeostatic, nonlethal stress response mechanism for recycling proteins to protect cells from low nutrient supply, autophagy has a central function in cell death mechanisms.
In mammals, autophagy is initiated by the activation of the ULK1/2 complex (the mammalian homolog of yeast ATG1), which is negatively regulated by mammalian target of rapamycin (mTOR) [111–113]. Autophagy begins with the formation of double-membrane vesicles, which subsequently engulf cytoplasmic components, including cytosolic proteins and organelles, to become autophagosomes (APs). APs fuse with lysosomes to form autolysosomes, in which components are degraded by lysosomal hydrolases [114].
The dysregulation of autophagic machinery is implicated in several diseases, including neurodegeneration and cancer [115–117].
The current consensus is that autophagy is a new component that governs neuronal fate and mediates the pathogenesis of chronic and acute neurological diseases [118, 119]. However, in contrast to chronic neurodegenerative disorders, for which much effort has been made in characterizing the molecular participants and effects of autophagy, the study of autophagy in acute brain damage is in its infancy. Regarding brain damage, after TBI and SCI, a marked autophagic activity is observed at the primary site of damage (for a review, see [118]), but few studies on autophagy in remote damage exist.
Recently, autophagy machinery has been implicated in neuronal cell fate decisions in paradigms of remote damage after corticovascular [120] and cerebellar [108] focal lesions. In both models, early activation of autophagy is observed in remote injured neurons. Specifically, LC3-1/2 lipidation and Beclin 1 (Becn1) expression increase, and autophagosomal vacuoles, secondary lysosomes, double-membrane structures, and multilamellar bodies form, based on EM findings [108, 120].
In autophagy, whether the activation of autophagy is protective or detrimental is debated [118, 119], as it is in remote damage models [108]. In HCb, the activation of autophagy has been interpreted as a reactive response that protects neurons by engulfing damaged mitochondria and thus neutralizing pro-apoptotic factors that favor internal homeostasis [108]. Conversely, in the corticovascular model, such activation triggers the apoptotic cascade [120] and thus encourages degeneration.
Although no conclusions can be drawn, based on the paucity of data, these findings implicate autophagy as a pathophysiological mechanism of remote damage, suggesting that drugs that target autophagy will reduce remote neurodegeneration.
Apoptosis and Autophagy
Autophagy and apoptosis are distinct processes with disparate biochemical and morphological features, but the protein networks that control their regulation and execution are undoubtedly connected [121–124]. In recent years, much effort has been directed toward determining the links between autophagy and apoptosis [124].
Depending on the circumstance, autophagy prevents apoptotic cell death or constitutes an alternative pathway of cell death—a process known as autophagic cell death; in certain cases, autophagic cell death and apoptosis occur in parallel and share regulatory mechanisms [123, 124]. Autophagy and apoptosis mediate remote cell death mechanisms, but their relationship in this context has not been examined extensively. HCb studies have linked the early stages of mitochondrial dysfunction to apoptotic cell death, identifying the times during which autophagy is active, and suggest that apoptosis begins only if the cyt c that is released exceeds the clearance by autophagy, indicating that autophagy is protective [109]. Also, data on the thalamus after corticovascular lesion implicate the existence of autophagy-apoptosis crosstalk, which has been interpreted to favor degeneration [120]. There is no evidence of a causal link between autophagic activity and the induction of apoptosis, necessitating ad hoc studies to examine the communication between autophagy and apoptosis.
Oxidative and Nitrosative Stress
The generation of ROS and oxidative damage is believed to mediate the pathogenesis of neurodegenerative disorders. Further, reactive nitrogen species (RNS), such as nitric oxide (NO), also damages neurons (for a review, see [125]. Oxidative stress results from an imbalance between ROS generation and defense by antioxidants, which can induce the degradation of proteins, lipids, and nucleic acids [126], resulting in cell death. However, whether oxidative stress causes neurodegeneration or is secondary to it is unknown [127].
Mitochondria are the major source of ROS, and mitochondrial dysfunction contributes to neuronal death. Mitochondria contain enzymes to combat ROS production, converting superoxide radicals into hydrogen peroxide with manganese superoxide dismutase (MnSOD). In some circumstances, however, this protective system fails, and ROS attacks polyunsaturated fatty acids in cell membranes, triggering free radical chain reactions and membrane lipid peroxidation (MLP), generating aldehydes [128].
NO is one of the most frequently studied trauma-generated ROS. NO is a small and highly diffusible molecule that is synthesized by three nitric oxidase synthase isoforms: neuronal (nNOS), endothelial (eNOS), and inducible (iNOS). The effects of NO are determined primarily by the active NOS isoform and NO concentration [129]: low levels of NO that is generated by nNOS or eNOS are associated with cell signaling [130], whereas high levels of NO that is generated by iNOS are associated with toxicity. The toxic effects of NO are mediated primarily by peroxynitrite, a highly RNS that is formed by a diffusion-controlled reaction between NO and the ROS superoxide anion (for a review, see [131]). Peroxynitrite interacts with proteins, lipids, and DNA through oxidative reactions, propelling cells toward death [131].
Oxidative and nitrosative stress mediates the pathogenesis of chronic and acute neurological diseases (for a review, see [132, 133]) and in remote damage, as shown in the HCb [96], OCA [27, 28, 30], and SCI [133] models. In the HCb paradigm, oxidative/nitrosative stress results from a vicious cycle between neurons and astrocytes (Fig. 2a). ROS that is released from injured neurons triggers chronic activation of and induces iNOS in astrocytes. iNOS-derived NO diffuses to neurons and reacts with the superoxide in them to form peroxynitrite. iNOS/NO that is synthesized by activated astrocytes diffuses in high concentrations into injured neurons, exacerbating the axotomy-induced mitochondrial damage and leading to neuronal death. This crosstalk establishes a perilous loop that accelerates remote degeneration [96].

Schematic of the positive interactions between endocannabinoid and nitrergic systems (a) and of the negative interactions between endocannabinoid and glucocorticoid systems (b) in axotomized neurons after hemicerebellectomy (HCb). a Positive interactions: HCb leads to marked upregulation of CB2 receptor and of neuronal nitric oxide synthase (nNOS) in axotomized neurons with production of nitric oxide (NO) (red arrows). Furthermore, HCb leads to persistent activation of astrocytes, which release amounts of NO, via induction of inducible nitric oxide synthase (iNOS) (red arrows). The high concentration of nitric oxide (NO/iNOS-derived) readily diffuses to neurons and reacts in these cells with the ROS produced from various sources (including mitochondria) to form a more reactive oxidant peroxynitrite (ONOO–), which eventually exacerbate the mitochondrial damage—already caused by HCb—with consequent cytochrome c release (cyt c) and caspase-3 activation (casp-3) that ultimately kills neurons by apoptosis (black arrows). However, the exact intracellular pathways activated by iNOS (and nNOS) are not known (on the left). In this scenario, pharmacological CB2 receptor stimulation (blue and light-blue arrows), probably through the PI3K/Akt signaling, restores the physiological redox state in injured neurons through different mechanisms: (1) increasing nNOS expression and activity in neurons, (2) reducing iNOS expression and activity in astrocytes, (3) attenuating oxidative/nitrative stress in neurons, (4) increasing the levels of proteins that mediate antioxidative (hsp70) and antiapoptotic (Bcl-2) mechanisms, and (5) promoting cell survival. Thus, the neuroprotective effect of CB2 receptor stimulation relies on the interactions with the NO system particularly controlling the nNOS-iNOS balance. b Negative interactions: in the HCb model of axotomy, single stimulation of CB2 receptor or of glucocorticoid receptor (GR) displays neuroprotective effects that are abolished by simultaneous stimulation of both receptors. The schema highlights negative interaction mechanisms between CB2 receptor and GR stimulations and shows the signaling pathways involved (on the left). GR activity is regulated by the balance of two splicing variants: GRα and GRβ. (1) GRα is part of an inactive heterocomplex—containing heat shock proteins hsp90 and hsp70—that becomes active after glucocorticoid (GC) binding and dissociation from the other components of the GR heterocomplex. (2) Once activated, GRα dimerizes, translocates to the nucleus, and activates the transcriptional responses. Conversely, GRβ does not bind to GC but functions as a dominant negative inhibitor of GRα-dependent or GRα-independent transcriptional activity. (3) CB2 receptor stimulation increases the expression of hsp70 and hsp90, impeding heterocomplex dissociation, as well as the level of (4) GRβ, inhibiting the GRα-induced transcriptional activity and GC neuroprotection (5)
In the OCA model, apoptotic dLGN neurons accumulate mitochondria, increase their intracellular Ca2+ levels, and enhance their production of ROS, including superoxide and NO, accompanied by greater formation of protein carbonyls, nitration, and S-nitrosylation [28]. In this model, NO overproduction is associated with nNOS activation in damaged neurons, and nNOS-derived NO compromises neuronal bioenergetics, inducing S-nitrosylation of mPTP proteins and peroxynitrite formation. Peroxynitrite, in turn, activates mitochondrial-dependent apoptosis and ultimately cell death [29].
In SCI, oxidative stress is a potential mechanism of axotomy-induced remote degeneration. After spinal cord hemisection, oxidative stress neuronal death occurs in the nucleus dorsalis (ND) but not the RN [133]. The constitutive antioxidative potential of RN neurons, due to the presence of MnSOD and Cu/Zn-SOD, has been implicated in shielding RN neurons from oxidative damage [133].
These findings demonstrate that as in other neuropathological conditions, mitochondrial dysfunction and oxidative stress are critical factors in remote degeneration, which must be considered when neuroprotection strategies are developed. As discussed, many neuropathological mechanisms can be active at a given time after a brain lesion, meriting the characterization of shared aspects between pathological mechanisms in examining neuroprotection.
The Retrograde Signaling After Axotomy
After brain damage, retrograde injury signals travelling through the axon back to the cell body are essential to allow injured neurons to initiate a proper response to damage and to promote successful survival/regeneration [134–136]. Different signaling mechanisms operate to account for the variety of the retrograde responses occurring after injury [134], all requiring the involvement of sophisticated mechanisms that link signaling systems with intracellular transport machineries [137]. Two temporal phases characterize retrograde signaling: an early phase, in which changes in ion fluxes are propagated along the axon, and a delayed phase, in which the injury signals are converted to a transcriptional response [138]. The changes in ion fluxes, due to rupture of the axonal membrane, can early signal to the soma [139]. These flux changes may have a crucial role in triggering calpains [140] and other retrograde injury signaling, such as the regulation of immediate-early genes, whose activation plays a key role in regulating neuronal response to axonal injury for axonal regeneration [141]. Interestingly, very recent data suggest that an early calcium wave from the injured axon can elicit epigenetic changes in the soma that controls neuronal axon regeneration [142].
However, although there has been intense interest in the mechanisms controlling ion flux in several neurodegenerative diseases [143], few details of the mechanisms by which transient injury-induced ion flux waves are propagated through the axons are available [139, 144].
The second delayed phase, in which the retrograde signaling is conveyed to the cell body, is mediated by importin-dependent signals [145] and by MAP kinase and associated molecules [146]. Kinases are quintessential signaling molecules, and the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3 kinase (PI3K) are the key elements in the axonal damage signaling and participate in the execution of the cell body response program [147]. MAPK is a family of serine/threonine protein kinases that transduce extracellular stimuli into intracellular posttranslational and transcriptional responses and includes extracellular signal-regulated protein kinase (ERK1/2), p38, c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) [148]. Activation of MAPKs and PI3K is considered a key retrograde signaling with a substantial role in determining cell fate by posttranslational modifications of target proteins and increase of gene transcription [141].
In addition, axonal injury signaling reaches the nucleus either by activation of transcription factors, including STAT3, or by mRNA translation mechanisms [149]. In this regard, axonal mRNA translation and de novo synthesis of proteins such as importin-β and vimentin have been suggested as the link between retrograde injury signaling and nuclear machinery [134–136].
In conclusion, several classes of injury signals coexist in the axotomized neuron and precise intracellular signal integration mechanisms ensure precise information flow to the cell body on the nature of the damage. This complex information flow is essential for correct cell body response to axonal injury [134]. Understanding the regulation and integration of these pathways will be important challenges for future research in the field.
Compensatory and Reactive Responses to Axonal Injury
This section is focused on the reactive and compensative mechanisms occurring in the remote axotomized neurons after damage. Notably, many neurotransmitter systems are activated after axonal injury, differing significantly between brain structures and within neuronal subsets in the same structure.
The Purinergic System
ATP is an important neurotransmitter in the peripheral and CNS that is released from neurons and glial cells. Purinergic receptors are subdivided into the P2X and P2Y subsets, based on their mechanism of action and pharmacology [150]. ATP can function as the sole transmitter or as a co-transmitter and has a wide range of effects [151, 152] in health and many human CNS pathologies, such as acute and chronic degeneration and inflammation [151]. Trauma and ischemia lead to a massive release of ATP from various cells of the CNS. Paradoxically, ATP signaling aggravates neuronal and glial damage and contributes to neuroprotection [153].
Although the function of purinergic signaling in CNS diseases has been studied frequently [154, 155], there is scant evidence on its role in remote degeneration. In the HCb model, Florenzano and colleagues [75] reported the time-dependent upregulation of P2X1 and P2X2 in axotomized neurons. Despite the lack of direct proof, this sustained expression and the absence of degenerative morphological features in purinergic positive neurons suggest that purinergic activation is pro-survival [75]. This hypothesis was confirmed, based on the association of purinergic activation and NOS expression in neurons with enhanced survival [26].
In conclusion, little evidence exists regarding the involvement of purinergic signaling in remote degeneration. Nevertheless, the extensive data on purinergic signaling in neurodegeneration and the specific, although scant, evidence in remote degeneration indicates the purinergic system as a worthy field in remote damage research.
The Nitrergic System
NO is generated by three NOS enzymes: eNOS and nNOS are constitutive, are Ca2+-responsive, and control basal NO levels, and iNOS is Ca2+-insensitive [156]. NO has many functions in the nervous system, acting as a neurotransmitter, neuromodulator, and vasodilator [157]. Recent studies have provided considerable evidence on the function of NO in many diseases, including Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis (ALS), and stroke [129].
Although the mechanism of NO-mediated neurodegeneration is unknown, there are several hypotheses regarding it. The S-nitrosylation of selected proteins, such as MMP-9, parkin, and GAPDH, and the interaction between NO and superoxide and the consequent formation of peroxynitrite have been hypothesized to be critical mechanisms through which NO becomes neurotoxic [129, 158]. In contrast, NO signaling is neuroprotective in certain contexts [129, 158]. NO has neuroprotective effects through several mechanisms. The S-nitrosylation of proteins, such as the NMDA receptor subunits NR1 and NR2 and caspase-3; the stimulation of the sGC/cGMP/PKG system; and the induction of Akt, heme oxygenase, and the Keap1/Nrf2/ARE pathway have been proposed to be critical mechanisms through which NO mediates neuroprotection [129, 158].
The protective and detrimental effects of NO have generated interest in the nitrergic system with regard to remote damage. Early studies have reported the induction of nNOS in injured neurons in various axotomy models [19, 159–163]. Modulation of the NO system—i.e., nNOS induction—has also been reported in all of the remote cell death models that we have discussed: OCA, SCI, and HCb. Nevertheless, the functional significance of this induction differs between the OCA and HCb models.
In the OCA model, cortical lesions upregulate nNOS in dLGN neurons but do not affect iNOS or eNOS expression. The induction in nNOS is accompanied by the aberrant accumulation of NO and superoxide and peroxynitrite formation, favoring apoptosis of the dLGN. The function of nNOS induction in life and death decisions has been confirmed by genetic deletion of nNOS and pharmacological inhibition of nNOS, both of which effect decreased remote death of the dLGN [28].
nNOS and iNOS are the principal effectors in the HCb model. HCb enhances nNOS expression in neurons and iNOS in astrocytes, with no effects on endothelial eNOS levels. Thus, after HCb, NO is produced by nNOS in axotomized neurons and by iNOS in reactive astrocytes, but these cases differ with regard to control mechanisms and rates of production. nNOS production is regulated by changes in intracellular calcium levels, whereas iNOS is selectively released and calcium-independent. Further, NO/nNOS is transiently produced in low amounts (in the nanomolar range), but NO/iNOS is synthesized continuously at higher rates (micromolar range). Furthermore, the type of origin labels the NO physiological effects. Whereas inhibition of nNOS activity negatively affects neuronal survival, nitrative/oxidative stress, and neurological improvement, inhibition of iNOS has the opposite effect on these parameters. In the HCb model, iNOS-derived NO from reactive astrocytes is cytotoxic, but neuronal nNOS-derived NO is neuroprotective [96].
The dual behavior of NO is well established [158, 164], but in remote damage paradigms, NO appears to be a multifaceted molecule. Its effects differ between NOS isoforms and the neuronal populations that are involved. NO/nNOS protects precerebellar neurons after HCb but neurotoxic to the dLGN after OCA. These findings add to the myriad factors that influence the effects of NO [129], complicating the development of neuroprotective drugs against the NO system further.
Overall, the NO system is a significant factor in the intricate network of intracellular signals that govern remotely damaged areas, as evidenced by its complex interaction with other systems. This aspect is expounded on in a separate section.
The Endocannabinoid System
The endocannabinoid system (ECS) is a ubiquitous lipid signaling system that has homeostatic functions. The ECS comprises at least two receptors, cannabinoid receptor type-1 (CB1) and type-2 (CB2); their endogenous ligands, the endocannabinoids (eCBs); and the proteins that mediate their transport, synthesis, and degradation [165, 166]. On binding to cannabinoid receptors, ECBs modulate neuronal, glial, and endothelial cell function and regulate many processes, including nociception, appetite, lipid metabolism, blood pressure, cardiovascular modulation, mood, motor control, and memory [167–171]. The ECS is altered in various pathological events, supporting that it is a compensatory and repair system in the brain [172–174].
In response to CNS insults, the brain overproduces eCBs. After TBI [175] and SCI [176], anandamide (AEA) and 2-arachidonoylglycerol (2-AG) levels rise in the primary lesion site. Specifically, AEA and 2-AG levels increase in the early and late stages after damage, respectively.
In addition to the primary site of damage, dynamic changes in the ECS occur in remote damage [89]. After HCb, cannabinoid receptor expression modulates in axotomized precerebellar nuclei. Notably, after such a lesion, when the number of axotomized neurons declines to half of the prelesional levels, de novo synthesis of CB2 receptor occurs in approximately half of all surviving neurons [107].
Many studies have implicated CB2 receptor in the neuroprotective activity of cannabinoids, which act primarily through a series of glia-dependent anti-inflammatory processes [177]. In various paradigms, CB2 receptor is upregulated in reactive microglial cells and astrocytes in primary lesion sites in response to inflammatory stimuli [178, 179]. Emerging research suggests that a CB2 receptor agonist is neuroprotective—an effect that has been attributed to immunomodulatory effects that, through decreased macrophage and microglial activation, indirectly protect neurons [178, 179].
In HCb-induced remote damage, the scenario is quite different. Although microglia and astrocytes become activated, they do not express CB2 receptor, which is exclusive to neurons [107]. In this model, treatment with a CB2 receptor agonist protects neurons directly by activating PI3K/Akt signaling, which prevents axotomy-dependent mitochondrial failure. Mitochondrial protection decreases cytochrome c release into the cytosol, impeding the apoptotic cascade [107]. Thus, in the HCb paradigm, the primary function of CB2 receptor agonists is to protect neurons by inhibiting the intrinsic mitochondrial apoptotic cascade, not to modulate inflammation.
Collectively, these findings indicate that the ECS is an endogenous protective system [180] and that its pharmacological modulation is a potential therapeutic approach in human brain diseases [181].
Interactions Between Systems
The physiopathology of many brain diseases involves many systems that can operate independently, antagonistically, or synergistically on cell populations. This pattern is also true for remote damage. As discussed, ECBs; NO; purinergic systems; and inflammatory, apoptotic, and autophagic mechanisms interact to intervene in the pathophysiology of remote damage—not in isolation. The study of these interactions remains elementary, but several aspects have been addressed.
Interactions between the ECS and neurotransmitters have been observed in several paradigms [182]. The interaction between the endocannabinoid and nitrergic systems has been documented in vitro and in vivo, wherein the activation of CB1 and CB2 receptors stimulates and inhibits NO production, differentially influencing the expression and activity of NOS isoforms [183, 184]. Recently, Oddi and colleagues reported ECS-NO interactions (Fig. 2a) in the HCb remote damage paradigm [96], in which CB2 receptor stimulation modulated NO production, altering the balance between nNOS and iNOS in neurons. Specifically, selective CB2 receptor activation concomitantly augmented nNOS expression in neurons and downregulated iNOS in astrocytes, improving cellular and neurological outcomes. The effects of CB2 receptor stimulation with simultaneous nNOS or iNOS inhibition confirmed this interaction. Inhibition of nNOS, but not iNOS, negates the neuroprotective effects of CB2 receptor stimulation. Thus, CB2 receptor-mediated neuroprotection in HCb is not a monosystemic phenomenon; it relies heavily on the NO system, particularly on the nNOS-iNOS balance. Notably, the balance between nNOS and iNOS is a critical pathophysiological control mechanism in various brain pathologies [129].
Complicating the pathophysiology of remote damage, the NO system does not interact solely with the ECS. In the HCb model, NO and ATP also interact. nNOS and ionotropic purinergic receptor expression is altered in neurons that survive longer, suggesting a positive functional interaction between NO and ATP [26, 185]. Notably, these neurons are the same in which de novo expression of CB2 receptor is observed [96]. Thus, complex CB2/NO/ATP crosstalk might exist. Interactions between systems are not always positive. Recently, Bisicchia and colleagues [186] reported negative interactions between CB2 receptor and glucocorticoid receptor alpha (GRα) in HCb (Fig. 2b). In this model, concomitant activation of CB2 receptor and GRα mitigated the neuroprotective effects of the activation of either receptor alone. The recession of neuroprotection on simultaneous activation is driven by at least two mechanisms: the GRα/GRβ ratio and GR heterocomplex signaling [186]. After HCb and concomitant CB2 receptor and GRα stimulation, GRβ is upregulated and the GRα/GRβ is altered, inhibiting GRα-induced transcriptional activity. Further, under the same conditions, hsp90 and hsp70 levels increase significantly, impeding activation of the GR heterocomplex and inducing glucocorticoid insensitivity [187, 188].
These findings confirm that remote damage is the result of complex pathophysiological mechanisms that involve interactions between systems. These aspects are critical and must be considered when exploring therapeutic options.
Therapeutic Approaches
Remote neuronal damage is a progressive event that continues for months to years after the initial brain damage. This delay in degeneration is significant in determining the overall clinical outcome in many CNS pathologies, including SCI and TBI [189, 190]. Because remote cell death mechanisms are long-lasting, they are a suitable target for pharmacological intervention. The availability of sophisticated neurochemical, histopathological, and molecular data on the basic mechanisms of life-death decisions in remote damage has prompted the development of several agents to improve functional recovery by reducing remote damage. Remote damage is never an isolate event and is always associated with some form of primary damage, and the pathophysiology of degeneration can differ between primary and remote lesions (see the “Remote Cell Death Mechanisms” section). Thus, primary and remote therapeutic approaches might differ. Data from remote models indicate that degeneration mechanisms are time- and context-dependent and are present in acute as well as chronic degenerative diseases. As experience has shown, almost in all CNS pathologies, it is very unlikely that a single therapy may protect from degeneration. Each drug might be effective on a pathway that would count only for a fraction of the overall degenerative mechanisms. Combinatory approaches have to be explored, and remote damage and its complexity have also to be taken into account in planning therapeutic strategies. In subsequent sections, we compare the neuroprotective efficacy of pharmacological agents on SCI and TBI primary lesion sites and remote damage to describe the complexity of these therapeutic approaches.
Glucocorticoids
The use of anti-inflammatory compounds in CNS pathologies is based on the rationale that reducing inflammation limits cell death and improves recovery. Glucocorticoids are the most frequently used anti-inflammatory drugs [191].
The effects of glucocorticoids are mediated by intracellular glucocorticoid receptor (GR), which functions as a hormone-activated transcription factor of target genes [192]. GR has two splice variants, GRα and GRβ. GRα resides in the cytoplasm as part of a complex that contains heat shock proteins that translocates to the nucleus on glucocorticoid binding [193]. Conversely, GRβ does not bind to glucocorticoids but modulates their responses by inhibiting GRα-induced transcriptional activity or through novel, intrinsic, and GRα-independent transcriptional activity [192, 193]. GRα is a critical factor in the regulatory network, blocking several inflammatory pathways through genomic and nongenomic mechanisms [194].
Methylprednisolone sodium succinate (MPSS) is the most commonly used glucocorticoid in experimental and clinical studies on neurological diseases. MPSS is used in clinical practice to treat acute SCI, and a high-dose regimen has become the standard, albeit debated, treatment [195, 196]. In several in vivo SCI models, MPSS treatment results in a long-lasting reduction in delayed inflammatory processes, including free radical formation, tissue edema, inflammation, and apoptosis, at least in the primary site of damage [197]. This evidence suggests that the central mechanisms of MPSS-mediated neuroprotection are linked to the inhibition of inflammation. Nevertheless, several groups have failed to report any effects of MPSS on functional outcomes after SCI [198–200].
In TBI, glucocorticoids were originally introduced to reduce brain edema, but several studies have failed to demonstrate any overall benefit [201]. Moreover, glucocorticoids promote posttraumatic apoptosis in the hippocampus, resulting in learning and memory deficits [202, 203]. Thus, glucocorticoid treatment is not considered an option for TBI [204].
Despite the conflicting findings regarding MPSS use after traumatic brain and spinal cord injury, it has been proposed to be efficacious in halting neuronal loss in a paradigm of remote damage [89]. After HCb, MPSS reduces inflammatory responses and remote neuronal death in precerebellar nuclei. Notably, MPSS-mediated protection lasts only as long as the treatment lasts—when administration of MPSS is interrupted, microglial and astrocyte responses and pro-inflammatory cytokines reactivate, and neuronal death resumes at rates that are comparable with those in untreated animals.
These studies demonstrate the complexity of inflammation in the pathophysiology of delayed damage. The future of MPSS as a neuroprotective agent relies on defining the therapeutic window for its use with regard to time (acute versus delayed) and location (primary versus remote).
Minocycline
Minocycline is a highly lipophilic, semisynthetic tetracycline compound that crosses the blood-brain barrier using various brain damage-related mechanisms and has anti-inflammatory, antiapoptotic, and antioxidative effects [94, 205]. However, its primary neuroprotective effect has been linked to the inhibition of microglial activation and proliferation [206].
Minocycline has had neuroprotective effects in several brain and spinal cord injury models. In rats, after SCI, minocycline inhibits microglial cell activation [207] and oligodendrocytes apoptosis, reduces lesion size [208] in the primary lesion site, and improves functional recovery [209]. In the same model, minocycline has longer and more effective neuroprotection than MPSS [210].
Similar results have been observed after TBI. Minocycline significantly reduces TBI-induced microglial activation, cerebral edema, and primary lesion size—effects that are associated with long-lasting improvements in neurological recovery [211–213] and have been attributed to the capacity to inhibit microglia/macrophage proliferation and activation [214].
However, the data on minocycline-induced neuroprotection are contradictory. Minocycline fails to halt neuronal loss or improve functional recovery in animal models [215–218]. Specifically, in MPTP primate and mouse models of Parkinson disease, minocycline exacerbates MPTP-induced damage, despite inhibiting microglial activation [215, 216].
The inability of minocycline to halt neuronal death has also been demonstrated in a model of remote damage [219]. In the HCb paradigm, consistent with its anti-inflammatory properties, minocycline induces marked and nearly total inhibition of microglial activation in precerebellar nuclei. Yet, minocycline fails to effect neuroprotection of precerebellar axotomized neurons. In general, the debate on minocycline role for neuroprotection still awaits an answer [220]. As already stated, dosage and delivery route, not to mention insult type and species, are all factors that have to be carefully analyzed [221, 222].
Rapamycin
Rapamycin is an immunosuppressant that inhibits mTOR, an intracellular serine/threonine protein kinase that mediates many processes, including cell growth and proliferation, protein synthesis, and autophagy [223]. Several studies have implicated rapamycin as a neuroprotective agent [224]. In neurodegenerative disease models, the principal mechanism by which rapamycin is neuroprotective is the enhancement of autophagic clearance of damaged organelles [225, 226].
After SCI, there is marked autophagic activity at the primary site of damage in rodents [227–230]. In this model, overactivation of autophagy by rapamycin reduces tissue damage, inflammation, and cell death at the lesion epicenter and effects neurological recovery [229, 230]; conversely, inhibition of autophagy by 3-methyladenine (3-MA) significantly increases the rate of apoptosis.
Whereas rapamycin is neuroprotective in SCI models, the results are conflicting in TBI models. Enhancement of autophagy by rapamycin [231] and its inhibition by 3-MA [232] increase neuronal survival, reduce lesion volume, and improve functional recovery in TBI models.
Despite the effort that has been made in determining the contribution of autophagy in neurodegenerative disorders, the first attempt to understand its function in remote damage paradigms has only recently been made [108]. Enhancement of autophagy by rapamycin is associated with greater neuronal survival and functional recovery in the HCb remote damage model (Fig. 3). Further, increased neuronal death and worse functional recovery have been observed after HCb in a genetically autophagy-impaired mouse strain [108].

Schematic of autophagy activation in remote damage. Axonal damage induces retrograde signaling toward the cell body (1) provoking mitochondrial damage (2). Mitochondrial damage, in turn, triggers autophagy machinery (3). Autophagy activation follows distinct stages, including (A) vesicle nucleation (formation of phagophore), (B) vesicle elongation (autophagosome formation), (C) vesicle completion (the edges of the phagophore fuse to form the autophagosome), and (D) vesicle degradation: fusion of the autophagosome with a lysosome to form an autophagolysosome where cytosolic components are degraded. Autophagy by engulfing suffering mitochondria (4) reduces the release of pro-apoptotic factors (cytochrome c release) and thus has a neuroprotective function (5) as confirmed by the protective action of rapamycin treatment (three asterisks)
Autophagy is a modulatory mechanism that maintains cellular homeostasis by controlling the state of intracellular waste. Dysregulation of this mechanism influences the life-death fate in various pathophysiological conditions. Data on autophagy mechanisms from animal models of TBI and SCI are guiding the development of autophagy-based neuroprotective drugs. Further, that autophagy is critical in affecting remote damage has drawn greater interest in such activity.
Cannabinoid-Based Drugs
Cannabinoids are a class of compounds that activate CB1 and CB2 receptors with great potency and selectivity. Cannabinoids include the endocannabinoids (produced endogenously), phytocannabinoids (produced by cannabis and other plants), and synthetic cannabinoids (manufactured industrially). The most extensively studied phytocannabinoids are the major psychoactive plant-derived ∆9-tetrahydrocannabinol (THC) and cannabidiol (CBD), the latter of which has no psychoactive effects. In recent years, many synthetic cannabinoids and cannabis analogs that selectively activate CB1 and CB2 have become available [233, 234].
The neuroprotective activity of cannabinoids is based primarily on the reduction of hypothermia, excitotoxicity, inflammation, and oxidative stress [174, 235], supporting the hypothesis that endocannabinoids constitute a new family of lipid mediators that form part of the brain's compensatory repair mechanism [236]. This evidence has studies to determine the potential of cannabinoids as therapeutic agents in acute and chronic neurological diseases [21, 172, 237–239].
Mounting in vivo findings have demonstrated the neuroprotective function of cannabinoids after TBI and SCI. After TBI, synthetic 2-AG reduces brain edema, infarct volume, inflammation, and BBB permeability and improves functional recovery [175]. The neuroprotective effects of 2-AG are mediated primarily by CB1 receptor signaling pathways that entail the inhibition of intracellular inflammatory signaling cascades [240] and rise in endogenous antioxidant levels [241].
Similarly, treatment with 2-AG protects against expansion of the lesion and white matter damage at the primary lesion site after SCI [242]. This early activity is not transient and is maintained for more than 4 weeks after injury, mediated presumably by CB1 receptor [242].
Although CB1 receptor has been considered the only receptor that mediates neuroprotection by cannabinoids, recent evidence has demonstrated that it involves CB2 receptor as well [177, 179–181]. Administration of a CB2 receptor agonist attenuates BBB disruption, cerebral edema, macrophage/microglial activation, and neuronal degeneration and promotes functional recovery after TBI [243, 244]. Further, after SCI, both CB1 and CB2 receptors promote neuroprotection by reducing excitotoxicity, calcium influx, inflammation, and oxidative stress at the primary lesion site [242, 245]. Positive effects on neurological recovery, lesion size, inflammatory indices, edema, and cell death have been reported after selective CB2 receptor treatment in SCI models [242, 245].
As discussed, CB2 receptor is activated in remote damage models, and stimulation of CB2 receptors has neuroprotective effects [107]. In the HCb model, selective CB2 receptor stimulation by JWH-015 protects neurons and improves neurological outcomes, whereas inhibition of CB2 receptor negatively influences cell survival and neurological outcomes.
CB2 receptor-related neuroprotective mechanisms act primarily through a series of glia-dependent anti-inflammatory processes in many neurodegeneration models [177]. Cannabinoid receptors trigger phosphorylation cascades that involve mitogen-activated protein kinases (including ERK1/2, JNK, and p38) and PI3K/Akt signaling [246, 247], through which they have their positive effects.
In remote damage, the mechanism through which CB2 receptor mediates neuroprotection differs. In the HCb model, consistent with the exclusively neuronal expression of CB2 receptor , CB2 receptor-mediated neuroprotection acts directly on neurons, inhibiting mitochondrial damage and the subsequent release of cytochrome c through a PI3K/Akt-dependent pathway [107].
These findings highlight the potential use of CB2 receptor-related drugs in glia- and neuron-dependent degeneration. Regarding the use of neuroprotective CB2 receptor-based drugs in several diseases for which steroids are routinely given, one must consider the negative interaction between cannabinoids and glucocorticoids in combining CB2 receptor and GR drugs.
Concluding Remarks
The significance of remote cell death in neurological disorders became evident several decades ago, and its importance in clinical outcomes is now well established. The characterization of the factors in remote cell death mechanisms has just begun and has yielded significant findings. Overall, remote damage is not simply repetition of the same pathogenic mechanisms occurring at the primary lesion sites on a smaller scale. Differences in neuronal and glial function, patterns of receptor activation, and sensitivity to treatments have been observed. Notably, many factors of remote damage players have been identified, including inflammatory, excitotoxic, apoptotic, autophagic, and oxidative mechanisms—a list that is likely to grow (Fig. 4).

Schematic of the molecules so far characterized in the hemicerebellectomy (HCb) model playing a key role in determining death/survival fate of axotomized neurons. Survival molecules (right side): upregulation of neuronal nitric oxide synthase (nNOS), cannabinoid receptor type 2 (CB2R), and purinergic receptor X1, X2 (P2X1, 2) and Beclin 1 and LC3II conversion—these latter two as markers of autophagic machinery—promote neuronal survival. Death molecules (left side): cytochrome c release (cyt c), caspase-3 activation (casp-3), inducible nitric oxide synthase (iNOS) upregulation, and IL-1β and reactive oxygen species (ROS) production cause neuronal death
The complexity of mechanisms of remote damage models allows us the opportunity to examine the functional interactions between factors and systems in vivo, which is particularly important, considering the negative interactions between potential neuroprotective agents. Targeting a single mechanism might be insufficient, because successful neuroprotection might be achieved by the associations between drugs that act at multiple levels. Recent successful combinatorial approaches of minocycline with drugs with complementary mechanisms of action clearly support this idea [220, 221]. Targeting more than one pathway appears the most effective therapeutic strategy. In particular, considering the number and complexity of the systems involved, this would be the right choice for supporting neuroprotection in remote damage. The search for interactions and combinatory therapy has already begun, demonstrating CB2 receptor-NOS and CB2 receptor-GR interactions in the HCb model. Although our knowledge of remote damage mechanisms remains poor, there is much potential in meeting the increasing demand for effective neuroprotective therapeutic agents.
References
Bartolomeo P (2011) The quest for the ‘critical lesion site’ in cognitive deficits: problems and perspectives. Cortex 47:1010–1012
Zhang J, Zhang Y, Xing S, Liang Z, Zeng J (2012) Secondary neurodegeneration in remote regions after focal cerebral infarction: a new target for stroke management? Stroke 43:1700–1705
Carter AR, Patel KR, Astafiev SV, Snyder AZ, Rengachary J et al (2012) Upstream dysfunction of somatomotor functional connectivity after corticospinal damage in stroke. Neurorehabil Neural Repair 26:7–19
Viscomi MT, Florenzano F, Latini L, Molinari M (2009) Remote cell death in the cerebellar system. Cerebellum 8:184–191
Park E, Velumian AA, Fehlings MG (2004) The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J Neurotrauma 21:754–774
Block F, Dihne M, Loos M (2005) Inflammation in areas of remote changes following focal brain lesion. Prog Neurobiol 75:34–365
Pinching AJ, Powell TP (1971) Ultrastructural features of transneuronal cell degeneration in the olfactory system. J Cell Sci 8:253–287
Sakai T, Matsuda H, Watanabe N, Kamei A, Takashima S (1994) Olivocerebellar retrograde trans-synaptic degeneration from the lateral cerebellar hemisphere to the medial inferior olivary nucleus in an infant. Brain Dev 16:229–232
Fitzek C, Fitzek S, Stoeter P (2004) Bilateral Wallerian degeneration of the medial cerebellar peduncles after ponto-mesencephalic infarction. Eur J Radiol 49:198–203
Yucel Y, Gupta N (2008) Glaucoma of the brain: a disease model for the study of transsynaptic neural degeneration. Prog Brain Res 173:465–478
Jindahra P, Petrie A, Plant GT (2012) The time course of retrograde trans-synaptic degeneration following occipital lobe damage in humans. Brain 135:534–541
Yamada K, Patel U, Shrier DA, Tanaka H, Chang JK et al (1998) MR imaging of CNS tractopathy: wallerian and transneuronal degeneration. AJR Am J Roentgenol 171:813–818
Faden AI (2002) Neuroprotection and traumatic brain injury: theoretical option or realistic proposition. Curr Opin Neurol 15:707–712
Sofroniew MV, Isacson O (1988) Distribution of degeneration of cholinergic neurons in the septum following axotomy in different portions of the fimbria-fornix: a correlation between degree of cell loss and proximity of neuronal somata to the lesion. J Chem Neuroanat 1:327–337
Fry FJ, Cowan WM (1972) A study of retrograde cell degeneration in the lateral mammillary nucleus of the cat, with special reference to the role of axonal branching in the preservation of the cell. J Comp Neurol 144:1–23
Lieberman AR (1971) The axon reaction: a review of the principal features of perikaryal responses to axon injury. Int Rev Neurobiol 14:49–124
Hendrickson ML, Ling C, Kalil RE (2012) Degeneration of axotomized projection neurons in the rat dLGN: temporal progression of events and their mitigation by a single administration of FGF2. PLoS One 7:e46918
Al-Abdulla NA, Martin LJ (2002) Projection neurons and interneurons in the lateral geniculate nucleus undergo distinct forms of degeneration ranging from retrograde and transsynaptic apoptosis to transient atrophy after cortical ablation in rat. Neuroscience 115:7–14
Buffo A, Fronte M, Oestreicher AB, Rossi F (1998) Degenerative phenomena and reactive modifications of the adult rat inferior olivary neurons following axotomy and disconnection from their targets. Neuroscience 85:587–604
Buffo A, Carulli D, Rossi F, Strata P (2003) Extrinsic regulation of injury/growth-related gene expression in the inferior olive of the adult rat. Eur J Neurosci 18:2146–2158
Viscomi MT, Oddi S, Latini L, Bisicchia E, Maccarrone M et al (2010) The endocannabinoid system: a new entry in remote cell death mechanisms. Exp Neurol 224:56–65
Al-Abdulla NA, Martin LJ (1998) Apoptosis of retrogradely degenerating neurons occurs in association with the accumulation of perikaryal mitochondria and oxidative damage to the nucleus. Am J Pathol 153:447–456
Barron KD, Doolin PF, Oldershaw JB (1967) Ultrastructural observations on retrograde atrophy of lateral geniculate body. I. Neuronal alterations. J Neuropathol Exp Neurol 26:300–326
Gage FH, Wictorin K, Fischer W, Williams LR, Varon S et al (1986) Retrograde cell changes in medial septum and diagonal band following fimbria-fornix transection: quantitative temporal analysis. Neuroscience 19:241–255
Ruigrok TJ, de Zeeuw CI, Voogd J (1990) Hypertrophy of inferior olivary neurons: a degenerative, regenerative or plasticity phenomenon. Eur J Morphol 28:224–239
Viscomi MT, Florenzano F, Conversi D, Bernardi G, Molinari M (2004) Axotomy dependent purinergic and nitrergic co-expression. Neuroscience 123:393–404
Al-Abdulla NA, Portera-Cailliau C, Martin LJ (1998) Occipital cortex ablation in adult rat causes retrograde neuronal death in the lateral geniculate nucleus that resembles apoptosis. Neuroscience 86:191–209
Martin LJ, Adams NA, Pan Y, Price A, Wong M (2011) The mitochondrial permeability transition pore regulates nitric oxide-mediated apoptosis of neurons induced by target deprivation. J Neurosci 31:359–370
Martin LJ, Kaiser A, Yu JW, Natale JE, Al-Abdulla NA (2001) Injury-induced apoptosis of neurons in adult brain is mediated by p53-dependent and p53-independent pathways and requires Bax. J Comp Neurol 433:299–311
Martin LJ, Price AC, McClendon KB, Al-Abdulla NA, Subramaniam JR et al (2003) Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J Neurochem 85:234–247
Tetzlaff W, Alexander SW, Miller FD, Bisby MA (1991) Response of facial and rubrospinal neurons to axotomy: changes in mRNA expression for cytoskeletal proteins and GAP-43. J Neurosci 11:2528–2544
Kwon BK, Liu J, Messerer C, Kobayashi NR, McGraw J et al (2002) Survival and regeneration of rubrospinal neurons 1 year after spinal cord injury. Proc Natl Acad Sci U S A 99:3246–3251
Kwon BK, Liu J, Oschipok L, Teh J, Liu ZW et al (2004) Rubrospinal neurons fail to respond to brain-derived neurotrophic factor applied to the spinal cord injury site 2 months after cervical axotomy. Exp Neurol 189:45–57
Molinari M, Viscomi MT, Leggio MG (2013) Hemicerebellectomy. In: Manto M, Schmahmann JD, Rossi F, Gruol DL, Koibuchi N (eds) Handbook of the cerebellum and cerebellar disorders. Springer, Netherlands, pp 1579–1594
Castro AJ (1978) Projections of the superior cerebellar peduncle in rats and the development of new connections in response to neonatal hemicerebellectomy. J Comp Neurol 178:611–627
Kolodziejak A, Dziduszko J, Niechaj A, Tarnecki R (2000) Influence of acute cerebellar lesions on somatosensory evoked potentials (SEPs) in cats. J Physiol Pharmacol 51:41–55
Barrionuevo G, Pechadre JC, Gautron M, Guiot F (1978) Negative effects of chronic hemicerebellectomy of epileptiform after-discharges elicited by focal cortical stimulation in baboons (Papio papio). Electroencephalogr Clin Neurophysiol 44:232–235
Bialowas J, Hassler R, Wagner A (1984) Types of synapses and degeneration in the thalamic nucleus ventralis oralis posterior after cerebellar lesions in the squirrel monkey. J Hirnforsch 25:417–437
Goto N, Kaneko M (1981) Olivary enlargement: chronological and morphometric analyses. Acta Neuropathol 54:275–282
Birbamer G, Buchberger W, Felber S, Aichner F (1992) MR appearance of hypertrophic olivary degeneration: temporal relationships. AJNR Am J Neuroradiol 13:1501–1503
Uchino A, Takase Y, Nomiyama K, Egashira R, Kudo S (2006) Brainstem and cerebellar changes after cerebrovascular accidents: magnetic resonance imaging. Eur Radiol 16:592–597
Soblosky JS, Song JH, Dinh DH (2001) Graded unilateral cervical spinal cord injury in the rat: evaluation of forelimb recovery and histological effects. Behav Brain Res 119:1–13
Tohda C, Kuboyama T (2011) Current and future therapeutic strategies for functional repair of spinal cord injury. Pharmacol Ther 132:57–71
Wang M, Zhai P, Chen X, Schreyer DJ, Sun X et al (2011) Bioengineered scaffolds for spinal cord repair. Tissue Eng Part B Rev 17:177–194
Raisman G, Barnett SC, Ramon-Cueto A (2012) Repair of central nervous system lesions by transplantation of olfactory ensheathing cells. Handb Clin Neurol 109:541–549
Hains BC, Black JA, Waxman SG (2003) Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. J Comp Neurol 462:328–341
Nielson JL, Sears-Kraxberger I, Strong MK, Wong JK, Willenberg R et al (2010) Unexpected survival of neurons of origin of the pyramidal tract after spinal cord injury. J Neurosci 30:11516–11528
Nielson JL, Strong MK, Steward O (2011) A reassessment of whether cortical motor neurons die following spinal cord injury. J Comp Neurol 519:2852–2869
Brown LT (1974) Rubrospinal projections in the rat. J Comp Neurol 154:169–187
Bregman BS, Broude E, McAtee M, Kelley MS (1998) Transplants and neurotrophic factors prevent atrophy of mature CNS neurons after spinal cord injury. Exp Neurol 149:13–27
Feringa ER, McBride RL, Pruitt JN 2nd (1988) Loss of neurons in the red nucleus after spinal cord transection. Exp Neurol 100:112–120
Fukuoka T, Miki K, Yoshiya I, Noguchi K (1997) Expression of beta-calcitonin gene-related peptide in axotomized rubrospinal neurons and the effect of brain derived neurotrophic factor. Brain Res 767:250–258
Kobayashi NR, Fan DP, Giehl KM, Bedard AM, Wiegand SJ et al (1997) BDNF and NT-4/5 prevent atrophy of rat rubrospinal neurons after cervical axotomy, stimulate GAP-43 and Talpha1-tubulin mRNA expression, and promote axonal regeneration. J Neurosci 17:9583–9595
Liu PH, Wang YJ, Tseng GF (2003) Close axonal injury of rubrospinal neurons induced transient perineuronal astrocytic and microglial reaction that coincided with their massive degeneration. Exp Neurol 179:111–126
Mori F, Himes BT, Kowada M, Murray M, Tessler A (1997) Fetal spinal cord transplants rescue some axotomized rubrospinal neurons from retrograde cell death in adult rats. Exp Neurol 143:45–60
McBride RL, Feringa ER, Garver MK, Williams JK Jr (1989) Prelabeled red nucleus and sensorimotor cortex neurons of the rat survive 10 and 20 weeks after spinal cord transection. J Neuropathol Exp Neurol 48:568–576
Novikova LN, Novikov LN, Kellerth JO (2000) Survival effects of BDNF and NT-3 on axotomized rubrospinal neurons depend on the temporal pattern of neurotrophin administration. Eur J Neurosci 12:776–780
Houle JD, Ye JH (1999) Survival of chronically-injured neurons can be prolonged by treatment with neurotrophic factors. Neuroscience 94:929–936
Novikova LN, Novikov LN, Kellerth JO (2002) Differential effects of neurotrophins on neuronal survival and axonal regeneration after spinal cord injury in adult rats. J Comp Neurol 452:255–263
Zhou L, Connors T, Chen DF, Murray M, Tessler A et al (1999) Red nucleus neurons of Bcl-2 over-expressing mice are protected from cell death induced by axotomy. Neuroreport 10:3417–3421
Xu XM, Martin GF (1990) The response of rubrospinal neurons to axotomy in the adult opossum, Didelphis virginiana. Exp Neurol 108:46–54
Wannier-Morino P, Schmidlin E, Freund P, Belhaj-Saif A, Bloch J et al (2008) Fate of rubrospinal neurons after unilateral section of the cervical spinal cord in adult macaque monkeys: effects of an antibody treatment neutralizing Nogo-A. Brain Res 1217:96–109
Liu Y, Himes BT, Murray M, Tessler A, Fischer I (2002) Grafts of BDNF-producing fibroblasts rescue axotomized rubrospinal neurons and prevent their atrophy. Exp Neurol 178:150–164
Xiao M, Klueber KM, Guo Z, Lu C, Wang H et al (2007) Human adult olfactory neural progenitors promote axotomized rubrospinal tract axonal reinnervation and locomotor recovery. Neurobiol Dis 26:363–374
Little JW, Halar E (1985) Temporal course of motor recovery after Brown-Sequard spinal cord injuries. Paraplegia 23:39–46
Roth EJ, Park T, Pang T, Yarkony GM, Lee MY (1991) Traumatic cervical Brown-Sequard and Brown-Sequard-plus syndromes: the spectrum of presentations and outcomes. Paraplegia 29:582–589
Khaing ZZ, Geissler SA, Jiang S, Milman BD, Aguilar SV et al (2012) Assessing forelimb function after unilateral cervical spinal cord injury: novel forelimb tasks predict lesion severity and recovery. J Neurotrauma 29:488–498
Dihne M, Grommes C, Lutzenburg M, Witte OW, Block F (2002) Different mechanisms of secondary neuronal damage in thalamic nuclei after focal cerebral ischemia in rats. Stroke 33:3006–3011
Ross DT, Ebner FF (1990) Thalamic retrograde degeneration following cortical injury: an excitotoxic process? Neuroscience 35:525–550
Braeuninger S, Kleinschnitz C (2009) Rodent models of focal cerebral ischemia: procedural pitfalls and translational problems. Exp Transl Stroke Med 1:8
Agarwala S, Kalil RE (1998) Axotomy-induced neuronal death and reactive astrogliosis in the lateral geniculate nucleus following a lesion of the visual cortex in the rat. J Comp Neurol 392:252–263
Lieberman AR, Webster KE (1974) Proceedings: terminal and retrograde degeneration in the dorsal lateral geniculate nucleus (LGd) of the rat following cortical lesions. J Anat 118:384–386
Matthews MA (1973) Death of the central neuron: an electron microscopic study of thalamic retrograde degeneration following cortical ablation. J Neurocytol 2:265–288
Briggs F, Usrey WM (2008) Emerging views of corticothalamic function. Curr Opin Neurobiol 18:403–407
Florenzano F, Viscomi MT, Cavaliere F, Volonte C, Molinari M (2002) Cerebellar lesion up-regulates P2X1 and P2X2 purinergic receptors in precerebellar nuclei. Neuroscience 115:425–434
Barron KD (2004) The axotomy response. J Neurol Sci 220:119–121
Lieberman AR, Webster KE (1974) Aspects of the synaptic organization of intrinsic neurons in the dorsal lateral geniculate nucleus. An ultrastructural study of the normal and of the experimentally deafferented nucleus in the rat. J Neurocytol 3:677–710
Tseng GF, Wang YJ, Lai QC (1996) Perineuronal microglial reactivity following proximal and distal axotomy of rat rubrospinal neurons. Brain Res 715:32–43
Wang YJ, Chen JR, Tseng GF (2002) Fate of the soma and dendrites of cord-projection central neurons after proximal and distal spinal axotomy: an intracellular dye injection study. J Neurotrauma 19:1487–1502
Ben Taib NO, Manto M, Pandolfo M, Brotchi J (2005) Hemicerebellectomy blocks the enhancement of cortical motor output associated with repetitive somatosensory stimulation in the rat. J Physiol 567:293–300
Oulad Ben Taib N, Manto M (2006) Hemicerebellectomy impairs the modulation of cutaneomuscular reflexes by the motor cortex following repetitive somatosensory stimulation. Brain Res 1090:110–115
Ben Taib NO, Manto M (2009) Trains of transcranial direct current stimulation antagonize motor cortex hypoexcitability induced by acute hemicerebellectomy. J Neurosurg 111:796–806
Missitzi J, Gentner R, Geladas N, Politis P, Karandreas N et al (2011) Plasticity in human motor cortex is in part genetically determined. J Physiol 589:297–306
Pearson-Fuhrhop KM, Burke E, Cramer SC (2012) The influence of genetic factors on brain plasticity and recovery after neural injury. Curr Opin Neurol 25:682–688
Hohlfeld R, Kerschensteiner M, Meinl E (2007) Dual role of inflammation in CNS disease. Neurology 68:S58–S63, discussion S91–56
Esiri MM (2007) The interplay between inflammation and neurodegeneration in CNS disease. J Neuroimmunol 184:4–16
Stoll G, Jander S, Schroeter M (2002) Detrimental and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system. Adv Exp Med Biol 513:87–113
Dihne M, Block F (2001) Focal ischemia induces transient expression of IL-6 in the substantia nigra pars reticulata. Brain Res 889:165–173
Viscomi MT, Florenzano F, Latini L, Amantea D, Bernardi G et al (2008) Methylprednisolone treatment delays remote cell death after focal brain lesion. Neuroscience 154:1267–1282
Kawano H, Kimura-Kuroda J, Komuta Y, Yoshioka N, Li HP et al (2012) Role of the lesion scar in the response to damage and repair of the central nervous system. Cell Tissue Res 349:169–180
Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32:638–647
Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69
Kim HS, Suh YH (2009) Minocycline and neurodegenerative diseases. Behav Brain Res 196:168–179
Lull ME, Block ML (2010) Microglial activation and chronic neurodegeneration. Neurotherapeutics 7:354–365
Oddi S, Latini L, Viscomi MT, Bisicchia E, Molinari M et al (2012) Distinct regulation of nNOS and iNOS by CB2 receptor in remote delayed neurodegeneration. J Mol Med (Berl) 90:371–387
Aloisi F (2001) Immune function of microglia. Glia 36:165–179
van Rossum D, Hanisch UK (2004) Microglia. Metab Brain Dis 19:393–411
Sofroniew MV (2005) Reactive astrocytes in neural repair and protection. Neuroscientist 11:400–407
Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87:99–163
Galluzzi L, Kepp O, Kroemer G (2012) Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol 13:780–788
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281:1309–1312
Kroemer G, Dallaporta B, Resche-Rigon M (1998) The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 60:619–642
Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305:626–629
Kroemer G (1999) Mitochondrial control of apoptosis: an overview. Biochem Soc Symp 66:1–15
Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G et al (2004) Toxic proteins released from mitochondria in cell death. Oncogene 23:2861–2874
Viscomi MT, Oddi S, Latini L, Pasquariello N, Florenzano F et al (2009) Selective CB2 receptor agonism protects central neurons from remote axotomy-induced apoptosis through the PI3K/Akt pathway. J Neurosci 29:4564–4570
Viscomi MT, D'Amelio M, Cavallucci V, Latini L, Bisicchia E et al (2012) Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy 8:222–235
Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721
Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463–477
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93
Inoue Y, Klionsky DJ (2010) Regulation of macroautophagy in Saccharomyces cerevisiae. Semin Cell Dev Biol 21:664–670
Mizushima N (2010) The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 22:132–139
Xie Z, Klionsky DJ (2007) Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9:1102–1109
Meijer AJ, Codogno P (2006) Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med 27:411–425
Nixon RA (2006) Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci 29:528–535
Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306:990–995
Puyal J, Ginet V, Grishchuk Y, Truttmann AC, Clarke PG (2012) Neuronal autophagy as a mediator of life and death: contrasting roles in chronic neurodegenerative and acute neural disorders. Neuroscientist 18:224–236
Viscomi MT, D'Amelio M (2012) The “Janus-faced role” of autophagy in neuronal sickness: focus on neurodegeneration. Mol Neurobiol 46:513–521
Xing S, Zhang Y, Li J, Zhang J, Li Y et al (2012) Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 8:63–76
Fimia GM, Piacentini M (2010) Regulation of autophagy in mammals and its interplay with apoptosis. Cell Mol Life Sci 67:1581–1588
Gordy C, He YW (2012) The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell 3:17–27
Gozuacik D, Kimchi A (2007) Autophagy and cell death. Curr Top Dev Biol 78:217–245
Rubinstein AD, Kimchi A (2012) Life in the balance—a mechanistic view of the crosstalk between autophagy and apoptosis. J Cell Sci 125:5259–5268
Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN et al (2012) Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci 322:254–262
Reynolds A, Laurie C, Mosley RL, Gendelman HE (2007) Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol 82:297–325
Souza JM, Peluffo G, Radi R (2008) Protein tyrosine nitration—functional alteration or just a biomarker? Free Radic Biol Med 45:357–366
Baldwin SA, Broderick R, Osbourne D, Waeg G, Blades DA et al (1998) The presence of 4-hydroxynonenal/protein complex as an indicator of oxidative stress after experimental spinal cord contusion in a rat model. J Neurosurg 88:874–883
Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA et al (2007) Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 8:766–775
Garthwaite J, Boulton CL (1995) Nitric oxide signaling in the central nervous system. Annu Rev Physiol 57:683–706
Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87:315–424
Nakamura T, Cho DH, Lipton SA (2012) Redox regulation of protein misfolding, mitochondrial dysfunction, synaptic damage, and cell death in neurodegenerative diseases. Exp Neurol 238:12–21
Xu M, Yip GW, Gan LT, Ng YK (2005) Distinct roles of oxidative stress and antioxidants in the nucleus dorsalis and red nucleus following spinal cord hemisection. Brain Res 1055:137–142
Abe N, Cavalli V (2008) Nerve injury signaling. Curr Opin Neurobiol 18:276–283
Rishal I, Fainzilber M (2010) Retrograde signaling in axonal regeneration. Exp Neurol 223:5–10
Rishal I, Fainzilber M (2013) Axon-soma communication in neuronal injury. Nat Rev Neurosci 14:278–291
Ibanez CF (2007) Message in a bottle: long-range retrograde signaling in the nervous system. Trends Cell Biol 17:519–528
Ambron RT, Walters ET (1996) Priming events and retrograde injury signals. A new perspective on the cellular and molecular biology of nerve regeneration. Mol Neurobiol 13:61–79
Mandolesi G, Menna E, Harauzov A, von Bartheld CS, Caleo M et al (2005) A role for retinal brain-derived neurotrophic factor in ocular dominance plasticity. Curr Biol 15:2119–2124
Vosler PS, Brennan CS, Chen J (2008) Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol 38:78–100
Sung YJ, Povelones M, Ambron RT (2001) RISK-1: a novel MAPK homologue in axoplasm that is activated and retrogradely transported after nerve injury. J Neurobiol 47:67–79
Cho Y, Sloutsky R, Naegle KM, Cavalli V (2013) Injury-Induced HDAC5 Nuclear Export Is Essential for Axon Regeneration. Cell 155:894–908
Bezprozvanny I (2009) Calcium signaling and neurodegenerative diseases. Trends Mol Med 15:89–100
Rigaud M, Gemes G, Weyker PD, Cruikshank JM, Kawano T et al (2009) Axotomy depletes intracellular calcium stores in primary sensory neurons. Anesthesiology 111:381–392
Perry RB, Doron-Mandel E, Iavnilovitch E, Rishal I, Dagan SY et al (2012) Subcellular knockout of importin beta1 perturbs axonal retrograde signaling. Neuron 75:294–305
Cavalli V, Kujala P, Klumperman J, Goldstein LS (2005) Sunday Driver links axonal transport to damage signaling. J Cell Biol 168:775–787
Lingor P, Koch JC, Tonges L, Bahr M (2012) Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res 349:289–311
Seger R, Krebs EG (1995) The MAPK signaling cascade. FASEB J 9:726–735
Perry RB, Fainzilber M (2009) Nuclear transport factors in neuronal function. Semin Cell Dev Biol 20:600–606
Burnstock G (2007) Purine and pyrimidine receptors. Cell Mol Life Sci 64:1471–1483
Burnstock G, Krugel U, Abbracchio MP, Illes P (2011) Purinergic signalling: from normal behaviour to pathological brain function. Prog Neurobiol 95:229–274
North RA, Barnard EA (1997) Nucleotide receptors. Curr Opin Neurobiol 7:346–357
Volonte C, Amadio S, Cavaliere F, D'Ambrosi N, Vacca F et al (2003) Extracellular ATP and neurodegeneration. Curr Drug Targets CNS Neurol Disord 2:403–412
Burnstock G (2008) Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov 7:575–590
Burnstock G (2013) Introduction to purinergic signalling in the brain. Adv Exp Med Biol 986:1–12
Mayer B, Hemmens B (1997) Biosynthesis and action of nitric oxide in mammalian cells. Trends Biochem Sci 22:477–481
Lipton SA, Singel DJ, Stamler JS (1994) Nitric oxide in the central nervous system. Prog Brain Res 103:359–364
Calabrese V, Cornelius C, Rizzarelli E, Owen JB, Dinkova-Kostova AT et al (2009) Nitric oxide in cell survival: a janus molecule. Antioxid Redox Signal 11:2717–2739
Xu M, Ng YK, Leong SK (2000) Neuroprotective and neurodestructive functions of nitric oxide after spinal cord hemisection. Exp Neurol 161:472–480
Yu Y, Matsuyama Y, Nakashima S, Yanase M, Kiuchi K et al (2004) Effects of MPSS and a potent iNOS inhibitor on traumatic spinal cord injury. Neuroreport 15:2103–2107
Chen S, Aston-Jones G (1994) Cerebellar injury induces NADPH diaphorase in Purkinje and inferior olivary neurons in the rat. Exp Neurol 126:270–276
Saxon DW, Beitz AJ (1994) Cerebellar injury induces NOS in Purkinje cells and cerebellar afferent neurons. Neuroreport 5:809–812
Saxon DW, Beitz AJ (1996) Induction of NADPH-diaphorase/nitric oxide synthase in the brainstem trigeminal system resulting from cerebellar lesions. J Comp Neurol 371:41–71
Thippeswamy T, McKay JS, Quinn JP, Morris R (2006) Nitric oxide, a biological double-faced janus—is this good or bad? Histol Histopathol 21:445–458
Bari M, Battista N, Fezza F, Gasperi V, Maccarrone M (2006) New insights into endocannabinoid degradation and its therapeutic potential. Mini Rev Med Chem 6:257–268
Di Marzo V (2008) Endocannabinoids: synthesis and degradation. Rev Physiol Biochem Pharmacol 160:1–24
Bermudez-Silva FJ, Viveros MP, McPartland JM, Rodriguez de Fonseca F (2010) The endocannabinoid system, eating behavior and energy homeostasis: the end or a new beginning? Pharmacol Biochem Behav 95:375–382
Di Marzo V, Matias I (2005) Endocannabinoid control of food intake and energy balance. Nat Neurosci 8:585–589
El Manira A, Kyriakatos A (2010) The role of endocannabinoid signaling in motor control. Physiology (Bethesda) 25:230–238
Pacher P, Batkai S, Kunos G (2005) Blood pressure regulation by endocannabinoids and their receptors. Neuropharmacology 48:1130–1138
Pacher P, Steffens S (2009) The emerging role of the endocannabinoid system in cardiovascular disease. Semin Immunopathol 31:63–77
Shohami E, Cohen-Yeshurun A, Magid L, Algali M, Mechoulam R (2011) Endocannabinoids and traumatic brain injury. Br J Pharmacol 163:1402–1410
Sanchez AJ, Garcia-Merino A (2012) Neuroprotective agents: cannabinoids. Clin Immunol 142:57–67
Galve-Roperh I, Aguado T, Palazuelos J, Guzman M (2008) Mechanisms of control of neuron survival by the endocannabinoid system. Curr Pharm Des 14:2279–2288
Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A et al (2001) An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413:527–531
Garcia-Ovejero D, Arevalo-Martin A, Petrosino S, Docagne F, Hagen C et al (2009) The endocannabinoid system is modulated in response to spinal cord injury in rats. Neurobiol Dis 33:57–71
Fernandez-Ruiz J, Pazos MR, Garcia-Arencibia M, Sagredo O, Ramos JA (2008) Role of CB2 receptors in neuroprotective effects of cannabinoids. Mol Cell Endocrinol 286:S91–S96
Stella N (2010) Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 58:1017–1030
Zarruk JG, Fernandez-Lopez D, Garcia-Yebenes I, Garcia-Gutierrez MS, Vivancos J et al (2012) Cannabinoid type 2 receptor activation downregulates stroke-induced classic and alternative brain macrophage/microglial activation concomitant to neuroprotection. Stroke 43:211–219
Pacher P, Mechoulam R (2011) Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res 50:193–211
Han S, Thatte J, Buzard DJ, Jones RM (2013) Therapeutic utility of cannabinoid receptor type 2 (CB2) selective agonists. J Med Chem 56:8224–8256
Fernandez-Ruiz J, Garcia C, Sagredo O, Gomez-Ruiz M, de Lago E (2010) The endocannabinoid system as a target for the treatment of neuronal damage. Expert Opin Ther Targets 14:387–404
Gonzalez C, Herradon E, Abalo R, Vera G, Perez-Nievas BG et al (2011) Cannabinoid/agonist WIN 55,212-2 reduces cardiac ischaemia-reperfusion injury in Zucker diabetic fatty rats: role of CB2 receptors and iNOS/eNOS. Diabetes Metab Res Rev 27:331–340
Maccarrone M, Fiori A, Bari M, Granata F, Gasperi V et al (2006) Regulation by cannabinoid receptors of anandamide transport across the blood-brain barrier and through other endothelial cells. Thromb Haemost 95:117–127
Florenzano F, Viscomi MT, Amadio S, D'Ambrosi N, Volonte C et al (2008) Do ATP and NO interact in the CNS? Prog Neurobiol 84:40–56
Bisicchia E, Chiurchiu V, Viscomi MT, Latini L, Fezza F et al (2013) Activation of type-2 cannabinoid receptor inhibits neuroprotective and antiinflammatory actions of glucocorticoid receptor alpha: when one is better than two. Cell Mol Life Sci 70:2191–2204
Kojika S, Sugita K, Inukai T, Saito M, Iijima K et al (1996) Mechanisms of glucocorticoid resistance in human leukemic cells: implication of abnormal 90 and 70 kDa heat shock proteins. Leukemia 10:994–999
Matysiak M, Makosa B, Walczak A, Selmaj K (2008) Patients with multiple sclerosis resisted to glucocorticoid therapy: abnormal expression of heat-shock protein 90 in glucocorticoid receptor complex. Mult Scler 14:919–926
Buffon F, Molko N, Herve D, Porcher R, Denghien I et al (2005) Longitudinal diffusion changes in cerebral hemispheres after MCA infarcts. J Cereb Blood Flow Metab 25:641–650
Herve D, Molko N, Pappata S, Buffon F, LeBihan D et al (2005) Longitudinal thalamic diffusion changes after middle cerebral artery infarcts. J Neurol Neurosurg Psychiatry 76:200–205
Barnes PJ (2011) Glucocorticosteroids: current and future directions. Br J Pharmacol 163:29–43
Chrousos GP, Kino T (2005) Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Sci STKE 2005:pe48
Kino T, Su YA, Chrousos GP (2009) Human glucocorticoid receptor isoform beta: recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci 66:3435–3448
Rhen T, Cidlowski JA (2005) Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 353:1711–1723
Sayer FT, Kronvall E, Nilsson OG (2006) Methylprednisolone treatment in acute spinal cord injury: the myth challenged through a structured analysis of published literature. Spine J 6:335–343
Lee HC, Cho DY, Lee WY, Chuang HC (2007) Pitfalls in treatment of acute cervical spinal cord injury using high-dose methylprednisolone: a retrospect audit of 111 patients. Surg Neurol 68(Suppl 1):S37–S41, discussion S41–32
Sharma A (2012) Pharmacological management of acute spinal cord injury. J Assoc Physicians India 60(Suppl):13–18
Mu X, Azbill RD, Springer JE (2000) Riluzole and methylprednisolone combined treatment improves functional recovery in traumatic spinal cord injury. J Neurotrauma 17:773–780
Weaver LC, Gris D, Saville LR, Oatway MA, Chen Y et al (2005) Methylprednisolone causes minimal improvement after spinal cord injury in rats, contrasting with benefits of an anti-integrin treatment. J Neurotrauma 22:1375–1387
Pereira JE, Costa LM, Cabrita AM, Couto PA, Filipe VM et al (2009) Methylprednisolone fails to improve functional and histological outcome following spinal cord injury in rats. Exp Neurol 220:71–81
Gomes JA, Stevens RD, Lewin JJ 3rd, Mirski MA, Bhardwaj A (2005) Glucocorticoid therapy in neurologic critical care. Crit Care Med 33:1214–1224
Chen X, Zhang KL, Yang SY, Dong JF, Zhang JN (2009) Glucocorticoids aggravate retrograde memory deficiency associated with traumatic brain injury in rats. J Neurotrauma 26:253–260
Chen X, Zhang B, Chai Y, Dong B, Lei P et al (2011) Methylprednisolone exacerbates acute critical illness-related corticosteroid insufficiency associated with traumatic brain injury in rats. Brain Res 1382:298–307
Bratton SL, Chestnut RM, Ghajar J, McConnell Hammond FF, Harris OA et al (2007) Guidelines for the management of severe traumatic brain injury. XV. Steroids. J Neurotrauma 24(Suppl 1):S91–S95
Yong VW, Wells J, Giuliani F, Casha S, Power C et al (2004) The promise of minocycline in neurology. Lancet Neurol 3:744–751
Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J (2001) Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21:2580–2588
Festoff BW, Ameenuddin S, Arnold PM, Wong A, Santacruz KS et al (2006) Minocycline neuroprotects, reduces microgliosis, and inhibits caspase protease expression early after spinal cord injury. J Neurochem 97:1314–1326
Yune TY, Lee JY, Jung GY, Kim SJ, Jiang MH et al (2007) Minocycline alleviates death of oligodendrocytes by inhibiting pro-nerve growth factor production in microglia after spinal cord injury. J Neurosci 27:7751–7761
Cho DC, Cheong JH, Yang MS, Hwang SJ, Kim JM et al (2011) The effect of minocycline on motor neuron recovery and neuropathic pain in a rat model of spinal cord injury. J Korean Neurosurg Soc 49:83–91
Wells JE, Hurlbert RJ, Fehlings MG, Yong VW (2003) Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain 126:1628–1637
Lam TI, Bingham D, Chang TJ, Lee CC, Shi J et al (2013) Beneficial effects of minocycline and botulinum toxin-induced constraint physical therapy following experimental traumatic brain injury. Neurorehabil Neural Repair 27:889–899
Homsi S, Federico F, Croci N, Palmier B, Plotkine M et al (2009) Minocycline effects on cerebral edema: relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res 1291:122–132
Homsi S, Piaggio T, Croci N, Noble F, Plotkine M et al (2010) Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: a twelve-week follow-up study. J Neurotrauma 27:911–921
Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB et al (2004) Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci 24:2182–2190
Diguet E, Fernagut PO, Wei X, Du Y, Rouland R et al (2004) Deleterious effects of minocycline in animal models of Parkinson's disease and Huntington's disease. Eur J Neurosci 19:3266–3276
Yang L, Sugama S, Chirichigno JW, Gregorio J, Lorenzl S et al (2003) Minocycline enhances MPTP toxicity to dopaminergic neurons. J Neurosci Res 74:278–285
Pinzon A, Marcillo A, Quintana A, Stamler S, Bunge MB et al (2008) A re-assessment of minocycline as a neuroprotective agent in a rat spinal cord contusion model. Brain Res 1243:146–151
Bye N, Habgood MD, Callaway JK, Malakooti N, Potter A et al (2007) Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp Neurol 204:220–233
Viscomi MT, Latini L, Florenzano F, Bernardi G, Molinari M (2008) Minocycline attenuates microglial activation but fails to mitigate degeneration in inferior olive and pontine nuclei after focal cerebellar lesion. Cerebellum 7:401–405
Chen X, Ma X, Jiang Y, Pi R, Liu Y et al (2011) The prospects of minocycline in multiple sclerosis. J Neuroimmunol 235:1–8
Plane JM, Shen Y, Pleasure DE, Deng W (2010) Prospects for minocycline neuroprotection. Arch Neurol 67:1442–1448
Xue M, Mikliaeva EI, Casha S, Zygun D, Demchuk A et al (2010) Improving outcomes of neuroprotection by minocycline: guides from cell culture and intracerebral hemorrhage in mice. Am J Pathol 176:1193–1202
Jung CH, Ro SH, Cao J, Otto NM, Kim DH (2010) mTOR regulation of autophagy. FEBS Lett 584:1287–1295
Bove J, Martinez-Vicente M, Vila M (2011) Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 12:437–452
Chen H, Qu Y, Tang B, Xiong T, Mu D (2012) Role of mammalian target of rapamycin in hypoxic or ischemic brain injury: potential neuroprotection and limitations. Rev Neurosci 23:279–287
Kanno H, Ozawa H, Sekiguchi A, Itoi E (2009) The role of autophagy in spinal cord injury. Autophagy 5:390–392
Kanno H, Ozawa H, Sekiguchi A, Itoi E (2009) Spinal cord injury induces upregulation of Beclin 1 and promotes autophagic cell death. Neurobiol Dis 33:143–148
Kanno H, Ozawa H, Sekiguchi A, Yamaya S, Itoi E (2011) Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine (Phila Pa 1976) 36:E1427–E1434
Sekiguchi A, Kanno H, Ozawa H, Yamaya S, Itoi E (2012) Rapamycin promotes autophagy and reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. J Neurotrauma 29:946–956
Chen HC, Fong TH, Hsu PW, Chiu WT (2013) Multifaceted effects of rapamycin on functional recovery after spinal cord injury in rats through autophagy promotion, anti-inflammation, and neuroprotection. J Surg Res 179:e203–e210
Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R (2007) Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis 26:86–93
Luo CL, Li BX, Li QQ, Chen XP, Sun YX et al (2011) Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience 184:54–63
Pertwee RG (2005) Pharmacological actions of cannabinoids. Handb Exp Pharmacol 168:1–51
Pertwee RG (2005) The therapeutic potential of drugs that target cannabinoid receptors or modulate the tissue levels or actions of endocannabinoids. AAPS J 7:E625–E654
van der Stelt M, Di Marzo V (2005) Cannabinoid receptors and their role in neuroprotection. Neuromolecular Med 7:37–50
Bahr BA, Karanian DA, Makanji SS, Makriyannis A (2006) Targeting the endocannabinoid system in treating brain disorders. Expert Opin Investig Drugs 15:351–365
Scotter EL, Abood ME, Glass M (2010) The endocannabinoid system as a target for the treatment of neurodegenerative disease. Br J Pharmacol 160:480–498
Velayudhan L, Van Diepen E, Marudkar M, Hands O, Suribhatla S et al (2013) Therapeutic potential of cannabinoids in neurodegenerative disorders: a selective review. Curr Pharm Des 33(3):755–766
Sagredo O, Pazos MR, Valdeolivas S, Fernandez-Ruiz J (2012) Cannabinoids: novel medicines for the treatment of Huntington's disease. Recent Pat CNS Drug Discov 7:41–48
Panikashvili D, Mechoulam R, Beni SM, Alexandrovich A, Shohami E (2005) CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J Cereb Blood Flow Metab 25:477–484
Panikashvili D, Shein NA, Mechoulam R, Trembovler V, Kohen R et al (2006) The endocannabinoid 2-AG protects the blood-brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol Dis 22:257–264
Arevalo-Martin A, Garcia-Ovejero D, Molina-Holgado E (2010) The endocannabinoid 2-arachidonoylglycerol reduces lesion expansion and white matter damage after spinal cord injury. Neurobiol Dis 38:304–312
Amenta PS, Jallo JI, Tuma RF, Elliott MB (2012) A cannabinoid type 2 receptor agonist attenuates blood-brain barrier damage and neurodegeneration in a murine model of traumatic brain injury. J Neurosci Res 90:2293–2305
Elliott MB, Tuma RF, Amenta PS, Barbe MF, Jallo JI (2011) Acute effects of a selective cannabinoid-2 receptor agonist on neuroinflammation in a model of traumatic brain injury. J Neurotrauma 28:973–981
Adhikary S, Li H, Heller J, Skarica M, Zhang M et al (2011) Modulation of inflammatory responses by a cannabinoid-2-selective agonist after spinal cord injury. J Neurotrauma 28:2417–2427
Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H et al (2003) Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci 23:2371–2382
Molina-Holgado F, Pinteaux E, Heenan L, Moore JD, Rothwell NJ et al (2005) Neuroprotective effects of the synthetic cannabinoid HU-210 in primary cortical neurons are mediated by phosphatidylinositol 3-kinase/AKT signaling. Mol Cell Neurosci 28:189–194
Acknowledgments
This work was supported by the Italian Ministry of Health (Ricerca Corrente - MM), by the Wings for Life Spinal Cord Research Foundation (M.T.V.), by the International Foundation for Research in Paraplegia (IFP) (M.T.V.), and by the program Young Researchers of Italian Ministry of Health (GR10.184; M.T.V.). We thank Prof. G. Bernardi for his continuous support and encouragement. The professional editorial work of Blue Pencil Science is also acknowledged.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Viscomi, M.T., Molinari, M. Remote Neurodegeneration: Multiple Actors for One Play. Mol Neurobiol 50, 368–389 (2014). https://doi.org/10.1007/s12035-013-8629-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8629-x