
Abstract
Sphingosine kinases (Sphk1/2) are crucial enzymes in regulation of the biostat between sphingosine-1-phosphate (S1P) and ceramide and play an important role in the pathogenesis/pathomechanism of Alzheimer’s disease (AD). These enzymes synthesise S1P, which regulates neurotransmission, synaptic function and neuron cell proliferation, by activating five G protein-coupled receptors (S1P1-5). However, S1P synthesised by Sphk2 could be involved in amyloid β (Aβ) release by stimulation of Aβ precursor protein degradation. The significance of this bioactive sphingolipid in the pathogenesis of Parkinson’s disease (PD) is unknown. The aim of our study was to investigate the expression level of Sphk1 and its role in human dopaminergic neuronal cell (SH-SY5Y) viability under oxidative stress, evoked by 1-methyl-4-phenylpyridinium (MPP+). Moreover, the mechanism of S1P action on the death signalling pathway in these experimental conditions was evaluated. Our study indicated marked downregulation of Sphk1 expression in this cellular PD model. Inhibition of Sphk1 decreased SH-SY5Y cell viability and concomitantly enhanced the reactive oxygen species (ROS) level. It was found that exogenous S1P (1 μM) exerted the neuroprotective effect by activation of Sphk1 and S1P1 receptor gene expression. Moreover, S1P downregulated Bax and harakiri, death protein 5 (Hrk/DP5) expression and enhanced cell viability in MPP+-treated cells. The neuroprotective mechanism of S1P is mainly dependent on S1P1 receptor signalling, which was indicated by using specific agonists and antagonists of S1P1 receptor. The results show that S1P and S1P1 receptor agonists protected a significant population of neuronal cells against death.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
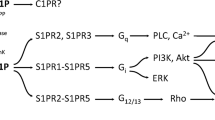
Sphingosine kinases 1 and 2 (Sphk1/2) are two conservative isoenzymes which generate a potent lipid mediator, sphingosine-1-phosphate (S1P), and play a significant role in the regulation of the sphingolipid biostat [1–4]. Although both isoforms use the same substrate, sphingosine, and generate the same product, they have a different location within the cell and different biochemical properties. The roles of S1P pools synthesised by Sphk1 and Sphk2 have been indicated as having different functions [5–7]. A wide variety of agonists, including growth factors, hormones and pro-inflammatory cytokines, activate Sphk1 and synthesise the S1P pool, which exerts mitogenic and anti-apoptotic effects [8–15]. In contrast, Sphk2 induces cell cycle arrest, suppresses growth and enhances apoptosis [16, 17]. Over the last two decades, S1P has been shown to act as a primary or secondary messenger. S1P modulates mitochondrial respiration via direct interaction with mitochondrial proteins and is also involved in the release of cytochrome c, probably by modulation of the Bak protein [18, 19]. Despite intracellular action, S1P can be transported out of the cell via a member of the ABC transporter family [20], and it can act as a primary messenger in the autocrine/paracrine manner through five specific G protein-coupled cell surface receptors (GPCR), termed S1P1–5 [21, 22]. S1P1–3 receptors mediate the signalling pathways via phosphoinositide-3-kinase Akt (PI3K/Akt), phospholipase C (PLC), extracellular signal-regulated kinases 1/2 (ERK1/2) or GTPases, and through receptor signalling pathways. S1P regulates fundamental cellular processes, such as growth, differentiation, motility, proliferation, angiogenesis and inflammation [9, 23–26]. In the past decade, a growing number of studies have focused on the role of Sphks/S1P in neurodegenerative disorders. The perturbed sphingomyelin metabolism is a fundamental event in the degeneration of neurons in Alzheimer’s disease (AD) [27–29]. Sphks inhibition could be responsible for an alteration of the S1P/ceramide biostat [1, 30, 31]. In aging and AD, there is excessive production and accumulation of ceramides, which have been shown to modify the structure, function and plasticity of neurons, as was described recently by Haughey et al. [29]. Recently, Takasugi et al. [7] indicated that SphK2, which is upregulated in the brains of AD patients, synthesised the S1P pool which is engaged in the modulation of β-amyloid precursor protein cleaving enzyme 1 (BACE1) activity. Through this mechanism, Sphk2 could be involved in Aβ release. Changes in sphingolipid metabolism were also reported in other neurological disorders, including HIV dementia, brain ischemia, hypoxia and inflammation [32–34], and in various human tumours [35–37]. However, until now, the role of Sphk disturbances and S1P receptor signalling in Parkinson’s disease (PD) remains unknown. Our experiments were carried out on human SH-SY5Y cells highly used in the cellular model for PD research because of their characteristics of dopaminergic neurons [38, 39]. SH-SY5Y cells have the ability to synthesise dopamine (DA) and noradrenalin (NA) and express dopamine transporter (DAT), a protein typical for dopaminergic neurons within the central nervous system [40, 41]. In this study, we investigated the expression/protein level of Sphk1 and its role in cell viability in an experimental PD model, evoked by 1-methyl-4-phenylpiridinum (MPP+) in SH-SY5Y. Moreover, the effect of exogenously added S1P on MPP+-evoked cell death was evaluated.
Materials and Methods
Cell Culture
The studies were carried out using the human neuroblastoma cell line SH-SY5Y (a kind gift from Prof. Anne Eckert, Neurobiology Laboratory for Brain Aging and Mental Health, Psychiatric University Clinics, University of Basel). Cells were cultured in MEM/F-12 Ham's Nutrient Mixtures (1:1) supplemented with 15 % heat-inactivated fetal bovine serum (FBS), 1 % penicillin/streptomycin and 2 mM glutamine. The cells were maintained at 37 °C in a humidified incubator containing 5 % CO2. For the experiment, confluent cells were sub-cultured into dishes or collagen-coated 96-well plates. The cells were used for experiments at 75–90 % confluence or 1 day after being plated in the 96-well plate. Prior to treatment, the cells were replenished with a low-serum (2 % FBS) medium.
Cell Treatment Protocols
SH-SY5Y cells were treated with 1-methyl-4-phenylpyridinium (MPP+) in various concentrations up to 3 mM or with 5 μM SKI II. In some experiments, the cells were cultured for 24 h with 3 mM MPP+ and were pre-incubated with the following reagents: 1 μM S1P, 10 μM SEW, 20 μM W123, and 100 pM P-FTY720. The corresponding compounds were added to the culture medium 1 h before MPP+ treatment.
Cell Viability Analysis
Mitochondrial function and cellular viability were evaluated by using 2-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). After 24 h incubation with appropriate compounds, MTT (2.5 mg/ml) was added to all of the wells. The cells were incubated at 37 °C for 2 h. Then, the cells were lysed in DMSO, and spectrophotometric measurement at 595 nm was performed.
Determination of Free Radicals Using the DCF Probe
The DCF fluorescence assay detects the level of hydrogen peroxide and other free radicals (ROS) in cells. Free radicals were determined based on reactive oxygen species-mediated conversion of 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) into fluorescent 2′,7′-dichlorofluorescein (DCF). The cell media was changed after 24 h incubation with appropriate compounds to Hanks’ buffer without phenol red (Sigma, H8264), and incubation was continued in the presence of 10 μM H2DCF-DA in DMSO for 50 min at 37 °C. DMSO was used at a final concentration of 0.05 %, and at this concentration, it had no effect on the free radical levels. Fluorescence of DCF was measured using a PerkinElmer LS 50B spectrofluorometer with excitation and emission wavelengths at 488 and 535 nm, respectively. DCF fluorescence is reported in a percentage of the control.
In Vitro Sphk Activity Assay
Sphk activity assay was performed according to a previous report [7]. After 24 h incubation, the cells were washed with iced PBS and lysed by the freeze–thaw cycle in 50 mM HEPES, pH 7.4, 10 mM KCl, 15 mM MgCl2, 0.1 % Triton X-100, 20 % glycerol, 2 mM orthovanadate, 2 mM dithiothreitol, 10 mM NaF, 1 mM deoxypyridoxine and EDTA-free complete protease inhibitor (Roche Applied Science). Lysates were cleared by centrifugation at 15,000 rpm for 5 min. The lysates and NBD-Sphingosine (10 μM final; Avanti Polar Lipids) were mixed in a reaction buffer (50 mM HEPES, pH7.4, 15 mM MgCl2, 0.5 mM KCl, 10 % glycerol and 2 mM ATP) and incubated for 30 min at 30 °C. The reactions were stopped by the addition of an equal amount of 1 M potassium phosphate, pH 8.5, followed by the addition of 2.5-fold chloroform/methanol (2:1), and then centrifuged at 15,000 rpm for 1 min. Only the reactant NBD-S1P, but not the substrate NBD-sphingosine, was collected in the alkaline aqueous phase. After the aqueous phase was combined with an equal amount of dimethylformamide, the fluorescence value was read.
Western Blot
After protein measurement according to Lowry, the homogenate of SH-SY5Y cells was mixed with a 5× Laemmli sample buffer and denatured for 5 min at 95 °C. Forty micrograms of protein was loaded per lane on 10 % acrylamide gels and examined by SDS-PAGE. The proteins were transferred onto PVDF membranes at 100 V. The membranes were incubated in 5 % dry milk in TBS with Tween 20 (TBS-T) for 1 h and exposed overnight to the following antibodies: anti-Sphk1 (1:250, from Cell Signalling) and anti-GAPDH (1:10,000, from Sigma-Aldrich). Proteins were identified in accordance with the molecular weight marker (from Sigma-Aldrich). After treatment for 1 h with the corresponding horseradish peroxidase-coupled secondary antibodies (anti-rabbit from Sigma-Aldrich), the protein bands were detected by ECL reagent (ThermoScientific).
Analysis of the mRNA Level Using Real-Time PCR
RNA was isolated using a TRI-reagent from Sigma-Aldrich. The isolated RNA was dissolved in RNAse-free water (Promega Corporation). The amount and purity of RNA were determined using spectrophotometric measurement at 260 and 280 nm. The OD260/OD280 ratio of the RNA samples ranged from 1.6 to 1.9. Isolated RNA (5 μg) was used in RT-PCR. The reverse transcription was performed by using a High Capacity cDNA Reverse Transcription Kit according to the manufacturer’s protocol (Applied Biosystems, Foster City, CA, USA). Quantitative PCR was performed by using pre-developed TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA): actb Hs99999903_m1, bax Hs00180269_m1, bcl-2 Hs00608023_m1, gpx4 Hs00989766_g1, hrk Hs02621354_s1, sod2 Hs00167309_m1, sphk1 Hs01116530_g1, s1pr1 Hs01922614_s1 and s1pr3 Hs00245464_s1 on an ABI PRISM 7500 apparatus according to the manufacturer’s instructions. Actb was selected and used in all of the studies as a reference gene. The relative level of mRNA was calculated by the ΔΔCt method.
Determination of Apoptosis Using Hoechst 33342
For morphological studies, SH-SY5Y cells were subjected for 24 h to oxidative stress evoked by MPP+ (3 mM). Moreover, the effects of S1P (1 μM) and the effect of SKI II (5 μM) were evaluated. Coverslips containing SH-SY5Y cells were collected and washed in PBS. The cells were fixed in −20 °C MetOH for 30 min in 4 °C. Nuclei were visualised with Hoechst 33342 (0.2 μg/ml, Riedel-de-Haën Germany) fluorescent staining. The cells were examined under a fluorescence microscope (Olympus BX51, Japan) and photographed with a digital camera (Olympus DP70, Japan). Cells with typical apoptotic nuclear morphology (nuclear shrinkage, condensation) were identified and counted. The results were expressed as percentages of apoptotic cells in the whole cell population.
Statistical Analysis
Statistical analyses between the two groups were conducted using Student’s t test. Analyses among multi-group data were conducted using one-way analysis of variance (ANOVA), followed by the Newman–Keuls post-hoc test. The data are given as means ± SEM. p values <0.05 were considered statistically significant.
Results
In the present study, using a cellular PD model induced by MPP+, we investigated the Sphk1 gene expression, the role of Sphk1 in cell viability and the effect of exogenous S1P on neuronal cell death. Human dopaminergic cells (SH-SY5Y) were exposed for 24 h to various concentrations of MPP+ (0–5 mM). It was apparent that MPP+ decreased the viability of SH-SY5Y cells in a concentration-dependent manner, i.e., by 50 % at 3 mM (Fig. 1b). By applying 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), we indicated that 3 mM MPP+ increased the free radical level by about 90 % of the control (Fig. 1a). Microscopic examination of cell nuclei stained with DNA-binding fluorochrome Hoechst 33342 showed typical apoptotic morphology (Fig. 1c). Under these stress conditions, the mRNA level of antioxidant enzymes such as superoxide dismutase (SOD) and glutathione peroxidase (GPx) significantly decreased (Fig. 1d, e). Then, our study indicated that Sphk1 gene expression/protein level and Sphks activity were reduced in MPP+-treated cells (Fig. 2a–c). Cell survival and ROS concentration analysis indicated that inhibition of Sphk1(by the Sphk1 inhibitor, 5 μM SKI II) enhanced the free radical level and suppressed the viability of SH-SY5Y cells (Fig. 2d, e). Microscopic examination of cell nuclei showed a higher number of apoptotic cells after exposition to the Sphk1 inhibitor (5 μM SKI II) (Fig. 2f). The product of Sphk1, S1P (1 μM), added extracellularly to the cultivation medium, protects SH-SY5Y neuronal cells against stress evoked by MPP+. S1P activates the Sphk1 gene expression/protein level that is observed 3 and 12 h after its action in the presence of MPP+. S1P alone had no effect on Sphk1 expression after 3 h. However, it significantly decreased Sphk1 gene expression after 12 h (Fig. 3a). The slight decrease of Sphk1 expression in this study has no significant effect on Sphk1 protein level after 24 h incubation with-S1P (Fig. 3b). Subsequently, we observed that S1P decreased oxidative stress in cells treated with MPP+ and protected the significant pool of SH-SY5Y cells against death evoked by MPP+ (Fig. 3c, d). The amount of apoptotic cells was significantly lower in the presence of S1P (Fig. 3e). S1P alone had no effect. For better elucidation of mechanisms by which S1P protects SH-SY5Y cells from MPP+-induced death, we examined its effect on pro- and anti-apoptotic Bcl-2 proteins. Our study indicated that MPP+ leads to activation of gene expression for pro-apoptotic Bcl-2 proteins: Bax and Hrk/DP5. In the presence of exogenous S1P (1 μM), the mRNA level of both of these pro-apoptotic proteins was significantly reduced (Fig. 4a, b). Anti-apoptotic Bcl-2 gene expression was activated by MPP+, but in the presence of exogenously added S1P, the mRNA level of Bcl-2 was not enhanced, i.e., it was even lower as compared to MPP+-treated cells (Fig. 4c). Finally, we investigated whether the S1P effect is receptor-mediated. Our data demonstrated that the mRNA level of S1P1 under stress caused by MPP+ was not changed. However, S1P alone significantly increased its expression (Fig. 5a). Gene expression analysis of S1P3 indicated that the mRNA level of S1P3 was reduced after MPP+ treatment (Fig. 5b). Using specific S1P receptor agonists and antagonists, we demonstrated that S1P can exert its neuroprotective effect by activation of the S1P1 receptor. Moreover, the sphingosine analogue (P-FTY720) also has neuroprotective effects (Fig. 5c).

The effect of MPP+ on ROS generation, SH-SY5Y cell viability, apoptosis and gene expression for anti-oxidative enzymes. SH-SY5Y cells were treated with 0.5–5 mM MPP+ for 24 h. ROS generation was determined using DCFH-DA (a) and cell viability by MTT assay (b). Data represent the mean value ± S.E.M for four independent experiments with four to six replications. *p < 0.05, **p < 0.01 and ***p < 0.001 versus control (non-treated) SH-SY5Y cells by one-way ANOVA followed by the Newman–Keuls post-hoc test. Cell nuclei in 3 mM MPP+-treated cells were visualised with Hoechst 33342 and observed under the fluorescence microscope. Representative pictures show typical morphological changes in cell nuclei after MPP+ treatment (c). The mRNA level for SOD (d) and GPx (e) after 3 mM MPP+ treatment was measured with real-time PCR. The value expresses the fold of the above gene stimulation normalized against Actb (β-actin). Data represent the mean value ± S.E.M for three separate experiments with three replications. The relative level of mRNA was calculated by ΔΔCt method. *p < 0.05, **p < 0.01 and ***p < 0.001 versus control (non-treated) SH-SY5Y cells by one-way ANOVA followed by the Newman–Keuls post-hoc test

Sphk1 activity, gene expression/protein level in MPP+-treated SH-SY5Y cell and the role of Sphk1 inhibition on ROS generation and cell viability. SH-SY5Y cells were treated with MPP+ (3 mM) for 3 h, 12 h (a) or 24 h (b, c). The mRNA level for Sphk1 was measured with real-time PCR. The value expresses the fold of the above gene stimulation normalized against Actb (β-actin). Data represent the mean value ± S.E.M for three separate experiments with three replications. The relative level of mRNA was calculated by ΔΔCt method. **p < 0.01 and ***p < 0.001 versus control (non-treated) SH-SY5Y cells by one-way ANOVA followed by the Newman–Keuls post-hoc test (a). Sphk1 (~68.35 kDa) immunoreactivity in the cell homogenate was measured. A representative Western blot from one typical experiment is shown below the graph. Data represent the mean value ± S.E.M for four independent experiments normalized against GAPDH (~36.1 kDa) (b). Fluorescence value of Sphk activity was measured. Data represent the mean value ± S.E.M for three independent experiments (c). **p < 0.01 versus control (non-treated) SH-SY5Y cells by Student's t test. SH-SY5Y cells were treated with 5 μM SKI II for 24 h (d, e, f). ROS generation was determined using DCFH-DA (d) and cell viability by MTT assay (e). Data represent the mean value ± S.E.M for four independent experiments with four to six replications. **p < 0.01 and ***p < 0.01 versus control (non-treated) SH-SY5Y cells by Student's t test. Cell nuclei in SKI II-treated cells were visualised with Hoechst 33342 and observed under the fluorescence microscope. The percentage of apoptosis was calculated. Data represent the mean value±S.E.M for three independent experiments, **p<0.01 versus control (nontreated) SH-SY5Y cells by Student's t test. Representative pictures show typical morphological changes in cell nuclei after SKI II treatment (f)

Effect of S1P on Sphk1 gene expression/protein level, ROS generation and SH-SY5Y cell viability under stress evoked by MPP+. SH-SY5Y cells were treated with 1 μM S1P for 1 h and then exposed to 3 mM MPP+ for 3 h, 12 h (a) or 24 h (b, c, d, e). The mRNA level for Sphk1 was measured with real-time PCR. The value expresses the fold of the above gene stimulation normalized against Actb (β-actin). Data represent the mean value ± S.E.M for three separate experiments with three replications. The relative level of mRNA was calculated by ΔΔCt method. ***p < 0.001 versus control (non-treated) SH-SY5Y cells; ### p < 0.001 versus MPP+-treated cells by one-way ANOVA followed by the Newman–Keuls post-hoc test (a). Sphk1 (~68.35 kDa) immunoreactivity in the cell homogenate was measured. A representative Western blot from one typical experiment is shown below the graph. Data represent the mean value ± S.E.M for four independent experiments normalized against GAPDH (~36.1 kDa). *p < 0.05 versus control (non-treated) SH-SY5Y cells; ## p < 0.01 versus MPP+-treated cells by one-way ANOVA followed by the Newman–Keuls post-hoc test (b). ROS generation was determined using DCFH-DA (c) and cell viability by MTT assay (d). Data represent the mean value ± S.E.M for four independent experiments with four to six replications. ***p < 0.001 versus control (non-treated) SH-SY5Y cells; ## p < 0.01 and ### p < 0.001 versus MPP+-treated cells by one-way ANOVA followed by the Newman–Keuls post-hoc test. Cell nuclei in MPP+-treated cells were visualised with Hoechst 33342 and observed under the fluorescence microscope. The percentage of apoptosis was calculated. Data represent the mean value±S.E.M for three independent experiments, ***p < 0.001 versus control (non-treated) SH-SY5Y cells; ### p < 0.001 versus MPP+-treated cells by one-way ANOVA followed by the Newman–Keuls post-hoc test. Representative pictures show typical morphological changes in cell nuclei after S1P and MPP+ treatment (e)

Effect of S1P on Bcl-2 protein expression in SH-SY5Y cells treated with MPP+. SH-SY5Y cells were treated with 1 μM S1P for 1 h and then exposed to 3 mM MPP+ for 12 h. The mRNA level for Bax (a), Hrk/DP5 (b) and Bcl-2 (c) was measured with real-time PCR. The value expresses the fold of the above gene stimulation normalized against Actb (β-actin). Data represent the mean value ± S.E.M for three separate experiments with three replications. The relative level of mRNA was calculated by ΔΔCt method. **p < 0.01 and ***p < 0.001 versus control (non-treated) SH-SY5Y cells; # p < 0.05 and ### p < 0.001—the difference is statistically significant compared to MPP+-treated cells by one-way ANOVA followed by the Newman–Keuls post-hoc test

S1P1 and S1P3 gene expression and the effect of S1P1 receptor agonists and antagonists on SH-SY5Y cell viability under stress conditions evoked by MPP+. SH-SY5Y cells were treated with 1 μM S1P for 1 h and then exposed to 3 mM MPP+ for 12 h. The mRNA level for S1P1 (a) and S1P3 (b) was measured with real-time PCR. The value expresses the fold of the above gene stimulation normalized against Actb (β-actin). Data represent the mean value ± S.E.M for three separate experiments with three replications. The relative level of mRNA was calculated by ΔΔCt method. *p < 0.05 and **p < 0.01—the difference is statistically significant compared to the control (non-treated) SH-SY5Y cells by one-way ANOVA followed by the Newman–Keuls post-hoc test. SH-SY5Y cells were treated for 1 h with following compounds: 1 μM S1P, 10 μM SEW, 100 pM P-FTY720 and 20 μM W123 and then exposed to 3 mM MPP+ for 24 h (c). Cell viability was determined by MTT assay. Data represent the mean value ± S.E.M for four independent experiments with four to six replications. ***p < 0.001 versus control (non-treated) SH-SY5Y cells; # p < 0.05 and ### p < 0.001—the difference is statistically significant compared to MPP+-treated cells; $$$ p < 0.001—the difference is statistically significant compared to S1P and MPP+-treated cells by one-way ANOVA followed by the Newman–Keuls post-hoc test
Discussion
For many years, it has been suggested that reactive oxygen species (ROS) play an important role in PD pathogenesis. However, the pathomechanism/pathogenesis of neurodegenerative disorders has not been elucidated. Our data indicated that MPP+, a widely used compound in experimental models of PD, decreased the Sphk1 gene expression/protein level. Sphk1 inhibition induces oxidative stress and apoptotic cell death. The product of Sphk1, exogenous S1P (1 μM), protected a significant pool of neuronal cells against death and decreased the free radical level. Some previous studies, also our own, have revealed that MPP+ induced oxidative stress and dopaminergic cell death [42–45]. Until now, there has been no data on the role of sphingolipid alteration in neurons in PD and in the experimental model of this disease. Previous studies demonstrated changes in the sphingolipids and the alteration of gene expression for enzymes involved in sphingolipid metabolism in AD [7, 28, 29]. Significant correlations were observed between a reduced level of S1P and an increased concentration of amyloid beta (Aβ) peptide and hyperphosphorylated tau protein in AD brains [27]. Moreover, Gomez-Brouchet et al. [30] have reported reduced Sphk1 activity and a decrease in the S1P level in SH-SY5Y cells in oxidative stress conditions evoked by Aβ treatment. Gomez-Brouchet et al. [30] have also shown that the antioxidant N-acetylcysteine abolished Sphk1 inhibition. Maceyka et al. [46] suggested that Sphk1 could be a crucial step in oxidative stress-mediated cell death. Moreover, ROS may subsequently lead to Sphk1 inhibition. Our data demonstrated reduced Sphks activity and downregulation of Sphk1 expression under oxidative stress evoked by MPP+. Moreover, our results indicated that inhibition of Sphk1 can lead to ROS formation and neuronal cell apoptosis. Similarly, Pchejetski et al. [47] have reported that oxidative stress leads to Sphk1 inhibition, generation of pro-apoptotic ceramide and decrease of pro-survival S1P. It is well established that Sphk1 activity is enhanced in tumour cell lineages, which may be associated with the role of Sphk1 in adaptation to hypoxic stress [48, 49]. Sphk1 inhibition or knockdown of gene enhanced ROS production in several cancer cell lines [50]. In our study, we examined the effect of exogenous S1P (1 μM) on cell survival and death in conditions of oxidative stress evoked by MPP+. This 1 μM concentration of S1P corresponds to approximately 10 nmol of S1P/mg of protein, reported in the human brain by He et al. [27]. Their data demonstrated S1P level at 5 nmol of S1P/mg of protein in the AD brains and about 12 nmol of S1P/mg of protein in the brain of age-matched control. Moreover, S1P exists in the blood at 0.1–1 μM concentration [51]. Our data showed that S1P reduces the ROS concentration in SH-SY5Y cells treated with MPP+ and increases the viability of a significant pool of cells. Nakahara et al. [52] indicated that S1P protects cells against apoptosis induced by H2O2. Abrahan et al. [53] showed that enhanced S1P synthesis protects photoreceptors against paraquat toxicity. In this study, we demonstrated that S1P exerts its inhibitory effect on apoptosis by abolishing the downregulation of Sphk1 expression and through modulation of the Bcl-2 protein family. Gene expression studies in SH-SY5Y neuroblastoma cells may have some limitations which is characteristic of cancer cell lines. However, SH-SY5Y cells have been used frequently, either in an undifferentiated state [54–56] or in a neuron-like differentiated state for the study of molecular process in MPP+ or in the other cellular PD model [57–59]. Because differentiating agents can induce overexpression or downregulation of anti- and pro-apoptotic proteins like Bcl-2, we have used non-differentiating cells in our study. We showed increased expression of pro-apoptotic Bcl-2 proteins in stress evoked by MPP+, which is consistent with the results published previously by Zhai et al., Liu et al., Zhu et al. and Zhang et al. [60–63]. Moreover, S1P induced downregulation of Bax and Hrk/DP5 expression in MPP+-treated cells. The effect of S1P on the expression of pro-apoptotic Bcl-2 proteins can be an important protective mechanism against MPP+ toxicity. The Bax protein is recognised as a direct effector of protein-permeable pore formation in the outer mitochondrial membrane, which could lead to the release of cytochrome c and probably also the apoptosis-inducing factor (AIF) into the cytosol [64]. The other pro-apoptotic protein, death-promoting protein 5 (DP5), named Harakiri (Hrk) exerts pro-apoptotic activity by interacting with pro-survival Bcl-xL and Bcl-2 proteins [65, 66]. In this way, in our study, mRNA analysis revealed that the level of the pro-survival Bcl-2 protein in SH-SY5Y cells increased after MPP+ treatment. Similar data were obtained by other researchers using the same cell line [67, 68]. This event could be an adaptive survival response to oxidative stress. We also observed that pro-survival Bcl-2 gene expression is less activated in the presence of exogenous S1P, as compared to MPP+-treated cells. This effect may be connected with simultaneous significant downregulation of both pro-apoptotic Bcl-2 proteins (Bax and Hrk/DP5) by S1P. In contrast to our results, positive correlations between Bcl-2 proteins and Sphk1 were presented by Limaye et al. [69], who indicated that overexpression of Sphk1 can upregulate Bcl-2, and by Bektas et al. [70], whose studies showed that Bcl-2 overexpression stimulates Sphk1 expression and activity. It seems that S1P regulates the level of gene expression and exerts the neuroprotective effect depending on the type of cells and the type of stress. The question arises if S1P regulated pro-survival signalling by its specific receptors in our experimental conditions. By using several agonists and antagonists in our study, the data reveal that the pro-survival action of S1P is mainly dependent on S1P1 receptor signalling. We demonstrated the protective effects of S1P1-specific agonists (SEW-2871). Moreover, the protective effect of S1P was abolished by a specific S1P1 antagonist (W123). A number of studies described that the S1P1 receptor promotes pro-survival signal through PI3K/Akt [71–73]. PI3K/Akt is able to regulate cell survival by transcription and activity of pro- as well as anti-apoptotic proteins [74, 75]. It is possible that the decreased levels of pro-apoptotic Bcl-2 proteins as described above could be regulated by activation of S1P1 signalling. We also showed the neuroprotective properties of S1P receptor modulator P-FTY720, which represents a bioavailable compound that, besides having anti-inflammatory properties, also has a neuroprotective effect [76]. Summarizing, our studies demonstrate the importance of Sphk alteration in MPP+ stress responses and suggest its significant role in PD pathogenesis. Moreover, our data showed that S1P and S1P1 agonists can offer a novel neuroprotective strategy.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid beta
- BACE 1:
-
β-amyloid precursor protein cleaving enzyme 1
- DA:
-
Dopamine
- DAT:
-
Dopamine transporter
- DCF:
-
2′,7′-dichlorofluorescein
- DCFH-DA:
-
2′,7′-dichlorodihydrofluorescein diacetate
- DMSO:
-
Dimethyl sulfoxide
- ERK1/2:
-
Extracellular signal-regulated kinases 1/2
- FBS:
-
Fetal bovine serum
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GPCR:
-
G protein-coupled cell surface receptors
- GPx:
-
Glutathione peroxidase
- Hrk/DP5:
-
Harakiri, death protein 5
- MEM:
-
Minimum essential medium
- MPP+:
-
1-Methyl-4-phenylpyridinium
- mRNA:
-
Messenger ribonucleic acid
- MTT:
-
2-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NA:
-
Noradrenalin
- PBS:
-
Phosphate-buffered saline
- PCR:
-
Polymerase chain reaction
- PD:
-
Parkinson’s disease
- P-FTY720:
-
Phospho fingolimod
- PI3K/Akt:
-
Phosphoinositide-3-kinase Akt
- PLC:
-
Phospholipase C
- PVDF:
-
Polyvinylidene fluoride
- RNA:
-
Ribonucleic acid
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- S1P:
-
Sphingosine-1-phosphate
- S1P1–5:
-
Sphingosine-1-phosphate receptors 1–5
- SDS-PAGE:
-
SDS-polyacrylamide gel electrophoresis
- SOD:
-
Superoxide dismutase
- Sphk1:
-
Sphingosine kinase 1
- Sphk2:
-
Sphingosine kinase 2
- TBS:
-
Tris-buffered saline
- TBS-T:
-
Tris-buffered saline with Tween
References
Van Brocklyn JR, Williams JB (2012) The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: oxidative stress and the seesaw of cell survival and death. Comp Biochem Physiol B Biochem Mol Biol 163:26–36
Pitson SM (2011) Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci 36:97–107
Fyrst H, Saba JD (2010) An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem Biol 6:489–497
Okada T, Kajimoto T, Jahangeer S, Nakamura S (2009) Sphingosine kinase/sphingosine 1-phosphate signalling in central nervous system. Cell Signal 21:7–13
Neubauer HA, Pitson SM (2013) Roles, regulation and inhibitors of sphingosine kinase 2. FEBS J 280:5317–5336
Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH Jr, Milstien S, Spiegel S (2005) SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem 280:37118–37129
Takasugi N, Sasaki T, Suzuki K, Osawa S, Isshiki H, Hori Y, Shimada N, Higo T, Yokoshima S, Fukuyama T, Lee VM, Trojanowski JQ, Tomita T, Iwatsubo T (2011) BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J Neurosci 31:6850–6857
Edsall LC, Cuvillier O, Twitty S, Spiegel S, Milstien S (2001) Sphingosine kinase expression regulates apoptosis and caspase activation in PC12 cells. J Neurochem 76:1573–1584
Lebman DA, Spiegel S (2008) Cross-talk at the crossroads of sphingosine-1-phosphate, growth factors, and cytokine signaling. J Lipid Res 49:1388–1394
Sukocheva O, Wadham C, Xia P (2009) Role of sphingolipids in the cytoplasmic signaling of estrogens. Steroids 74:562–567
Melendez AJ (2008) Sphingosine kinase signalling in immune cells: potential as novel therapeutic targets. Biochim Biophys Acta 1784:66–75
Pitman MR, Pitson SM (2010) Inhibitors of the sphingosine kinase pathway as potential therapeutics. Curr Cancer Drug Targets 10:354–367
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9:139–150
Xia P, Wadham C (2011) Sphingosine 1-phosphate, a key mediator of the cytokine network: juxtacrine signaling. Cytokine Growth Factor 22:45–53
Gandy KA, Obeid LM (2013) Regulation of the sphingosine kinase/sphingosine 1-phosphate pathway. Handb Exp Pharmacol 216:275–303
Weigert A, Cremer S, Schmidt MV, von Knethen A, Angioni C, Geisslinger G, Brüne B (2010) Cleavage of sphingosine kinase 2 by caspase-1 provokes its release from apoptotic cells. Blood 115:3531–3540
Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura S (2003) Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem 278:46832–46839
Strub GM, Paillard M, Liang J, Gomez L, Allegood JC, Hait NC, Maceyka M, Price MM, Chen Q, Simpson DC, Kordula T, Milstien S, Lesnefsky EJ, Spiegel S (2011) Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J 25:600–612
Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, Green DR (2012) Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148:988–1000
Takabe K, Kim RH, Allegood JC, Mitra P, Ramachandran S, Nagahashi M, Harikumar KB, Hait NC, Milstien S, Spiegel S (2010) Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J Biol Chem 285:10477–10786
Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH (2010) International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol Rev 62:579–587
Cuvillier O (2012) Sphingosine 1-phosphate receptors: from biology to physiopathology. Med Sci (Paris) 28:951–957
Meyer zu Heringdorf D, Jakobs KH (2007) Lysophospholipid receptors: signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim Biophys Acta 1768:923–940
Spiegel S, Milstien S (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4:397–407
Olivera A, Spiegel S (2001) Sphingosine kinase: a mediator of vital cellular functions. Prostaglandins 64:123–134
Pyne S, Pyne NJ (2000) Sphingosine 1-phosphate signalling in mammalian cells. Biochem J 349:385–402
He X, Huang Y, Li B, Gong CX, Schuchman EH (2010) Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging 31:398–408
Katsel P, Li C, Haroutunian V (2007) Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: a shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem Res 32:845–856
Haughey NJ, Bandaru VV, Bae M, Mattson MP (2010) Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim Biophys Acta 1801:878–886
Gomez-Brouchet A, Pchejetski D, Brizuela L, Garcia V, Altié MF, Maddelein ML, Delisle MB, Cuvillier O (2007) Critical role for sphingosine kinase-1 in regulating survival of neuroblastoma cells exposed to amyloid-beta peptide. Mol Pharmacol 72:341–349
Maceyka M, Payne SG, Milstien S, Spiegel S (2002) Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim Biophys Acta 1585:193–201
Haughey NJ, Steiner J, Nath A, McArthur JC, Sacktor N, Pardo C, Bandaru VV (2008) Converging roles for sphingolipids and cell stress in the progression of neuro-AIDS. Front Biosci 13:5120–5130
Wei Y, Yemisci M, Kim HH, Yung LM, Shin HK, Hwang SK, Guo S, Qin T, Alsharif N, Brinkmann V, Liao JK, Lo EH, Waeber C (2011) Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann Neurol 69:119–129
Grin’kina NM, Karnabi EE, Damania D, Wadgaonkar S, Muslimov IA, Wadgaonkar R (2012) Sphingosine kinase 1 deficiency exacerbates LPS-induced neuroinflammation. PLoS One 7:e36475
Pyne NJ, Pyne S (2010) Sphingosine 1-phosphate and cancer. Nat Rev Cancer 10:489–503
Li J, Guan HY, Gong LY, Song LB, Zhang N, Wu J, Yuan J, Zheng YJ, Huang ZS, Li M (2008) Clinical significance of sphingosine kinase-1 expression in human astrocytomas progression and overall patient survival. Clin Cancer Res 14:6996–7003
Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW (2005) Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J Neuropathol Exp Neurol 64:695–705
Korecka JA, van Kesteren RE, Blaas E, Spitzer SO, Kamstra JH, Smit AB, Swaab DF, Verhaagen J, Bossers K (2013) Phenotypic characterization of retinoic acid differentiated SH-SY5Y cells by transcriptional profiling. PLoS One 8:e63862
Xie HR, Hu LS, Li GY (2010) SH-SY5Y human neuroblastoma cell line: in vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin Med J (Engl) 123:1086–1092
Oyarce AM, Fleming PJ (1991) Multiple forms of human dopamine beta-hydroxylase in SH-SY5Y neuroblastoma cells. Arch Biochem Biophys 290:503–510
Takahashi T, Deng Y, Maruyama W, Dostert P, Kawai M, Naoi M (1994) Uptake of a neurotoxin-candidate, (R)-1,2-dimethyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline into human dopaminergic neuroblastoma SH-SY5Y cells by dopamine transport system. J Neural Transm Gen Sect 98:107–118
Chalimoniuk M, Stolecka A, Ziemińska E, Stepień A, Langfort J, Strosznajder JB (2009) Involvement of multiple protein kinases in cPLA2 phosphorylation, arachidonic acid release, and cell death in in vivo and in vitro models of 1-methyl-4-phenylpyridinium-induced parkinsonism—the possible key role of PKG. J Neurochem 110:307–317
Qin R, Li X, Li G, Tao L, Li Y, Sun J, Kang X, Chen J (2011) Protection by tetrahydroxystilbene glucoside against neurotoxicity induced by MPP+: the involvement of PI3K/Akt pathway activation. Toxicol Lett 202:1–7
Chetsawang B, Kooncumchoo P, Govitrapong P, Ebadi M (2008) 1-Methyl-4-phenyl-pyridinium ion-induced oxidative stress, c-Jun phosphorylation and DNA fragmentation factor-45 cleavage in SK-N-SH cells are averted by selegiline. Neurochem Int 53:283–288
Zhou J, Sun Y, Zhao X, Deng Z, Pu X (2013) 3-O-demethylswertipunicoside inhibits MPP+-induced oxidative stress and apoptosis in PC12 cells. Brain Res 1508:53–62
Maceyka M, Milstien S, Spiegel S (2007) Shooting the messenger: oxidative stress regulates sphingosine-1-phosphate. Circ Res 100:7–9
Pchejetski D, Kunduzova O, Dayon A, Calise D, Seguelas MH, Leducq N, Seif I, Parini A, Cuvillier O (2007) Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ Res 100:41–49
Ader I, Brizuela L, Bouquerel P, Malavaud B, Cuvillier O (2008) Sphingosine kinase 1: a new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res 68:8635–8642
Jin ZQ, Goetzl EJ, Karliner JS (2004) Sphingosine kinase activation mediates ischemic preconditioning in murine heart. Circulation 110:1980–1989
Huwiler A, Kotelevets N, Xin C, Pastukhov O, Pfeilschifter J, Zangemeister-Wittke U (2011) Loss of sphingosine kinase-1 in carcinoma cells increases formation of reactive oxygen species and sensitivity to doxorubicin-induced DNA damage. Br J Pharmacol 162:532–543
Sensken SC, Bode C, Nagarajan M, Peest U, Pabst O, Graler MH (2010) Redistribution of sphingosine 1-phosphate by sphingosine kinase 2 contributes to lymphopenia. J Immunol 184:4133–4142
Nakahara T, Iwase A, Nakamura T, Kondo M, Bayasula KH, Takikawa S, Manabe S, Goto M, Kotani T, Kikkawa F (2012) Sphingosine-1-phosphate inhibits H2O2-induced granulosa cell apoptosis via the PI3K/Akt signaling pathway. Fertil Steril 98:1001–1008
Abrahan CE, Miranda GE, Agnolazza DL, Politi LE, Rotstein NP (2010) Synthesis of sphingosine is essential for oxidative stress-induced apoptosis of photoreceptors. Investig Ophthalmol Vis Sci 51:1171–1180
Conn KJ, Ullman MD, Larned MJ, Eisenhauer PB, Fine RE (2003) cDNA microarray analysis of changes in gene expression associated with MPP+ toxicity in SH-SY5Y cells. Neurochem Res 28:1873–1881
Brill LB, Bennett JP (2003) Dependence on electron transport chain function and intracellular signaling of genomic responses in SH-SY5Y cells to the mitochondrial neurotoxin MPP(+). Exp Neurol 181:25–38
Tiong CX, Lu M, Bian JS (2010) Protective effect of hydrogen sulphide against 6-OHDA-induced cell injury in SH-SY5Y cells involves PKC/PI3K/Akt pathway. Br J Pharmacol 161:467–480
Presgraves SP, Ahmed T, Borwege S, Joyce JN (2004) Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotox Res 5:579–598
Cheung YT, Lau WK, Yu MS, Lai CS, Yeung SC, So KF, Chang RC (2008) Effects of all-trans-retinoic acid on human SH-SY5Y neuroblastoma as in vitro model in neurotoxicity research. Neurotoxicology 30:127–135
Beck KE, De Girolamo LA, Griffin M, Billett EE (2006) The role of tissue transglutaminase in 1-methyl-4-phenylpyridinium (MPP+)-induced toxicity in differentiated human SH-SY5Y neuroblastoma cells. Neurosci Lett 405:46–51
Zhai A, Zhu X, Wang X, Chen R, Wang H (2013) Secalonic acid A protects dopaminergic neurons from 1-methyl-4-phenylpyridinium (MPP+)-induced cell death via the mitochondrial apoptotic pathway. Eur J Pharmacol 713:58–67
Liu L, Xu H, Jiang H, Wang J, Song N, Xie J (2010) Ghrelin prevents 1-methyl-4-phenylpyridinium ion-induced cytotoxicity through antioxidation and NF-kappaB modulation in MES23.5 cells. Exp Neurol 222:25–29
Zhu Q, Wang J, Zhang Y, Sun SJ (2012) Mechanisms of MPP+-induced PC12 cell apoptosis via reactive oxygen species. J Huazhong Univ Sci Technol Med Sci 32:861–866
Zhang ZG, Wu L, Wang JL, Yang JD, Zhang J, Zhang J, Li LH, Xia Y, Yao LB, Qin HZ, Gao GD (2012) Astragaloside IV prevents MPP+-induced SH-SY5Y cell death via the inhibition of Bax-mediated pathways and ROS production. Mol Cell Biochem 364:209–216
Basañez G, Soane L, Hardwick JM (2012) A new view of the lethal apoptotic pore. PLoS Biol 10:e1001399
Imaizumi K, Morihara T, Mori Y, Katayama T, Tsuda M, Furuyama T, Wanaka A, Takeda M, Tohyama M (1999) The cell death-promoting gene DP5, which interacts with the BCL2 family, is induced during neuronal apoptosis following exposure to amyloid beta protein. J Biol Chem 274:7975–7981
Barrera-Vilarmau S, Obregón P, de Alba E (2011) Intrinsic order and disorder in the bcl-2 member harakiri: insights into its proapoptotic activity. PLoS One 6:e21413
Veech GA, Dennis J, Keeney PM, Fall CP, Swerdlow RH, Parker WD Jr, Bennett JP Jr (2000) Disrupted mitochondrial electron transport function increases expression of anti-apoptotic bcl-2 and bcl-X(L) proteins in SH-SY5Y neuroblastoma and in Parkinson disease cybrid cells through oxidative stress. J Neurosci Res 61:693–700
Itano Y, Nomura Y (1995) 1-methyl-4-phenyl-pyridinium ion (MPP+) causes DNA fragmentation and increases the Bcl-2 expression in human neuroblastoma, SH-SY5Y cells, through different mechanisms. Brain Res 704:240–245
Limaye V, Li X, Hahn C, Xia P, Berndt MC, Vadas MA, Gamble JR (2005) Sphingosine kinase-1 enhances endothelial cell survival through a PECAM-1-dependent activation of PI-3K/Akt and regulation of Bcl-2 family members. Blood 105:3169–3177
Bektas M, Jolly PS, Müller C, Eberle J, Spiegel S, Geilen CC (2005) Sphingosine kinase activity counteracts ceramide-mediated cell death in human melanoma cells: role of Bcl-2 expression. Oncogene 24:178–187
Biswas K, Yoshioka K, Asanuma K, Okamoto Y, Takuwa N, Sasaki T, Takuwa Y (2013) Essential role of class II phosphatidylinositol-3-kinase-C2α in sphingosine 1-phosphate receptor-1-mediated signaling and migration in endothelial cells. J Biol Chem 288:2325–2339
Taha TA, Argraves KM, Obeid LM (2004) Sphingosine-1-phosphate receptors: receptor specificity versus functional redundancy. Biochim Biophys Acta 1682:48–55
Takuwa Y, Okamoto Y, Yoshioka K, Takuwa N (2012) Sphingosine-1-phosphate signaling in physiology and diseases. Biofactors 38:329–337
Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11:297–305
Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCubrey JA (2003) Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia 17:590–603
Lee KD, Chow WN, Sato-Bigbee C, Graf MR, Graham RS, Colello RJ, Young HF, Mathern BE (2009) FTY720 reduces inflammation and promotes functional recovery after spinal cord injury. J Neurotrauma 26:2335–2344
Acknowledgments
The studies were supported by NCN Grant 5870/B/PO1/2011/40.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pyszko, J., Strosznajder, J.B. Sphingosine Kinase 1 and Sphingosine-1-Phosphate in Oxidative Stress Evoked by 1-Methyl-4-Phenylpyridinium (MPP+) in Human Dopaminergic Neuronal Cells. Mol Neurobiol 50, 38–48 (2014). https://doi.org/10.1007/s12035-013-8622-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8622-4