
Abstract
There are still no approved treatments for the prevention or of cure of diabetic neuropathy, and only symptomatic pain therapies of variable efficacy are available. Inflammation is a cardinal pathogenic mechanism of diabetic neuropathy. The relationships between inflammation and the development of diabetic neuropathy involve complex molecular networks and processes. Herein, we review the key inflammatory molecules (inflammatory cytokines, adhesion molecules, chemokines) and pathways (nuclear factor kappa B, JUN N-terminal kinase) implicated in the development and progression of diabetic neuropathy. Advances in the understanding of the roles of these key inflammatory molecules and pathways in diabetic neuropathy will facilitate the discovery of the potential of anti-inflammatory approaches for the inhibition of the development of neuropathy. Specifically, many anti-inflammatory drugs significantly inhibit the development of different aspects of diabetic neuropathy in animal models and clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
On September 14, 2011, the International Diabetes Federation announced that 336 million people worldwide had type 2 diabetes and that this disease was responsible for 4.6 million deaths each year. Diabetic neuropathy is the most common complication of type 2 diabetes and affects approximately 50 % of patients over the course of their disease [1]. Diabetic neuropathy has typical characteristics such as pain, numbness, tingling, weakness, and difficulties with balance. Ultrathin sections of the sciatic nerve reveal that the thickness of myelin sheaths is reduced in small, medium-sized, and large axons, and the basement membrane of endoneural microvessels is thickened in db/db mice, a model of peripheral diabetic neuropathy [2]. Diabetic neuropathy can develop even after the initiation of strict blood glucose control due to a phenomenon termed “metabolic memory” [3]. In the absence of more specific drugs, early diagnosis and intensive glycemic control can, however, significantly reduce the incidence of neuropathy in diabetic subjects.
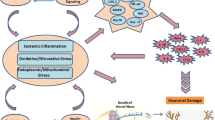
Hyperglycemia, dyslipidemia, and insulin resistance are involved in diabetic neuropathy [4]. Before the development of overt hyperglycemia and diabetes in patients with the metabolic syndrome, the presence of the accumulation of sorbitol, oxidative stress, and 12/15-lipoxygenase activation indicate that additional factors must link diabetes and peripheral neuropathy (Fig. 1) [5]. Biochemical factors resulting from the diabetic state contribute to oxidative stress and nerve damage in diabetic neuropathy. Glucose and lipoproteins interact with various receptors on neurons and microvascular endothelial cells. Diabetes-modified (oxidized and glycated) proteins and lipoproteins bind additional receptors. These receptors include transporters that internalize glucose and lipids, which can accumulate intracellularly and disrupt mitochondrial metabolic pathways. The receptors also initiate inflammatory signaling mechanisms that directly result in oxidative stress and increase the expression and activity of oxidative and nitrosative enzymes. Oxidative stress has been considered to be the final common pathway of cellular injury in hyperglycemia [6], but the mechanisms leading to diabetic neuropathy are more complex and include damage to mitochondria and other cellular components [7].

Summary illustration of the mechanisms that cause diabetic neuropathy. LDL low-density lipoprotein
Non-diabetic neuropathy is a general term for disorders of the peripheral nervous system that are not caused by diabetes. According to the Neuropathy Association, 30 % of neuropathies evolve from diabetes, 30 % from unknown causes, and 40 % from infections, autoimmune disorders, genetic factors, nutrient imbalances, tumors, or toxins. The first line of medications used to treat mild symptoms includes over-the-counter pain medications. In more severe cases, doctors may prescribe opiates, other narcotic medications, anti-seizure medications, lidocaine patches, or anti-depressants to relieve symptoms. However, modern medicine cannot reverse neuropathy once it has developed; thus, it is essential to treat the disease as early as possible symptoms of neuropathy are experienced. Anti-tumor necrosis factor-α (TNF-α) therapy or interferon-α therapy may have potential as treatment options for a spectrum of immune-mediated neuropathies, and useful guidelines exist for dose modifications that take advantage of the effects of chemotherapy and reduce neurotoxicity [8]. Chemotherapy-induced peripheral neuropathy and isolated suprascapular neuropathy can initially be treated with broad-spectrum analgesic medications such as non-steroidal anti-inflammatory drugs [9–11], and non-steroidal anti-inflammatory drugs are also used to treat gouty arthritis of the hand and wrist [12].
Recent reviews have focused on the influences of metabolic factors, the interactions of various mechanisms, progress in clinical trials, and how current knowledge should be applied in future therapeutic interventions [4]. This review expands on a previous paper that reviewed many of the inflammatory mediators involved in diabetic neuropathic conditions and provided an overview of the therapeutics that target inflammation [13]. Here, we review the related features of inflammation, the role of inflammation in the pathogenesis of diabetic neuropathy, and new strategies for the treatment of diabetic neuropathy based on agents that target inflammatory pathways.
Molecules and Pathways of Inflammation in Diabetic Neuropathy
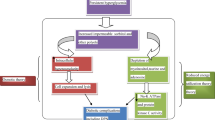
Diabetic neuropathy exhibits features of low-grade chronic, subclinical inflammation [4]. Proinflammatory cytokines, such as TNF-α, interleukin (IL)-1, IL-6, IL-8, monocyte chemoattractant protein-1, and C-reactive protein, are mainly produced by activated immune cells but are also produced by resident macrophages and adipocytes and play a role in amplifying inflammatory reactions. Circulating and locally produced intracellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1, and E-selectin, which reflect low-grade chronic inflammation, have been associated with diabetic complications [14]. Chemokines released from infected/injured tissue activate the endothelium to increase the expression of adhesion molecules and chemokines. Here, we discuss some of the potential mechanisms involved in the inflammatory response in this disease (Fig. 2).

Potential mechanisms involved in the inflammatory response in diabetic neuropathy. JNK JUN N-terminal kinase
Hypoxia
Reductions in blood flow lead to nerve hypoxia and result in diabetic neuropathy. Hypoxia induces the expression of numerous pro-angiogenic and proinflammatory genes in macrophages [15]. Alterations in microvessels are observed in peripheral nerves [16]. In human neuropathy, occlusion of the capillaries that supply the nerves causes ischemic nerve fiber damage and perineural capillary luminal occlusion that is due primarily to both endothelial cell hypertrophy and hyperplasia. Transient increases in the expression of hypoxia-inducible factor-1α in nerves and similar trends in the expression of several hypoxia-inducible factor-1α target genes are also observed [17]. The exposure of normal rats to hypoxic conditions leads to reduced nerve conduction velocity [18]. The block of potassium channels seen in hyperglycemic hypoxia is attributable to intra-axonal acidification by anaerobic glycolysis and may contribute to the pathogenesis of diabetic neuropathy [19]. Neuronal death (apoptosis) is associated with the upregulation of markers associated with DNA damage and aberrant entry into the G1 phase of the cell cycle [20]. Greater impairments of nerve function are observed in the more ischemic legs of diabetic patients, but reversal of hypoxia does not improve nerve function [21].
Receptor for Advanced Glycation End Products
Deletion of receptor for advanced glycation end products (RAGE) in diabetic mice causes greater myelinated fiber densities and conduction velocities subsequent to acute sciatic nerve crush compared to wild-type controls. Reconstitution of diabetic wild-type mice with RAGE-null versus wild-type bone marrow improves axonal regeneration and restoration of function. After crush, diabetic RAGE-null mice display higher numbers of invading macrophages with greater M2 polarization in the nerve segments compared to wild-type animals. In vitro, treatment of wild-type bone marrow-derived macrophages with advanced glycation end products (which accumulate in diabetic nerve tissue) increases M1 and decreases M2 gene expression in RAGE-dependent manners [22]. Blockade of RAGE improves axonal regeneration in superimposed acute peripheral nerve injury that is attributable to tissue-damaging inflammatory responses.
TNF-α
TNF-α is a potent proinflammatory cytokine involved in the pathogenesis of diabetic neuropathy. In contrast to retinopathy and nephropathy, the association between TNF-α and neuropathy appears to be stronger than the associations of IL-6 and C-reactive protein with neuropathy [23]. Diabetic TNF-α−/− mice show no evidence of abnormal nerve function compared to non-diabetic mice. A single injection of infliximab to inhibit TNF-α in diabetic TNF-α+/+ mice suppresses increased serum TNF-α and ameliorates the electrophysiological and biochemical deficits [24]. Animal models of neuropathic pain have persistently indicated that TNF-α has pivotal roles at both peripheral and central levels of sensitization [25].
IL-1
In db/db type 2 diabetes mice, activated astrocytes in the spine increase IL-1β expression, which may induce N-methyl-d-aspartic acid receptor phosphorylation in spinal dorsal horn neurons and enhance pain transmission [26]. TNF-α, IL-1β, IL-6, and monocyte chemoattractant protein-1 levels are increased in the spinal dorsal horns of db/db mice, and these increases are inhibited by anti-high-mobility group box 1 [27]. IL-1β and TNF-α mRNA expression are enhanced in the spinal cords of streptozotocin-diabetic rats [28]. Activation of p38 in spinal microglia increases the synthesis and release of TNF-α, IL-1β, and IL-6 [29]. Glucose-induced IL-1β production involves the NOD-, leucine-rich repeats- and pyrin domain-containing 3 inflammasome [30]. IL-1β induces the production of a wide range of cytokines and chemokines through nuclear factor-kappa B (NF-κB) activation, which is enhanced by free fatty acid-induced activation of Toll-like receptor 2 or Toll-like receptor 4 and leads to the recruitment of macrophages.
IL-6
IL-6, a member of the neuropoietic cytokine family, participates in neural development and has neurotrophic activity. IL-6 is a sensitive marker for diabetic nephropathy that predicts progression and severity of type 1 diabetes [31]. IL-6 improves aspects of small and large nerve fibers in diabetic rats. The functional benefits of IL-6 are related to increased nerve blood flow via a mechanism that involves endothelium-derived hyperpolarizing factor [32]. Increased levels of IL-6, IL-1, TNF-α, and transforming growth factor-β are correlated with the progression of nerve degeneration in diabetic neuropathy [33]. These proinflammatory cytokines affect glial cells and neurons to set the pathological process of diabetic neuropathy in motion. However, TNF-α and IL-6 may not be involved in the development of diabetic peripheral neuropathic pain [34].
Interferon-γ and IL-10
The interferon-γ and IL-10 genes, but not the TNF-α gene, are associated with peripheral neuropathy in South Indian type 2 diabetic patients. The “high-producer” IL-10 genotype and the “low-producer” interferon-γ genotype may be responsible for the downregulation of the immune responses that lead to inflammation in these circumstances [35]. Interferon-γ plays an obligatory role in the development of neuropathy because interferon-γ deficiency is accompanied by complete protection from disease, and infiltration into the nerves of NOD mice deficient for the co-stimulatory molecule B7-2 is also blocked [36]. IL-10 is upregulated in the early phase of Guillain–Barre syndrome and associated with axonal damage, but interferon-γ is not involved in the pathogenesis of Guillain–Barre syndrome [37].
C-Reactive Protein
There is a strong relationship between C-reactive protein and the impairments observed in diabetic neuropathy [38]. High-sensitivity C-reactive protein is a sensitive marker for diabetic nephropathy that predicts the progression and severity of type 1 diabetics [31]. C-reactive protein levels are higher in type 2 diabetic patients with peripheral neuropathy than those without neuropathy [39, 40]. Diabetic subjects with various grades of diabetic foot ulcers exhibit increased high-sensitivity C-reactive protein levels compared to with diabetic patients without foot ulcers [41]. Reduced C-reactive protein levels may serve as a major predictor of the success of percutaneous transluminal angioplasty in diabetic patients with infected foot ulcers [42]. The measurement of C-reactive protein levels may be valuable for distinguishing between infected and non-infected foot ulcers in subgroups of diabetic patients [43].
Adhesion Molecules
The levels of the adhesion molecules P-selectin, E-selectin, and ICAM-1 are higher at baseline in patients with neuropathy compared to non-neuropathic patients [44]. Diabetic peripheral neuropathy patients have higher ICAM-1 levels [45]. Poorly controlled type 2 diabetes patients have higher sE-selectin and soluble vascular cell adhesion molecule-1 levels [46]. Along with P-selectin, the levels of soluble ICAM-1 are higher in patients with neuropathy compared to those with no complications, and these levels correlate with nerve conduction velocities and vibration perception thresholds [47]. Painful neuropathy patients have higher serum level of sE-selectin [33]. ICAM-1 expression is upregulated in diverse zones of the cerebrum and cerebellum of diabetic rats [48].
Chemokines
Chemokines have a crucial role in the infiltration of tissue by immune cells in type 2 diabetes. Regarding the CXC family of chemokines, serum CXCL1 levels are higher in patients with demyelinating forms of diabetic neuropathy [49]. In B7-2-deficient NOD mice that have developed spontaneous autoimmune polyneuropathy, CXCL10 mRNA levels are increased in the sciatic nerve [50]. Members of the CC family of chemokines, which includes CCL2 [50, 51], CCL5 [52], CCL3, and CCL20 [51, 53], are increased in diabetic ZDF rats.
Adipokines
Adipokines are produced mainly or exclusively by adipocytes and have potential immunomodulatory effects. Neuropathic diabetic patients have higher serum levels of leptin and TNF-α [33], and plasma TNF-α affects nerve function [54]. Only sensory conduction velocities are related to TNF-α and leptin concentrations. Diabetic subjects with a diabetic foot exhibit higher plasma IL-6 and resistin levels and lower plasma adiponectin level than diabetics without a diabetic foot. Adiponectin and neuropathy are negatively correlated, and IL-6 and resistin are positively correlated with neuropathy [55]. Serum leptin levels are higher in women with neuropathy than in women without neuropathy, and leptin levels correlate with the degree of neuropathy in subjects with diabetes [56]. Leptin-deficient ob/ob mice develop both large motor and sensory fiber peripheral diabetic neuropathy and small sensory fiber peripheral diabetic neuropathy and respond to pathogenetic treatments [57].
The NF-κB and JNK Pathways
In streptozotocin-diabetic rats with neuropathy, the expressions of NF-κB, IκB-α, and phosphorylated IκB-α are elevated in the sciatic nerve as are the nuclear translocation of the p65/p50 subunit and the levels of TNF-α, IL-6, iNOS, and cyclooxygenase-2 (COX-2) [58, 59]. In liver, adipose, muscle, and hypothalamic tissues, IKKβ–NF-κB activation affects insulin resistance indirectly through changes in body weight, as opposed to the more direct effects on insulin resistance that result from the activation of this pathway in the liver, adipose tissue, and leukocytes. NF-κB is also activated in islet β cells through the actions of glucose and IL-1β, and inhibition of NF-κB protects β cells from various insults. The NF-κB and JUN N-terminal kinase (JNK) pathways are thus activated in multiple tissues in type 2 diabetes and have central roles in promoting tissue inflammation [60]. Accordingly, reducing the activity of these pathways may be therapeutically beneficial.
Reducing Inflammation as a New Therapeutic Strategy
The immense physical, psychological, and economic costs of diabetic neuropathy underscore the need for causally targeted therapies. The Diabetes Control and Complications Trial for type 1 diabetes showed the importance of early, intensive glucose control in the prevention of complications of diabetes, including neuropathy, and these conclusions were underscored by the subsequent Epidemiology of Diabetes and its Complications [61]. The UK Prospective Diabetes Study showed that intensive control of glycemia is associated with increases in severe myocardial events and is not recommended as standard care for type 2 diabetes [62]. Pain is the most severe consequence of neuropathy in terms of patient quality of life, yet pain remains undertreated. In addition, pain therapies remain variable, and only one third of patients report at least a 50 % reduction in pain with therapy [63].
A selection of the most promising strategies and a summary of compounds in development were provided by a recent review [4]. Most clinical trials have produced disappointing results, but these trials have often been confounded by high rates of improvement in the placebo group or other unanticipated effects [64]. Furthermore, the failures of new drugs in the long term are probably the results of the multiple mechanisms that contribute to neuronal injury in diabetes. Inflammatory mechanisms may have a central role in neuropathy. Therefore, we predict that successful treatment or prevention of diabetic neuropathy will require the prevention of inflammation at the systemic and cellular levels. Consistent with the supposition that inflammation has a key role in diabetic neuropathy, sciatic and sural nerve blood flows and conduction velocities are protected by the anti-inflammatory effects of erythropoietin in diabetic rats [65]. Activation of receptor-mediated inflammatory signaling by advanced glycation end products and oxidized lipoproteins [66] leads to oxidative and nitrosative stress, which can cause microvascular disease. The therapeutic drugs, and the targets of those drugs, that are used to treat diabetic neuropathy by reducing inflammation are shown in Table 1.
NF-κB Inhibitors
In rats with diabetic neuropathy, melatonin improves motor nerve conduction velocities and nerve blood flow, reduces the elevated expression of NF-κB, IκB-α, and phosphorylated IκB-α, and reduces the elevated levels of proinflammatory cytokines (TNF-α and IL-6), iNOS, and COX-2 in sciatic nerves [58, 67]. JSH-23 reverses nerve conduction and nerve blood flow deficits and partially corrects reductions in mechanical pain thresholds. JSH-23 inhibits protein expression of nuclear translocation of p65/p50 subunit and lowers the elevated IL-6, TNF-α, COX-2, and iNOS levels/expression in the sciatic nerve [59]. Inhibition of NF-κB by JSH-23 partially reverses the functional, behavioral, and biochemical deficits that accompany diabetic neuropathy.
p38 MAPK Inhibitors
The pain pathways in diabetic neuropathy involve the inflammatory mediator p38 mitogen-activated protein kinase (MAPK). Recent clinical trials of a novel p38 MAPK inhibitor produce rapid relief of neuropathic pain and decreased systemic inflammation [68]. However, extended treatment with a different p38 MAPK inhibitor does not effectively block systemic or chronic inflammation in patients with rheumatoid arthritis [69]. These results suggest that the role of this kinase in cellular inflammation is complex.
COX-2 Enzyme Inhibitors
Upregulation of the activity of the COX-2 pathway has been implicated in the pathogenesis of diabetic neuropathy. Selective inhibition of the proinflammatory enzyme COX-2 prevents cardiac autonomic neuropathy in type 1 diabetic mice [70], which provides further support for the use of anti-inflammatory agents to prevent neuropathy. Diabetic COX-2-deficient mice are protected against the functional and biochemical deficits of experimental diabetic peripheral neuropathy and protected against nerve fiber loss. In diabetic rats, selective COX-2 inhibition replicates this protection [71], which suggests that selective COX-2 inhibition is beneficial for diabetic neuropathy.
Transient Receptor Potential Vanilloid 1 Agonists
Streptozotocin directly induces its effects through the expression and function of the transient receptor potential vanilloid 1 (TRPV1) channel in sensory neurons and results in thermal hyperalgesia even in non-diabetic streptozotocin-treated mice. A proportion of streptozotocin-treated rats are normoglycemic but still exhibit thermal hyperalgesia and mechanical allodynia. Streptozotocin causes microglial activation, increases TRPV1 expression in the spinal dorsal horn, and increases levels of proinflammatory mediators (IL-1α, IL-6, and TNF-α) in spinal cord tissue. Capsaicin-stimulated release of the calcitonin gene-related peptide is elevated in the spinal cords of streptozotocin-treated animals. Intrathecal administration of resiniferatoxin, a potent TRPV1 agonist, attenuates thermal hyperalgesia but not mechanical allodynia and also prevents increases in TRPV1-mediated neuropeptide release in spinal cord tissues [72].
Anti-inflammatory Mesenchymal Stem Cells
Anti-inflammatory mesenchymal stem cells are prepared with optimized for the anti-inflammatory effects of mesenchymal stem cells. Anti-inflammatory mesenchymal stem cell-treated mice show improvement in radiant heat assays and mechanical stimuli tests and exhibit lower serum levels of many proinflammatory cytokines compared to vehicle- and mesenchymal stem cell-treated groups [73]. Thus, anti-inflammatory mesenchymal stem cell-based therapy represents a new anti-inflammatory treatment that should be considered for the management of the painful diabetic peripheral neuropathy that is induced by streptozotocin.
Neuronal Nicotinic Receptor Agonists
Compounds that act at nicotinic acetylcholine receptors have antinociceptive activities. Among these compounds, tebanicline (a potent nicotinic acetylcholine receptor agonist) has demonstrated analgesic effects across a broad range of preclinical models of nociceptive and neuropathic pain. Another nicotinic acetylcholine receptor agonist, A-366833 (at 1, 3, or 6 mg/kg) exerts antinociceptive activity and reduces mechanical hyperalgesia in diabetes-induced neuropathic models. A-366833 dose dependently attenuates mechanical hyperalgesia in the complete Freund’s adjuvant-induced inflammatory pain model [74].
Microglial Activation Inhibitors
Both neuronal cells and non-neuronal cells, particularly microglia, have roles in the development of neuronal hypersensitivity. Minocycline, a selective inhibitor of microglial activation, attenuates the development of diabetic neuropathy (as measured by cold allodynia and thermal and chemical hyperalgesia), reduces IL-1β, TNF-α, lipid peroxidation, and nitrite levels, and improves antioxidant defense in the spinal cords of rats [75]. The beneficial effects of minocycline are partly mediated by its anti-inflammatory effects (which are the result of reductions in the levels of proinflammatory cytokines) and partly mediated by its modulation of oxidative and nitrosative stress in the spinal cord; these latter modulatory effects might be involved in minocycline’s ability to attenuate the development of behavioral hypersensitivity in diabetic rats. Minocycline has no effects on acute peritoneal inflammation or nociception, and the chronic administration of minocycline before peripheral nerve injury attenuates and delays the development of neuropathic pain [76]. Minocyclineinocycline, an inhibitor of microglial activation, inhibits the development of neuropathic pain by preventing the release of proinflammatory cytokines [28].
Cartilage Oligomeric Matrix Protein-Angiopoietin-1
In leptin-deficient ob/ob mice, cartilage oligomeric matrix protein-angiopoietin-1 reduces fasting blood glucose and plasma cholesterol concentrations, upregulates neurofilament 68 and growth-associated protein 43 expression, improves the expression of gap junction proteins (including connexin 32 and 26), suppresses TNF-α and connexin 43 expression, decreases macrophage and T cell infiltration into the sciatic nerve, regenerates small-diameter endoneural microvessels, and increases the phosphorylation of Akt and p38 MAPK upon stimulation of the angiopoietin receptor Tie-2 [77].
Dipeptidyl Peptidase IV Inhibitors
PKF275-055 is a novel, selective, potent, orally bioavailable, and long-acting dipeptidyl peptidase IV inhibitor. PKF275-055 improves body and muscle weight, and glucose metabolism under prevention, protection, and treatment schedules for diabetic neuropathy in streptozotocin-induced diabetic rats. When tested in prevention and protection experiments, PKF275-055 completely prevents decreases in Na/K-ATPase activity and partially counteracts the nerve conduction velocity deficits that are observed in untreated diabetic rats, but has no effects on abnormal mechanical or thermal sensitivity. Therapeutic treatment with PKF275-055 corrects the alterations in Na/K-ATPase activity and nerve conduction velocity that are present in untreated diabetics. PKF275-055 treatment increases mechanical sensitivity thresholds by approximately 50 % and progressively improves alterations in thermal responsiveness. PKF275-055 has an anabolic effect, improves oral glucose tolerance, and counteracts the alterations in Na/K-ATPase activity, nerve conduction velocity, and nociceptive thresholds observed in diabetic rats [78].
Nonsteroidal Anti-inflammatory Agents
A study of 18 male outpatients compares the efficacies of the nonsteroidal anti-inflammatory drugs ibuprofen and sulindac in the treatment of painful diabetic peripheral neuropathy. In this study, discomfort is characterized and rated with a subjective neuropathy score. The responses to both ibuprofen and sulindac are better than the response to placebo across the entire group. There are no changes in glucose control or renal function [79].
Flavonoids
Phytoestrogen has immunomodulatory and anti-inflammatory activities because it reduces the peripheral and central overactivations of NF-κB, the nitric oxide system, and proinflammatory cytokines. In a mouse model of mononeuropathy induced by chronic constriction injury of the sciatic nerve, genistein prevents both neuropathic pain symptoms and the overexpression of IL-1β and IL-6 in the sciatic nerve, dorsal root ganglia, and spinal cord [80]. Genistein prevents increases of TNF-α, IL-1β, and IL-6 in mice with diabetic neuropathy [81]. Naringin is a flavanone with potential antioxidant, antiapoptotic, and disease-modifying properties that are mediated via the modulation of endogenous biomarkers that inhibit diabetes-induced neuropathic pain [82]. Baicalein alleviates nerve conduction deficits and small sensory nerve fiber dysfunction but does not affect diabetic hyperglycemia. Baicalein also counteracts diabetes-associated p38 MAPK phosphorylation, oxidative-nitrosative stress, and 12/15-lipoxygenase overexpression and activation but does not affect intraepidermal nerve fiber loss or the accumulation of glucose and sorbitol pathway intermediates in diabetic mice [83].
Antioxidants
The antioxidant coenzyme Q10 (CoQ10) inhibits body weight loss in diabetic mice but does not affect blood glucose levels. Low dose and long-term administration of CoQ10 prevent the development of neuropathic pain. CoQ10 inhibits mechanical allodynia and thermal hyperalgesia in diabetic mice in a dose-dependent manner. CoQ10 decreases lipid peroxidation in dorsal root ganglia, sciatic nerve, and spinal cord tissues of diabetic mice. CoQ10 reduces proinflammatory factor levels in the peripheral and central nervous system [84]. These results suggest that CoQ10 may be a low-risk and high-reward drug for protection against diabetic neuropathic pain due to its abilities to inhibit oxidative stress and reduce inflammation through the downregulation of proinflammatory factors.
Emblica officinalis (a potent natural antioxidant) aqueous extract dose-dependently attenuates decreases in tail-flick latency and paw withdrawal thresholds and dose-dependently attenuates increases in oxidative stress, nitrite levels and cytokine levels (TNF-α, IL-1β, and TGF-β1) both in the serum and sciatic nerves of diabetic rats. Insulin, in combination with E. officinalis extract, attenuates the diabetic condition and reverses neuropathic pain through modulation of oxidative-nitrosative stress [85]. Metanx, a product containing l-methylfolate, pyridoxal 5′-phosphate, and methylcobalamin (for the management of endothelial dysfunction) increase intraepidermal nerve fiber density and improve multiple parameters of peripheral nerve function in ZDF rats [86].
Conclusions
Inhibition of the inflammatory response is effective in the prevention of diabetic neuropathy. The mechanism by which the inflammatory cascade is initiated and maintained in diabetic neuropathy is currently unknown as are the individual roles of inflammatory molecules in diabetic neuropathy. Common downstream mechanisms of inflammation in diabetic neuropathy include the activation of the NF-κB and JNK pathways and cytokine and chemokine release. These downstream mechanisms lead to the recruitment of immune cells. Inflammatory molecules have redundant functions because many of these molecules cause the activation of similar downstream targets such as NF-κB and several chemokines that share the same receptor. It is important to understand the changes in the inflammatory cascade that occur after blocking an individual inflammatory molecule and to select several candidates for combination therapy. Alternatively, drugs targeting several mechanisms may be more efficient than those that target a single mechanism.
References
Edwards JL, Vincent AM, Cheng HT, Feldman EL (2008) Diabetic neuropathy: mechanisms to management. Pharmacol Ther 120(1):1–34
Nowicki M, Kosacka J, Serke H, Bluher M, Spanel-Borowski K (2012) Altered sciatic nerve fiber morphology and endoneural microvessels in mouse models relevant for obesity, peripheral diabetic polyneuropathy, and the metabolic syndrome. J Neurosci Res 90(1):122–131
Pop-Busui R, Herman WH, Feldman EL, Low PA, Martin CL, Cleary PA, Waberski BH, Lachin JM, Albers JW, Group DER (2010) DCCT and EDIC studies in type 1 diabetes: lessons for diabetic neuropathy regarding metabolic memory and natural history. Curr Diab Rep 10(4):276–282
Vincent AM, Callaghan BC, Smith AL, Feldman EL (2011) Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat Rev Neurol 7(10):573–583
Obrosova IG, Ilnytska O, Lyzogubov VV, Pavlov IA, Mashtalir N, Nadler JL, Drel VR (2007) High-fat diet induced neuropathy of pre-diabetes and obesity: effects of "healthy" diet and aldose reductase inhibition. Diabetes 56(10):2598–2608
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414(6865):813–820
Tecilazich F, Dinh T, Lyons TE, Guest J, Villafuerte RA, Sampanis C, Gnardellis C, Zuo CS, Veves A (2013) Postexercise phosphocreatine recovery, an index of mitochondrial oxidative phosphorylation, is reduced in diabetic patients with lower extremity complications. J Vasc Surg 57(4):997–1005
Toyooka K, Fujimura H (2009) Iatrogenic neuropathies. Curr Opin Neurol 22(5):475–479
Kaley TJ, Deangelis LM (2009) Therapy of chemotherapy-induced peripheral neuropathy. Br J Haematol 145(1):3–14
Freehill MT, Shi LL, Tompson JD, Warner JJ (2012) Suprascapular neuropathy: diagnosis and management. Phys Sportsmed 40(1):72–83
Boykin RE, Friedman DJ, Higgins LD, Warner JJ (2010) Suprascapular neuropathy. J Bone Joint Surg Am 92(13):2348–2364
Tang CY, Fung B (2011) The last defence? Surgical aspects of gouty arthritis of hand and wrist. Hong Kong Med J 17(6):480–486
Douglas EW (2011) Inflammatory mediators in diabetic neuropathy. J Diabetes Metab S5–004
Bluher M, Unger R, Rassoul F, Richter V, Paschke R (2002) Relation between glycaemic control, hyperinsulinaemia and plasma concentrations of soluble adhesion molecules in patients with impaired glucose tolerance or Type II diabetes. Diabetologia 45(2):210–216
Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, Lewis CE (2003) Hypoxia-induced gene expression in human macrophages: implications for ischemic tissues and hypoxia-regulated gene therapy. Am J Pathol 163(4):1233–1243
Zent R, Pozzi A (2007) Angiogenesis in diabetic nephropathy. Semin Nephrol 27(2):161–171
Chavez JC, Almhanna K, Berti-Mattera LN (2005) Transient expression of hypoxia-inducible factor-1 alpha and target genes in peripheral nerves from diabetic rats. Neurosci Lett 374(3):179–182
Doss DJ, Kuruvilla R, Bianchi R, Peterson RG, Eichberg J (1997) Effects of hypoxia and severity of diabetes on Na, K-ATPase activity and arachidonoyl-containing glycerophospholipid molecular species in nerve from streptozotocin diabetic rats. J Peripher Nerv Syst 2(2):155–163
Grafe P, Bostock H, Schneider U (1994) The effects of hyperglycaemic hypoxia on rectification in rat dorsal root axons. J Physiol 480(Pt 2):297–307
Honma H, Gross L, Windebank AJ (2004) Hypoxia-induced apoptosis of dorsal root ganglion neurons is associated with DNA damage recognition and cell cycle disruption in rats. Neurosci Lett 354(2):95–98
Veves A, Donaghue VM, Sarnow MR, Giurini JM, Campbell DR, LoGerfo FW (1996) The impact of reversal of hypoxia by revascularization on the peripheral nerve function of diabetic patients. Diabetologia 39(3):344–348
Juranek JK, Geddis MS, Song F, Zhang J, Garcia J, Rosario R, Yan SF, Brannagan TH, Schmidt AM (2013) RAGE deficiency improves postinjury sciatic nerve regeneration in type 1 diabetic mice. Diabetes 62(3):931–943
Empl M, Renaud S, Erne B, Fuhr P, Straube A, Schaeren-Wiemers N, Steck AJ (2001) TNF-alpha expression in painful and nonpainful neuropathies. Neurology 56(10):1371–1377
Yamakawa I, Kojima H, Terashima T, Katagi M, Oi J, Urabe H, Sanada M, Kawai H, Chan L, Yasuda H, Maegawa H, Kimura H (2011) Inactivation of TNF-alpha ameliorates diabetic neuropathy in mice. Am J Physiol Endocrinol Metab 301(5):E844–852
Leung L, Cahill CM (2010) TNF-alpha and neuropathic pain—a review. J Neuroinflammation 7:27
Liao YH, Zhang GH, Jia D, Wang P, Qian NS, He F, Zeng XT, He Y, Yang YL, Cao DY, Zhang Y, Wang DS, Tao KS, Gao CJ, Dou KF (2011) Spinal astrocytic activation contributes to mechanical allodynia in a mouse model of type 2 diabetes. Brain Res 1368:324–335
Ren PC, Zhang Y, Zhang XD, An LJ, Lv HG, He J, Gao CJ, Sun XD (2012) High-mobility group box 1 contributes to mechanical allodynia and spinal astrocytic activation in a mouse model of type 2 diabetes. Brain Res Bull 88(4):332–337
Talbot S, Chahmi E, Dias JP, Couture R (2010) Key role for spinal dorsal horn microglial kinin B1 receptor in early diabetic pain neuropathy. J Neuroinflammation 7(1):36
Wen YR, Tan PH, Cheng JK, Liu YC, Ji RR (2011) Microglia: a promising target for treating neuropathic and postoperative pain, and morphine tolerance. J Formos Med Assoc 110(8):487–494
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11(2):136–140
Shelbaya S, Amer H, Seddik S, Allah AA, Sabry IM, Mohamed T, El Mosely M (2012) Study of the role of interleukin-6 and highly sensitive C-reactive protein in diabetic nephropathy in type 1 diabetic patients. Eur Rev Med Pharmacol Sci 16(2):176–182
Cotter MA, Gibson TM, Nangle MR, Cameron NE (2010) Effects of interleukin-6 treatment on neurovascular function, nerve perfusion and vascular endothelium in diabetic rats. Diabetes Obes Metab 12(8):689–699
Doupis J, Lyons TE, Wu S, Gnardellis C, Dinh T, Veves A (2009) Microvascular reactivity and inflammatory cytokines in painful and painless peripheral diabetic neuropathy. J Clin Endocrinol Metab 94(6):2157–2163
Yu LN, Yang XS, Hua Z, Xie W (2009) Serum levels of pro-inflammatory cytokines in diabetic patients with peripheral neuropathic pain and the correlation among them. Zhonghua Yi Xue Za Zhi 89(7):469–471
Kolla VK, Madhavi G, Pulla Reddy B, Srikanth Babu BM, Yashovanthi J, Valluri VL, Ramesh J, Akka J (2009) Association of tumor necrosis factor alpha, interferon gamma and interleukin 10 gene polymorphisms with peripheral neuropathy in South Indian patients with type 2 diabetes. Cytokine 47(3):173–177
Bour-Jordan H, Thompson HL, Bluestone JA (2005) Distinct effector mechanisms in the development of autoimmune neuropathy versus diabetes in nonobese diabetic mice. J Immunol 175(9):5649–5655
Press R, Deretzi G, Zou LP, Zhu J, Fredman P, Lycke J, Link H (2001) IL-10 and IFN-gamma in Guillain–Barre syndrome. Network Members of the Swedish Epidemiological Study Group. J Neuroimmunol 112(1–2):129–138
Herder C, Lankisch M, Ziegler D, Rathmann W, Koenig W, Illig T, Doring A, Thorand B, Holle R, Giani G, Martin S, Meisinger C (2009) Subclinical inflammation and diabetic polyneuropathy: MONICA/KORA Survey F3 (Augsburg, Germany). Diabetes Care 32(4):680–682
Papanas N, Katsiki N, Papatheodorou K, Demetriou M, Papazoglou D, Gioka T, Maltezos E (2011) Peripheral neuropathy is associated with increased serum levels of uric acid in type 2 diabetes mellitus. Angiology 62(4):291–295
Azenabor A, Ogbera AO, Adejumo NE, Adejare AO (2011) Acute phase reactant dynamics and incidence of microvascular dysfunctions in type 2 diabetes mellitus. J Res Med Sci 16(10):1298–1305
Zubair M, Malik A, Ahmad J (2012) Plasma adiponectin, IL-6, hsCRP, and TNF-alpha levels in subject with diabetic foot and their correlation with clinical variables in a North Indian tertiary care hospital. Indian J Endocrinol Metab 16(5):769–776
Lin CW, Hsu LA, Chen CC, Yeh JT, Sun JH, Lin CH, Chen ST, Hsu BR, Huang YY (2010) C-reactive protein as an outcome predictor for percutaneous transluminal angioplasty in diabetic patients with peripheral arterial disease and infected foot ulcers. Diabetes Res Clin Pract 90(2):167–172
Jeandrot A, Richard JL, Combescure C, Jourdan N, Finge S, Rodier M, Corbeau P, Sotto A, Lavigne JP (2008) Serum procalcitonin and C-reactive protein concentrations to distinguish mildly infected from non-infected diabetic foot ulcers: a pilot study. Diabetologia 51(2):347–352
Jude EB, Abbott CA, Young MJ, Anderson SG, Douglas JT, Boulton AJ (1998) The potential role of cell adhesion molecules in the pathogenesis of diabetic neuropathy. Diabetologia 41(3):330–336
Zakareia FA (2008) Electrophysiological changes, plasma vascular endothelial growth factor, fatty acid synthase, and adhesion molecules in diabetic neuropathy. Neurosciences 13(4):374–379
Albertini JP, Valensi P, Lormeau B, Aurousseau MH, Ferriere F, Attali JR, Gattegno L (1998) Elevated concentrations of soluble E-selectin and vascular cell adhesion molecule-1 in NIDDM. Effect of intensive insulin treatment. Diabetes Care 21(6):1008–1013
Hussain MJ, Peakman M, Gallati H, Lo SS, Hawa M, Viberti GC, Watkins PJ, Leslie RD, Vergani D (1996) Elevated serum levels of macrophage-derived cytokines precede and accompany the onset of IDDM. Diabetologia 39(1):60–69
Vargas R, Rincon J, Pedreanez A, Viera N, Hernandez-Fonseca JP, Pena C, Mosquera J (2012) Role of angiotensin II in the brain inflammatory events during experimental diabetes in rats. Brain Res 1453:64–76
Michalowska-Wender G, Adamcewicz G, Wender M (2007) Impact of cytokines on the pathomechanism of diabetic and alcoholic neuropathies. Folia Neuropathol 45(2):78–81
Kim HJ, Jung CG, Jensen MA, Dukala D, Soliven B (2008) Targeting of myelin protein zero in a spontaneous autoimmune polyneuropathy. J Immunol 181(12):8753–8760
Galloway C, Chattopadhyay M (2013) Increases in inflammatory mediators in DRG implicate in the pathogenesis of painful neuropathy in type 2 diabetes. Cytokine 63(1):1–5
Bhangoo S, Ren D, Miller RJ, Henry KJ, Lineswala J, Hamdouchi C, Li B, Monahan PE, Chan DM, Ripsch MS, White FA (2007) Delayed functional expression of neuronal chemokine receptors following focal nerve demyelination in the rat: a mechanism for the development of chronic sensitization of peripheral nociceptors. Mol Pain 3:38
Rothman SM, Ma LH, Whiteside GT, Winkelstein BA (2011) Inflammatory cytokine and chemokine expression is differentially modulated acutely in the dorsal root ganglion in response to different nerve root compressions. Spine 36(3):197–202
Matsuda M, Kawasaki F, Inoue H, Kanda Y, Yamada K, Harada Y, Saito M, Eto M, Matsuki M, Kaku K (2004) Possible contribution of adipocytokines on diabetic neuropathy. Diabetes Res Clin Pract 66(Suppl 1):S121–123
Tuttolomondo A, La Placa S, Di Raimondo D, Bellia C, Caruso A, Lo Sasso B, Guercio G, Diana G, Ciaccio M, Licata G, Pinto A (2010) Adiponectin, resistin and IL-6 plasma levels in subjects with diabetic foot and possible correlations with clinical variables and cardiovascular co-morbidity. Cardiovasc Diabetol 9:50
Gottsater A, Ahren B, Sundkvist G (1999) The relationship between leptin and the insulin resistance syndrome is disturbed in type 2 diabetic subjects with parasympathetic neuropathy. Diabetes Care 22(11):1913–1914
Drel VR, Mashtalir N, Ilnytska O, Shin J, Li F, Lyzogubov VV, Obrosova IG (2006) The leptin-deficient (ob/ob) mouse: a new animal model of peripheral neuropathy of type 2 diabetes and obesity. Diabetes 55(12):3335–3343
Negi G, Kumar A, Sharma SS (2011) Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: effects on NF-kappaB and Nrf2 cascades. J Pineal Res 50(2):124–131
Kumar A, Negi G, Sharma SS (2011) JSH-23 targets nuclear factor-kappa B and reverses various deficits in experimental diabetic neuropathy: effect on neuroinflammation and antioxidant defence. Diabetes Obes Metab 13(8):750–758
Donath MY, Shoelson SE (2011) Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11(2):98–107
Martin CL, Albers J, Herman WH, Cleary P, Waberski B, Greene DA, Stevens MJ, Feldman EL, Group DER (2006) Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes Care 29(2):340–344
NA (1998) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 352 (9131):837–853
Jensen TS, Backonja MM, Hernandez Jimenez S, Tesfaye S, Valensi P, Ziegler D (2006) New perspectives on the management of diabetic peripheral neuropathic pain. Diab Vasc Dis Res 3(2):108–119
Tesfaye S, Tandan R, Bastyr EJ 3rd, Kles KA, Skljarevski V, Price KL, Ruboxistaurin Study G (2007) Factors that impact symptomatic diabetic peripheral neuropathy in placebo-administered patients from two 1-year clinical trials. Diabetes Care 30(10):2626–2632
Loesch A, Tang H, Cotter MA, Cameron NE (2010) Sciatic nerve of diabetic rat treated with epoetin delta: effects on C-fibers and blood vessels including pericytes. Angiology 61(7):651–668
Bierhaus A, Nawroth PP (2009) Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 52(11):2251–2263
Negi G, Kumar A, Kaundal RK, Gulati A, Sharma SS (2010) Functional and biochemical evidence indicating beneficial effect of melatonin and nicotinamide alone and in combination in experimental diabetic neuropathy. Neuropharmacology 58(3):585–592
Anand P, Shenoy R, Palmer JE, Baines AJ, Lai RY, Robertson J, Bird N, Ostenfeld T, Chizh BA (2011) Clinical trial of the p38 MAP kinase inhibitor dilmapimod in neuropathic pain following nerve injury. Eur J Pain 15(10):1040–1048
Genovese MC, Cohen SB, Wofsy D, Weinblatt ME, Firestein GS, Brahn E, Strand V, Baker DG, Tong SE (2011) A 24-week, randomized, double-blind, placebo-controlled, parallel group study of the efficacy of oral SCIO-469, a p38 mitogen-activated protein kinase inhibitor, in patients with active rheumatoid arthritis. J Rheumatol 38(5):846–854
Kellogg AP, Converso K, Wiggin T, Stevens M, Pop-Busui R (2009) Effects of cyclooxygenase-2 gene inactivation on cardiac autonomic and left ventricular function in experimental diabetes. Am J Physiol Heart Circ Physiol 296(2):H453–461
Kellogg AP, Wiggin TD, Larkin DD, Hayes JM, Stevens MJ, Pop-Busui R (2007) Protective effects of cyclooxygenase-2 gene inactivation against peripheral nerve dysfunction and intraepidermal nerve fiber loss in experimental diabetes. Diabetes 56(12):2997–3005
Bishnoi M, Bosgraaf CA, Abooj M, Zhong L, Premkumar LS (2011) Streptozotocin-induced early thermal hyperalgesia is independent of glycemic state of rats: role of transient receptor potential vanilloid 1(TRPV1) and inflammatory mediators. Mol Pain 7:52
Waterman RS, Morgenweck J, Nossaman BD, Scandurro AE, Scandurro SA, Betancourt AM (2012) Anti-inflammatory mesenchymal stem cells (MSC2) attenuate symptoms of painful diabetic peripheral neuropathy. Stem Cells Transl Med 1(7):557–565
Nirogi R, Jabaris SL, Jayarajan P, Abraham R, Shanmuganathan D, Rasheed MA, Royapalley PK, Goura V (2011) Antinociceptive activity of alpha4beta2* neuronal nicotinic receptor agonist A-366833 in experimental models of neuropathic and inflammatory pain. Eur J Pharmacol 668(1–2):155–162
Pabreja K, Dua K, Sharma S, Padi SS, Kulkarni SK (2011) Minocycline attenuates the development of diabetic neuropathic pain: possible anti-inflammatory and anti-oxidant mechanisms. Eur J Pharmacol 661(1–3):15–21
Padi SS, Kulkarni SK (2008) Minocycline prevents the development of neuropathic pain, but not acute pain: possible anti-inflammatory and antioxidant mechanisms. Eur J Pharmacol 601(1–3):79–87
Kosacka J, Nowicki M, Kloting N, Kern M, Stumvoll M, Bechmann I, Serke H, Bluher M (2012) COMP-angiopoietin-1 recovers molecular biomarkers of neuropathy and improves vascularisation in sciatic nerve of ob/ob mice. PLoS One 7(3):e32881
Bianchi R, Cervellini I, Porretta-Serapiglia C, Oggioni N, Burkey B, Ghezzi P, Cavaletti G, Lauria G (2012) Beneficial effects of PKF275-055, a novel, selective, orally bioavailable, long-acting dipeptidyl peptidase IV inhibitor in streptozotocin-induced diabetic peripheral neuropathy. J Pharmacol Exp Ther 340(1):64–72
Cohen KL, Harris S (1987) Efficacy and safety of nonsteroidal anti-inflammatory drugs in the therapy of diabetic neuropathy. Arch Intern Med 147(8):1442–1444
Valsecchi AE, Franchi S, Panerai AE, Sacerdote P, Trovato AE, Colleoni M (2008) Genistein, a natural phytoestrogen from soy, relieves neuropathic pain following chronic constriction sciatic nerve injury in mice: anti-inflammatory and antioxidant activity. J Neurochem 107(1):230–240
Valsecchi AE, Franchi S, Panerai AE, Rossi A, Sacerdote P, Colleoni M (2011) The soy isoflavone genistein reverses oxidative and inflammatory state, neuropathic pain, neurotrophic and vasculature deficits in diabetes mouse model. Eur J Pharmacol 650(2–3):694–702
Kandhare AD, Raygude KS, Ghosh P, Ghule AE, Bodhankar SL (2012) Neuroprotective effect of naringin by modulation of endogenous biomarkers in streptozotocin induced painful diabetic neuropathy. Fitoterapia 83(4):650–659
Stavniichuk R, Drel VR, Shevalye H, Maksimchyk Y, Kuchmerovska TM, Nadler JL, Obrosova IG (2011) Baicalein alleviates diabetic peripheral neuropathy through inhibition of oxidative-nitrosative stress and p38 MAPK activation. Exp Neurol 230(1):106–113
Zhang YP, Eber A, Yuan Y, Yang Z, Rodriguez Y, Levitt RC, Takacs P, Candiotti KA (2013) Prophylactic and antinociceptive effects of coenzyme Q10 on diabetic neuropathic pain in a mouse model of type 1 diabetes. Anesthesiology 118(4):945–954
Tiwari V, Kuhad A, Chopra K (2011) Emblica officinalis corrects functional, biochemical and molecular deficits in experimental diabetic neuropathy by targeting the oxido-nitrosative stress mediated inflammatory cascade. Phytother Res 25(10):1527–1536
Shevalye H, Watcho P, Stavniichuk R, Dyukova E, Lupachyk S, Obrosova IG (2012) Metanx alleviates multiple manifestations of peripheral neuropathy and increases intraepidermal nerve fiber density in Zucker diabetic fatty rats. Diabetes 61(8):2126–2133
Acknowledgments
The work was supported by grants from the National Natural Science Foundation of China (no. 81100597), the Natural Science Foundation Project of the Chongqing Science and Technology Commission (CSTC 2009BA5012), and the Natural Science Foundation of the Third Military Medical University (2012XJQ17, 2009XQN34).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhou, J., Zhou, S. Inflammation: Therapeutic Targets for Diabetic Neuropathy. Mol Neurobiol 49, 536–546 (2014). https://doi.org/10.1007/s12035-013-8537-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8537-0