
Abstract
Evidence is accumulating that an imbalance between pathways for degeneration or survival in motor neurons may play a central role in mechanisms that lead to neurodegeneration in amyotrophic lateral sclerosis (ALS). We and other groups have observed that downregulation, or lack of induction, of the PI3K/Akt prosurvival pathway may be responsible for defective response of motor neurons to injury and their consequent cellular demise. Some of the neuroprotective effects mediated by growth factors may involve activation of Akt, but a proof of concept of Akt as a target for therapy is lacking. We demonstrate that specific expression of constitutively activated Akt3 in motor neurons through the use of the promoter of homeobox gene Hb9 prevents neuronal loss induced by SOD1.G93A both in vitro (in mixed neuron/astrocyte cocultures) and in vivo (in a mouse model of ALS). Inhibition of ASK1 and GSK3beta was involved in the neuroprotective effects of activated Akt3, further supporting the hypothesis that induction of Akt3 may be a key step in activation of pathways for survival in the attempt to counteract motor neuronal degeneration in ALS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating disease affecting motor neurons in motor cortex, brain stem, and spinal cord, leading to axon degeneration, muscle atrophy, paralysis, and ultimately death of the patient usually within 2–5 years from diagnosis. For most cases (90 %), the origin of the disease is not known (sporadic ALS), whereas remarkable progress has been made in the field of familial ALS based on genetic discoveries of dominant mutations which cover more than 50 % of the familial cases (reviewed in [1]). Notably, sporadic and familial ALS have similar pathological hallmarks, suggesting that the two forms of ALS may share similar pathogenic mechanisms. One of the most common cause of inherited ALS which account for 20 % of the familial cases has been associated, 20 years ago, with mutations in the gene encoding the Cu/Zn superoxide dismutase (SOD1). This discovery led rapidly to creation of transgenic mice overexpressing the human gene encoding for SOD1 mutated in Gly93-Ala (SOD1G93A). SOD1G93A mice recapitulate several aspects of the disease and, therefore, they provide a powerful model system to identify pathophysiological mechanisms associated with ALS and to screen potential therapeutics [2–4]. Recently, numerous attempts have been made to generate new animal models of ALS based on the discovery of other genes linked to ALS, particularly those encoding TDP-43 and FUS/TLS [5, 6]. However, these new models that overexpress the wild type gene or its mutated form, or even those lacking the gene, show a pathological phenotype that, in most cases, is far from the pathological features of human ALS, making their validity questionable. Though the causes of ALS are still unknown, several mechanisms are implied in ALS pathogenesis, such as excitotoxicity, protein aggregation, neuroinflammation, or defects in the delivery of trophic factors from muscles to the perykarion due to axonal transport impairment or energy failure. These alterations may perturb homeostasis of the motor neuronal cell, thus creating an unbalance between the intracellular signaling pathways of degeneration and survival, eventually causing cell death. Several efforts have been made to counteract the degenerative intracellular signaling in ALS motor neurons, while less attention has been paid to the modulation of survival and antiapoptotic pathways, such as the phosphatidylinositol-3 kinase (PI3K)/Akt.
In the adult nervous system, the PI3K/Akt pathway plays a major role in the transduction of survival signals in neurons and in mediating protection against toxic insults. This is particularly evident in some acute neurological disorders, including epilepsy [7], transient ischemia of the spinal cord [8], and cerebral ischemia, in which Akt induction is a physiological response of surviving neurons to acute injury [9–11]. In contrast, in chronic neurodegenerative diseases, such as Huntington's disease (HD) [12], Alzheimer's disease (AD) [13–15], and ALS [16–20], a lack of or no change in the induction of the prosurvival PI3K/Akt pathway has been reported.
PI3K and Akt are key players in the transduction of signals from membrane receptors to intracellular and nuclear targets. Upon activation, the catalytic subunit of PI3K phosphorylates phosphoinositides (PI) at the 3-position of the inositol ring to generate phosphatidylinositol-3-phosphate (PI3P), PI(3,4)P2, and PI(3,4,5)P3. PIP3 is recognized by a pleckstrin homology (PH) domain in Akt structure, which mediates translocation of Akt from the cytoplasm (where Akt usually resides in an inactive state) to the plasma membrane (where Akt undergoes phosphorylation at Thr308 and Ser473 mediated by phosphoinositide-dependent kinases 1 and 2). These phosphorylations induce a conformational change that determines Akt activation and its dissociation from the plasma membrane to the cytoplasm and nucleus, where it phosphorylates many different substrates leading to activation of the appropriate physiological response [21, 22].
PI3K activities and protein levels were higher in the membrane fractions of spinal cord from ALS patients than controls. However, the downstream targets, such as Akt and S6K, were not upregulated in ALS tissues, suggesting that this signal transduction cascade may be impaired in the disease [16]. In a subsequent study, the same group reported a reduction of PI3K levels and activity, with no change in Akt, in the spinal cord of a pmn mouse model of ALS [17]. In spinal cord homogenates of late onset SOD1.G93A transgenic mice, PI3K and total Akt protein levels were low even before the onset of symptoms [18]. Downregulation of phospho-Akt has also been detected in the spinal motor neurons of presymptomatic SOD1.G93A mice, and this was confirmed by immunohistochemistry in postmortem samples from ALS patients [20].
A gene expression profiling study comparing laser-captured motor neurons from patients with SOD1-related ALS compared with neurologically normal control cases identified multiple genes involved in the PI3K signaling cascade that were expressed differentially in motor neurons that survived the disease. The authors suggested that the RhoA/PI3K/PTEN and Akt3/protein kinase C epsilon pathways could play a role in preventing neuronal cell death in these motor neuron populations [23]. These data are in line with the observation that Akt3, one of the three isoforms of Akt, displays specific neuroprotective properties in NSC34 cells, antagonizing mutant SOD1 toxicity, whereas Akt1 appears to have a more general antiapoptotic effect [24]. The same group provided further evidence of a role of Akt3 in neuroprotection with the identification of a novel Akt3-interacting protein, BTBD10. Overexpression of BTBD10 in NSC34 cells inhibited PP2A activity, preventing Akt3 dephosphorylation and leading to suppression of mutant SOD1-induced cell death [25], downregulation of BTBD10 led to motor neuron death in vitro, and motor neuron loss and locomotor deficits in Caenorabditis elegans [26].
Based on these findings, we proposed to generate a constitutively activated mutant protein construct derived from the fusion of Akt3 with a myristylation recognition sequence [27–29] to be delivered specifically to motor neurons from SOD1.G93A mice. We tested its effect on motor neuron survival, providing proof of concept of Akt3 as a potential therapeutic target for neuroprotection in ALS.
Materials and Methods
Plasmid Constructs
Myr.AKT1 and pAKT3 plasmids were kindly provided by Dr. Brian Hemmings (FMI, Switzerland). Myr.AKT1 is derived from pcDNA3.1H+ (Invitrogen) by cloning the cDNA of human AKT1 (accession number NM_001014432), fused in frame to HA-tag and a myristylation sequence. pAKT3 derives from pCMV5 by cloning the cDNA of human AKT3 gene (accession number NM_181690.1) fused at the N-terminal with HA-tag. The Myr.AKT3 construct was generated by PCR, using pAKT3 plasmid as template, a forward primer containing 39-bp coding for a myristilation sequence (5′-GGGGTACCGCCGCCACCATGGGCTGCGTTTGCTCGTCGAATCCCGAGGACGACGCTGCTTACCCATACGATGTTCCAG–3′) and a reverse primer annealing to AKT3 C-terminus (5′-GGTCTAGATTATTCTCGTCCACTT-GCAGAGT-3′) with a proofreading Pfx DNA polymerase (Invitrogen). The myristylation sequence is in bold; the nucleotides annealing to HA.AKT3 sequence are underlined. The amplicon was then cut by KpnI and XbaI and ligated into plasmid pMyr.AKT1, after KpnI/XbaI excision of the Myr.AKT1 construct. The construct was finally sequence-verified using T7 as sequencing primer and then excised from pcDNA3.1H+ by KpnI/XbaI cut and religated into PmeI/SpeI sites of pWPXLd lentiviral vector, after excision of green fluorescent protein (GFP) (PmeI and SpeI sites destroyed). This vector was called WPX.Myr.AKT3. As a final step, the enhancer elements of Hb9, fused to beta-globin minimal promoter, were excised from the Bg.Hb9_1.6Kb plasmid (described in [30]) by SalI/SbfI digestion and then religated into SalI/SwaI sites of WPX.Myr.AKT3 (SbfI and SwaI sites destroyed), after excision of the EF1-alpha ubiquitous promoter. The new W.1.6.Myr.AKT3 viral vector construct was then used to generate high-titer viral vectors for delivery in vivo.
Transfection of the HEK293 Cell Line
One day before transfection cells were passaged in six-well plates at 50 % confluence. The next day, cells were transfected with purified DNA constructs (Myr.AKT1 or Myr.AKT3) using Effectene transfection reagent (Quiagen) according to the manufacturer's instructions. DNA (0.5 and 1.0 mg) was used for each construct to ensure maximum efficiency of expression. Medium was replaced 16 h after transfection, and then 72-h post-transfection cells were pelleted and lysed with radioimmunoprecipitation assay (RIPA) buffer for 30 min on ice. Lysates were cleared by centrifugation at 15,000g for 15 min at 4 °C, and protein concentration was measured with the bicinchoninic acid assay (BCA) method (Pierce). Thirty micrograms of proteins from each sample was separated by 10 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and blotted on nitrocellulose membrane. Immunoblotting experiments were run according to published protocols [19] with primary antibodies directed to P-Akt (rabbit polyclonal specific for phospho-Ser473 of AKT, 1:500 dilution, Cell Signaling), P-GSK3beta (rabbit polyclonal specific for phospho-Ser9 of GSK3beta, Cell Signaling), P-ASK1 (rabbit polyclonal specific for phospho-Ser83 of ASK1, Cell Signaling), HA-tag (mouse monoclonal, 1:100 dilution, Cell Signaling), or actin (mouse monoclonal, 1:5000 dilution, Millipore).
Viral Vectors
High-titer lentiviral vectors were produced according to published protocols [30]. Since the viral vectors carried motor neuron-specific promoters, the titer of the viral stocks was assayed as described [31], in parallel with vectors of known titer-carrying EF1a ubiquitous promoter cassettes, by infecting neuronal cultures instead of HEK293 cell line. The titer of viral vectors was in the range of 1.0–1.2 × 109 TU/mL.
Infection of SOD1.G93A Neuron/Astrocyte Cocultures and Quantitative Assessment of Motor Neuron Survival
Primary spinal neurons were prepared from 14- to 15-day-old SOD1.G93A or NTg mouse embryos (C57/BL6, Harlan, Italy). Spinal cords were isolated by dissection and the meninges removed. Spinal cords were then mechanically dissociated using a fire-polished glass Pasteur pipette in Hank's Balanced Salt Solution (HBSS, Ca2+, and Mg2+ free) supplemented with glucose (33 mM). The cell suspension was layered onto a 4 % bovine serum albumin (BSA) cushion, centrifuged at 1,000 rpm for 10 min, and the cells from the pellet were resuspended in culture medium composed of Neurobasal (Gibco) supplemented with 10 % inactivated horse serum, 16.5 mM glucose, 1 ng/mL brain-derived neurotrophic factor (BDNF), 2 mM glutamine, 100 mg/mL streptomycin, and 60 mg/mL penicillin. A mixture of hormones and salts composed of insulin (25 mg/mL), transferrin (100 mg/mL), putrescine (60 mM), progesterone (20 nM), and sodium selenate (30 nM) (all from Sigma-Aldrich) was also added to the culture medium. Cells were plated (one spinal cord per eight wells) in 48-well Nunc multiwell plates that had been previously coated with a confluent monolayer of astrocytes, prepared according to a published protocol [32]. Cells were cultured at 37 °C in a humidified atmosphere of 95 % air and 5 % CO2. Cultures were infected 1 day after plating of neurons, with concentrated viral stocks (2 × 106 TU/well), without araC. One day after infection fresh medium was added to the cells and araC was added to a final concentration of 10 mM to inhibit glial cell proliferation. Cells were then kept in culture for five more days, and finally, paraformaldehyde (PFA) was fixed and processed for immunocytochemistry with anti-SMI32 antibody (Covance, 1:1,000 dilution) to highlight motor neuronal profiles, anti-NeuN primary antibody (Chemicon, 1:250 dilution) to identify the whole neuronal population, and Hoechst-33258 dye to highlight nuclei [30]. Motor neurons and total neurons were measured by counting SMI32-positive cells with typical motor neuronal morphology and large cell bodies (diameter ≥ 20 μm) and NeuN-positive cells, respectively, in 20 adjacent frames per well at 10× magnification. Data were then expressed as the ratio of the number of motor neurons to the total neurons in culture. The difference between groups was analyzed by two-way ANOVA followed by Bonferroni's post hoc test, using Prism 4 for Windows, version 4.03 (GraphPad Software Inc.).
129SV SOD1.G93A Mice
In vivo experiments were done in female 129Sv mice overexpressing human SOD1 mutated in G93A. They were generated in the laboratory by more than ten backcrosses of SOD1.G93A C57BL/6 males with nontransgenic female 129SV mice. These mice develop the first signs of neuropathology at the motor neuronal level around 4 weeks of age, and the first symptoms of muscular dysfunction appear around 14 weeks, with progressive reduction in the extension reflex of the hind limbs when the mice are raised by the tail. At about 15 weeks, the mice show a progressive muscular weakness, with increasing difficulty in staying on a rotating bar. At 18 weeks, the animals have complete hindlimb paralysis and cannot recover if laid on one side [33].
Intraspinal Injections
129SV SOD1.G93A female mice at 12 weeks of age were used. Thirty minutes before surgery, animals received an injection of ampicillin (0.03 mg/kg, s.c.) and buprenorphine (0.05 mg/kg, s.c.). All surgical procedures were then done under deep anesthesia with 2.5 % Avertin (2,2,2 tribromoethanol in 2-methyl-2-butanol, 400 mg/kg, i.p.). A laminectomy of L1 vertebra was done to uncover lumbar spinal cord at levels L2–L4, as described [30]. Using a glass capillary (40 ± 5 mm diameter), viral solution was injected bilaterally in the spinal cord in two sites separated 2 mm along the spinal cord (1.5 μL/site; flow rate of 0.2 μL/min). Stereotaxic coordinates were referred to the midline of the dorsal horn of the spinal cord. The needle was positioned ±0.5 mm aside from the midline and deep into the parenchyma to 0.8 mm below the pia; the injector was left in place for 1 min and then retracted for 0.2 mm before starting the delivery. After completion of the injection, the needle was left in place for another 2 min and then gently withdrawn. Dorsal muscles were juxtaposed with absorbable sutures, and skin sutured and disinfected. After surgery, animals were kept on a warm pad for 30 min and then placed in separate cages for recovery. The day after operation, the mice were checked for motor deficits due to accidental damage to the spinal cord. Mice with any signs of motor impairment 1 day after the surgical intervention were not considered further in the analysis.
One week after the operation, the mice were tested for motor performances (grip strength and latency to stay on the rotarod bar), as described [33]. Tests were repeated twice a week until the end stage of the disease. Body weight was recorded before each test session. Mice at the end stage of the disease that were unable to right themselves within 30 s after being placed on one side were euthanized by a high dose of anesthetic and perfused for immunohistochemical analyses, as described [19]. Neuromuscular impairment was analyzed by two-way ANOVA for repeated measures followed by Bonferroni post hoc test for comparison of groups. The survival time of the treated groups was analyzed by the logrank test.
Procedures involving animals and their care were conducted in accordance with the institutional guidelines, which are in compliance with national (D.L. no. 116, G.U. suppl. 40, Feb. 18, 1992, Circolare No.8, G.U., 14 luglio 1994) and international laws and policies (EEC Council Directive 86/609, OJ L 358, 1 DEC.12, 1987; NIH guide for the Care and use of Laboratory Animals, US National Research Council, 1996). The animals were housed under standard conditions (22 ± 1 °C, 60 % relative humidity, 12 h light/dark schedule), three per cage, with free access to food (Altromin, MT, Rieper) and water.
Histology and Immunohistochemistry
Surviving motor neurons was measured by NISSL staining of 24 spinal cord sections 30 μm thick, 300 μm apart, and collected between spinal cord levels L2 and L4. NISSL-stained samples prepared for quantitative analysis were examined under an Olympus BX61 light microscope, and images of the ventral spinal cord were collected with a camera, using analySIS software (Soft Imaging Systems, ver. 3.2). Areas and numbers of cells with clear nucleus and nucleolus in the ventral horn spinal cord were measured using ImageJ (free domain software) on the previously collected calibrated images. Motor neurons were defined as cells with area larger than 250 μm2 similarly to other studies [33, 34]. The mean number of surviving neurons/hemisection for each animal was then calculated. The mean of three to four animals for each experimental group was used in the analysis. The difference between groups was evaluated by one-way ANOVA followed by Tukey Kramer's post hoc test and confirmed by Kruskal–Wallis nonparametric test.
Immunohistochemistry for P-Akt (activated Akt), microtubule-associated protein 2 (MAP2) (neuronal stain), and HA-tag (tag fused to myr.AKT3 construct) was done on 30-μm-thick coronal sections obtained from the lumbar spinal cord of PFA-perfused NTg mice or SOD.1G93A mice killed at different stages of disease progression. Free-floating sections were incubated with 5 % normal goat serum (NGS), 0.05 % Triton in PBS 1 h at room temperature (RT); endogenous biotin was blocked by Avidin–Biotin Blocking solution (Vector). Subsequently, sections were probed with the appropriate primary antibody (anti-P-Akt rabbit monoclonal antibody, 1:500 dilution, cat. no. 3787 from Cell Signaling and anti-MAP2 rabbit polyclonal antibody, 1:4,000 dilution from Millipore); anti-HA-tag mouse monoclonal antibody, 1:100 dilution, from Cell Signaling) in 5 % NGS 0.1 % Triton in PBS O/N at 4 °C and then washed in PBS. For P-Akt or MAP2 staining, the signal was detected by incubation for 1 h at RT in 1 % NSG, PBS with a secondary biotinylated antibody (anti-rabbit, 1:200 dilution, from Vector) followed by tyramide amplification (PerkinElmer) [35]. For HA-tag staining, the signal was detected by incubation for 1 h at RT in 1 % NGS and PBS with an anti-mouse antibody, 1:500 dilution, and conjugated to Alexa.488 fluorophore (Invitrogen). No signal was detected in negative-control sections, incubated without the primary antibody.
Results
Activated Akt3 Protects SOD1G93A Motor Neurons in Primary Astrocyte/Neurons Cocoltures
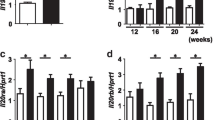
Myr.Akt3 was prepared by fusion of a myristylation sequence to the N-terminal of human HA.Akt3, a fusion protein containing HA-tag at its N-terminal (Fig. 1a). The function of this construct was first tested in vitro, in comparison with Myr.Akt1, a similar fusion protein previously validated [28]. Efficient induction of the Akt pathway was detected with both Myr.Akt1 and Myr.Akt3, as demonstrated by increased levels of Akt phosphorylated at Ser473, and a corresponding increase of GSK3beta phosphorylated at Ser9 (a specific downstream target of Akt) in transfected HEK293 cell line (Fig. 1b). The Myr.Akt3 construct, without the CMV promoter, was then subcloned into a lentiviral vector cassette, under a motor neuron-specific promoter [30], and used to infect murine primary neuron/astrocyte cocultures and for in vivo studies. Western blot experiments on lysates from these cultures 5 days after infection, confirmed that the Myr.Akt3 construct markedly increased P-Akt levels (Fig. 2a). We then verified whether the Myr.Akt3 construct counteracted motor neuronal death in neuron/astrocyte cocultures from SOD1.G93A animals (Fig. 2b). Cells were infected 1 day after plating using lentiviral vectors expressing Myr.AKT3 under Hb9-derived motor neuron-specific promoter (group AKT3 in Fig. 2). As control groups, cells were infected with GFP-expressing lentiviral vectors (GFP) or not treated (NT). There was a significant and selective reduction of motor neuron (MN) viability in the cocultures expressing SOD1.G93A without any treatment. Surviving MNs in the SOD1.G93A group (G93A) were 47.34 ± 7.01 % of those counted in nontransgenic (NTg) cultures. Infection of NTg cultures with lentiviral vectors tended to reduce the number of surviving MNs, compared to untreated NTg cultures. MN counts in GFP or AKT3-infected NTg cultures were 81.48 ± 4.28 and 83.95 ± 6.53 %, respectively, compared to untreated NTg cultures, and this effect was not significant. However, when SOD1.G93A cultures were infected with GFP, a significant 20 % reduction of viable MNs was reported (59.01 ± 4.98 % MNs compared to GFP-infected NTg), whereas in AKT3-infected G93A cultures the MNs were significantly rescued (81.31 ± 2.90 % viable MNs compared to AKT3-infected NTg cultures). Immunocytochemistry of Myr.Akt3 expression indicated that the construct had been expressed by 62.47 ± 5.12 % of the surviving MNs in AKT3-infected G93A cultures. Immunocytochemistry also showed that expression of the Myr.Akt3 construct in murine primary neuron/astrocyte cocultures increased the phosphorylation of two of the downstream targets of Akt, GSK3beta (specifically, phosphorylated at Ser9) (Fig. 2c) and ASK1 (specifically, phosphorylated at Ser83) (Fig. 2d). In fact, Akt-mediated phosphorylation of GSK3beta and ASK1 is involved in neuronal survival.

Generation of a constitutively active AKT3 construct and in vitro tests. a Schematic representation of the main features in the sequence of Myr.AKT1 and Myr.AKT3 constructs. The sequences coding for the myristylation signal and HAtag have been fused, in frame, to the cDNA for AKT1 and AKT3. KpnI and XbaI restriction sites used for subcloning the constitutively activated AKT constructs are indicated on the scheme; b representative immunoblots performed on HEK293 cell lysates after transfection with Myr.AKT1 or Myr.AKT3 (μg of DNA used for transfection are indicated) or with an empty control vector (CV), compared to the nontransfected (NT) condition. There was a strong increase of Akt phosphorylation (at Ser473) with Myr.AKT1 and Myr.AKT3 constructs. This was paralleled by increased phosphorylation of GSK3b at Ser9, one of the downstream targets of activated Akt

Effect of Myr.AKT3 on motor neuron survival in neuron–astrocyte cocultures from SOD1.G93A mice. a Representative immunoblots on lysates from neuron astrocyte cocultures after infection with Myr.AKT3, or with a control GFP-expressing lentiviral vector, compared to the uninfected (NT no treatment) condition. Phosphorylation of Akt (at Ser473) was increased with Myr.AKT3 construct. b Quantitative assessment of motor neuron survival in neuron astrocyte cocultures from untransgenic (NTg) or SOD1.G93A (G93A) mice. Cells were infected with lentiviral vectors expressing Myr.Akt3 (AKT3) or GFP reporter gene (GFP), or left untreated (NT) as control. Each column shows the number of viable motor neurons (as a percentage of NTg-NT samples). Each value is the mean ± S.E.M. of four to eight samples. Motor neuron viability was significantly reduced in untreated (NT) or GFP-expressing (GFP) SOD1.G93A cultures. The effect was completely rescued in Myr.Akt3 (AKT3)-expressing SOD1.G93A cultures. $ = p < 0.05 vs. respective NTg; * = p < 0.01 vs. GFP-G93A; # = p < 0.001 vs. NT-G93A; two-way ANOVA followed by Bonferroni's post hoc test. c Laser scanning confocal microphotographs of phosphorylated (Ser9) GSK3b (red signal) and GFP reporter gene or HAtag fused to Myr.AKT3 (green signal) in mixed neuron astrocyte cocultures transfected with empty vector-expressing GFP or a vector expressing Myr.AKT3 under Hb9-derived motor neuron-specific promoter. The phospho-GSK3b signal is increased in cells expressing Myr.AKT3, reflecting induction of the Akt pathway. Scale bar 50 μm. d Laser scanning confocal microphotographs of phosphorylated (Ser83) ASK1 (red signal) and GFP reporter gene or HAtag fused to Myr.AKT3 (green signal) in mixed neuron astrocyte cocultures transfected with empty vector-expressing GFP or a vector-expressing myr.AKT3 under Hb9-derived motor neuron-specific promoter. The phospho-ASK1 signal is increased in cells expressing Myr.AKT3, reflecting induction of the Akt pathway. Scale bar 20μm
Activated Akt3 Reduces the Spinal Motor Neuron Loss but Does not Ameliorate the Course of the Disease in SOD1G93A Mice
Given the neuroprotective effect of Myr.Akt3 on SOD1.G93A motor neurons in vitro, lentiviral vectors expressing Myr.Akt3 were tested by intraspinal injection in 12-week-old SOD1.G93A mice. At this stage of the disease, the body weight of SOD1.G93A animals starts to differentiate from that of nontransgenic littermates and the hind limbs show reduced abduction, although no deficits are reported in motor tasks tests yet. The mice were divided into groups as follows: (1) intraspinal injection of lentiviral vectors expressing Myr.Akt3 (AKT3, n = 7 mice), (2) intraspinal injection of a vector expressing GFP (EV, n = 6), and (3) surgical laminectomy without injection (CTR, n = 5). The expression of the transgene was first visualized by immunohistochemistry through staining of the HA-tag (at 2 and 5 weeks after injection, corresponding, respectively, to symptom onset and late stages of the disease). Myr.Akt3 was clearly expressed in spinal cord motor neurons of animals injected with Myr.Akt3 lentivectors, as highlighted by colocalization of HA-tag staining with MAP2 neuronal marker; expression was maintained until the late stages of disease progression (Fig. 3b and c). HA-tag was mainly localized at the submembrane and cytoplasmic level, with a weak signal in the nucleus. This is consistent with prevalent membrane targeting of the fusion protein due to the myristylation sequence.

Expression of Myr.AKT3 in ventral horn spinal cord motor neurons in vivo. Laser scanning confocal photomicrographs of Myr.AKT3 expression (HAtag, green signal) and MAP2 neuronal stain (red signal) in lumbar spinal cord neurons of control mice at 14 weeks of age (a–c), 14–15 weeks SOD1.G93A mice (d–f) and SOD1.G93A mice at 17–18 weeks (g–h). Viral vectors expressing the Myr.AKT3 construct under Hb9-derived promoter were injected at 12 weeks. Myr.AKT3 is expressed in large and very large ventral horn neurons in control mice, confirmed by colocalization between HAtag and MAP2 neuronal marker (c). Myr.AKT3 expression is also maintained in SOD1.G93A at the symptomatic (f) and advanced stages of the disease (i). Scale bar in i applies to a–h 50 μm
To analyze the effects of Myr.Akt3 expression on motor neuronal survival and disease progression, we analyzed treated animals twice a week for motor performance, and they were euthanized at the end stage of the disease for histological examination. NISSL staining was used to highlight neuronal profiles in spinal cord sections collected from treated animals. The number of large-sized motor neurons (with an area larger than 250 μm2) was considered in the analysis. The mean number of surviving cells in the Myr.AKT3-treated group (AKT3, 16.53 ± 0.86 cells per hemisection) was significantly higher than the empty vector-treated group (EV, 12.67 ± 1.06 cells per hemisection), and the control group (CTR, 11.81 ± 0.27 cells per hemisection) (Fig. 4a and c–e). The number of motor neurons in NTg littermates was 20.0 ± 0.89 cells per hemisection (data not shown). No difference between groups was reported for the number of surviving small-sized neurons with area range: 200–250 μm2 (Fig. 4b), supporting a specific effect of Myr.AKT3 on motor neuronal cells.

Effect of Myr.AKT3 on motor neuron survival in vivo in SOD1.G93A mice. a–b Median number of surviving large motor neurons (a; area higher than 250 μm2) and small area neurons (b; area range: 200–250 μm2) in the lumbar spinal cord of SOD.1G93A mice at the end stage of disease after treatment with Myr.AKT3 (AKT3; n = 5), compared to animals injected with empty vector (EV; n = 3) or sham-operated mice (CTR; n = 3). * = p < 0.05; Kruskal–Wallis nonparametric test for comparison of medians. c–e Representative images of NISSL staining performed on spinal cord sections from sham-operated (CTR) mice, mice given empty vector (EV) or Myr.AKT3 (AKT3) at the end stage of the disease. Scale bar in e applies to c and d: 100 μm. f–k Laser scanning confocal microphotographs of P-Akt (red) or HAtag (green) immunostaining in SOD1G93A animals treated with Myr.AKT3-expressing lentivirus (i–k), as compared to mice injected with empty GFP-expressing lentivector (f–h). Strong spotlike P-Akt immunoreactivity was detected throughout the grey and white matter in both treated groups, probably reflecting activation of the pathway in glial cells (asterisks in f–h and j, and k). Motor neurons of animals injected with empty vector showed faint P-Akt staining (arrows in f–h); in contrast, immunoreactivity for P-Akt was intense in motor neurons expressing Myr.AKT3 (arrowheads in i–k). Scale bar in k applies to f–j: 50 μm
Motor neurons infected with Myr.Akt3 displayed more intense P-Akt immunoreactivity than in the control group (arrows in Fig. 4f–i), indicating that expression of the transgene had indeed increased Akt activation. However, this signal was only partially colocalized with HA-tag, suggesting that the basal level of Akt phosphorylation may be higher in surviving motor neurons of SOD1.G93A animals (arrowheads in Fig. 4j–k).
Despite the neuroprotective effect of Myr.Akt3 in SOD1.G93A, treatment did not significantly alter the disease progression and survival of SOD1.G93A mice, compared to the control groups (Fig. 5). We could not detect any difference between the three groups for body weight loss or deficits in rotarod performance. Only in the grid test, Myr.Akt3-treated mice performed better than sham-operated and EV-treated control groups during the initial symptomatic phase. With disease progression, Myr.Akt3-treated mice showed a tendency to perform better in the grid test only in comparison with the sham-operated group, while their motor performance seemed worse than the group given only EV. Disease progression in animals that received EV was not significantly different from the control sham-operated group, except for a tendency to a less steep loss of motor performance in the grid test, starting from 111 days of age. Despite these subtle differences in the Myr.Akt3-treated group at the early phases of the pathology, the onset and progression of disease was not significantly delayed by the Myr.Akt3 treatment.

Effect of Myr.AKT3 on motor performance and survival in vivo in SOD1.G93A mice. a Survival curves for AKT3 (Myr.AKT3 treatment) and EV (injected with empty vector and CTR (sham operated) groups. Logrank analysis of probabilities showed no significant difference. b–d Body weight (b) and performance on grid test (c) and on rotarod (d) of Myr.AKT3-treated mice (AKT3; n = 7), compared to control sham-operated mice (CTR; n = 5) and to those injected with empty vector (EV; n = 6). In the grid test, the AKT3 group tended to have a less steep loss of motor performance starting from 111 days of age. On the same test, group EV had slightly better performance than the control group at 107 days of age. Two-way ANOVA for repeated measures showed no significant effect for any group in any test
Discussion
Activation of survival and antiapoptotic pathways, such as the PI3K/Akt, is an essential process triggered by neurons to counteract the induction of degenerative pathways under stressful and toxic stimuli [22]. The disruption of this balance in favor of degenerative signaling cascades may lead to motor neuronal demise, as in ALS. Our and other groups have reported sustained activation of proapoptotic signals mediated, for example, by p38MAPK in the motor neurons of mouse models and patients with ALS [35–37], whereas there was no change or even a decrease in survival factors like Akt [19, 20]. Thus, induction of the PI3K/Akt pathway may be crucial to counteracting motor neuron demise in ALS.
Several experiments on SOD1.G93A transgenic mice support this view. Delivery of insulin-like growth factor 1 (IGF-I) to SOD1.G93A mice through motor neuron-targeted viral vectors [38], or by intrathecal infusion [39], partially reduced motor neuron loss and delayed disease progression, and this was paralleled by increases of phoshorylated (activated) Akt (P-Akt) in the spinal cord. Similarly, a glial cell-derived neurotrophic factor (GDNF)-expressing adenovirus injected into hindlimb muscles of the same animal model produced sustained induction of P-Akt in the motor neurons of the spinal cord [40]; moreover, intracerebroventricular injection of VEGF in SOD1.G93A rats improved motor performance, prolonged survival, and raised P-Akt levels in the spinal cord [20, 41].
IGF-I receptors alpha and beta are highly oxidized in the motor neurons of symptomatic SOD1.G93A mice; this impairs IGF-I-mediated signal transduction, contributing to the impairment of Akt-mediated survival signaling in these cells [42], with potential drawbacks for treatments based on IGF-I delivery. Moreover, downregulation of BTB10D, an inhibitor of PP2A, may be responsible for reduced Akt phosphorylation in ALS patients and mouse models [26]. This points to insufficient phosphorylation of Akt as affecting the signaling downstream of Akt, leading to motor neuron demise.
The present study is the first to address the possibility of inducing Akt activation selectively in spinal cord motor neurons by an approach based on a lentiviral vector carrying motor neuron-specific regulatory sequences derived from the promoter of homeobox gene Hb9, which we recently generated [30]. The main advantage is the use of the activated selective neuronal Akt (Myr-Akt3) which may overcome possible defects in signal transduction to induce Akt phosphorylation. In this way, we provided direct proof of concept that specific and sustained induction of activated Akt in motor neurons can protect these cells from toxic stimuli both in vitro and in vivo. In fact, there was complete rescue of motor neuron loss in SOD1.G93A neuron/astrocyte cocultures and a 41.6 % increase of surviving motor neurons at the end stage of disease in SOD1.G93A animals treated with Hb9-Myr.Akt3 lentiviral vectors. The effect in vivo is in general agreement with reports of viral vector-mediated delivery of other survival factors, such as bcl-2 or IGF-I, to motor neurons of SOD1.G93A mice (65.2 and 34 % increase, respectively) [38, 43]. Akt induction might mirror the actions of bcl-2 as both are expected to exert antiapoptotic effects due to inhibition of Bad and subsequent inhibition of cytochrome c release from the mitochondria.
In cell cultures, Myr.Akt3 mediated phosphorylation of ASK1 (at Ser83) and GSK3 beta (at Ser9). Phosphorylation of ASK1 in Ser83, unlike that on Thr845, has been associated with inhibition of the activity of this kinase [44]. Under physiological conditions, Ser83 of ASK1 is fully phosphorylated and inactive, while upon oxidative stress, Ser83 of ASK1 is dephosphorylated, leading to restoration of ASK1 activity [45].
On the other hand, we reported a remarkable increase of phosphorylated Thr845 (activated) ASK1 upstream of the p38MAPK death pathway in the motor neurons of SOD1G93A mice [46]. The activation of ASK1, induced by ER stress and triggered by the specific interaction of Derlin-1 with mutant SOD1, seems to be vital for disease progression in SOD1 mutant mice [47]. Deletion of ASK1 from these mice reduced the motor neuron loss and extended their life span [47]. ASK1 negatively regulates the proteasome function through inhibition of 19S proteasome ATPase [48]. This supports the hypothesis from our and other groups of proteasomal malfunction as a key pathogenic mechanism responsible for the motor neuron loss in SOD1G93A mice [49–54].
GSK3b, another substrate of Akt, is involved in ALS disease process [55]. Treatment with a GSK3b inhibitor (starting from the presymptomatic stage) significantly delayed symptom onset in SOD1.G93A mice. However, once clinical symptoms developed, this neuroprotective effect was no longer significant due to activation of GSK3b-unrelated degenerative pathways (such as caspase-8 and Fas) [56]. Thus, the inhibition of GSK3b and ASK1 after expression of constitutively active Akt3 in vitro suggests that targeting Akt3 may be a promising approach for more effective and sustained neuroprotective effects. This warrants further investigation in in vivo models of ALS.
Despite significant protection of motor neuron in the lumbar spinal cord, we did not observe any beneficial effect on disease progression with Hb9-Myr.AKT3 treatment. Other preclinical studies on rodent models of ALS have reported similar protection of motor neuronal cell bodies under different conditions, with little or no effect on disease progression and survival [57–59]. This was explained with an insufficient protection of the peripheral axons and distal synapses. In fact, distal axonal degeneration has been reported in the early stages of disease in SOD1G93A mice [60]. It is possible that also, in our mice, there is a lack of protection of the motor axon terminals. In fact, factors others than Akt3 have been reported to be essential for the integrity of the distal axonal connections, for example, the acetylated α-tubulin, which levels were increased after histone deacetylase 6 (Hdac6) deletion in association with the axonal protection and increased survival of SOD1G93A mice [61]. However, this aspect deserves further analysis.
Other hypotheses may also account for the lack of effect on motor dysfunction and survival. For example, there are evidence that muscle fibers are themselves a target of SOD1G93A-mediated pathology [62, 63].
From this point of view, recent studies showed that PI3K was decreased in the gastrocnemius of SOD1G93A mice before the symptoms onset, and these changes were followed by structural and regulatory cytoskeletal alterations leading to a dramatic muscle wasting [64]. On the contrary, PI3K was increased in the triceps, which may reflect an important compensatory response mechanism that helps forelimb muscles to maintain normal function longer than hindlimbs [64]. In addition, reduced levels of Akt have been described in muscles from ALS patient [65]. Therefore, altogether, these data suggest that activating the Akt pathway in muscle too may be vital to counteract the progressive atrophy of this tissue and to ameliorate the disease course.
In line with this hypothesis, the delivery of IGF-1-expressing lentiviral vector to the peripheral muscles was sufficient to increase the survival of SOD1G93A mice, although a higher efficacy was observed with the delivery to both the muscle and the spinal cord. This effect was associated to increased levels of P-AktSer 473 into the spinal cord [38]. In addition, while direct injection of GDNF-expressing viral vectors into the lumbar spinal cord of SOD1.G93A mice did not prevent motor neuron loss and denervation, the delivery of GDNF into the muscles was able to maintain intact axonal projections to the periphery and protect motor neurons [66, 67].
Conclusions
Overall, our findings provide compelling evidence that induction of the Akt pathway, particularly the Akt3, could be a potential strategy to sustain motor neuron survival in the spinal cord. However, it seems that potential therapeutic interventions could need to combine a protective effect on neuronal cell body with preservation of the axonal projections and the integrity of peripheral muscles.
References
Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, de Carvalho M, Ince PG, Lin C, Miller RG, Mitsumoto H, Nicholson G, Ravits J, Shaw PJ, Swash M, Talbot K, Traynor BJ, Van den Berg LH, Veldink JH, Vucic S, Kiernan MC (2013) Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol 12(3):310–322
Bendotti C, Carri MT (2004) Lessons from models of SOD1-linked familial ALS. Trends Mol Med 10(8):393–400
Carri MT, Grignaschi G, Bendotti C (2006) Targets in ALS: designing multidrug therapies. Trends Pharmacol Sci 27(5):267–273
Turner BJ, Talbot K (2008) Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol 85(1):94–134
Tsao W, Jeong YH, Lin S, Ling J, Price DL, Chiang PM, Wong PC (2012) Rodent models of TDP-43: recent advances. Brain Res 1462:26–39
Lanson NA Jr, Pandey UB (2012) FUS-related proteinopathies: lessons from animal models. Brain Res 1462:44–60
Henshall DC, Araki T, Schindler CK, Lan JQ, Tiekoter KL, Taki W, Simon RP (2002) Activation of Bcl-2-associated death protein and counter-response of Akt within cell populations during seizure-induced neuronal death. J Neurosci 22(19):8458–8465
Sakurai M, Hayashi T, Abe K, Itoyuama Y, Tabayashi K (2001) Induction of phosphatidylinositol 3-kinase and serine-threonine kinase-like immunoreactivity in rabbit spinal cord after transient ischemia. Neurosci Lett 302(1):17–20
Ito Y, Sakagami H, Kondo H (1996) Enhanced gene expression for phosphatidylinositol 3-kinase in the hypoglossal motoneurons following axonal crush. Brain Res Mol Brain Res 37(1–2):329–332
Murashov AK, Ul Haq I, Hill C, Park E, Smith M, Wang X, Goldberg DJ, Wolgemuth DJ (2001) Crosstalk between p38, Hsp25 and Akt in spinal motor neurons after sciatic nerve injury. Brain Res Mol Brain Res 93(2):199–208
Namikawa K, Honma M, Abe K, Takeda M, Mansur K, Obata T, Miwa A, Okado H, Kiyama H (2000) Akt/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. J Neurosci 20(8):2875–2886
Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F (2002) The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell 2(6):831–837
Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O'Connor R, O'Neill C (2005) Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem 93(1):105–117
Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease—is this type 3 diabetes? J Alzheimers Dis 7(1):63–80
Lee HK, Kumar P, Fu Q, Rosen KM, Querfurth HW (2009) The insulin/Akt signaling pathway is targeted by intracellular beta-amyloid. Mol Biol Cell 20(5):1533–1544
Wagey R, Pelech SL, Duronio V, Krieger C (1998) Phosphatidylinositol 3-kinase: increased activity and protein level in amyotrophic lateral sclerosis. J Neurochem 71(2):716–722
Wagey R, Lurot S, Perrelet D, Pelech SL, Sagot Y, Krieger C (2001) Phosphatidylinositol 3-kinase activity in murine motoneuron disease: the progressive motor neuropathy mouse. Neuroscience 103(1):257–266
Warita H, Manabe Y, Murakami T, Shiro Y, Nagano I, Abe K (2001) Early decrease of survival signal-related proteins in spinal motor neurons of presymptomatic transgenic mice with a mutant SOD1 gene. Apoptosis 6(5):345–352
Peviani M, Cheroni C, Troglio F, Quarto M, Pelicci G, Bendotti C (2007) Lack of changes in the PI3K/AKT survival pathway in the spinal cord motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci 34(4):592–602
Dewil M, Lambrechts D, Sciot R, Shaw PJ, Ince PG, Robberecht W, Van den Bosch L (2007) Vascular endothelial growth factor counteracts the loss of phospho-Akt preceding motor neurone degeneration in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol 33(5):499–509
Toker A (2000) Protein kinases as mediators of phosphoinositide 3-kinase signaling. Mol Pharmacol 57(4):652–658
Rodgers EE, Theibert AB (2002) Functions of PI 3-kinase in development of the nervous system. Int J Dev Neurosci 20(3–5):187–197
Kirby J, Ning K, Ferraiuolo L, Heath PR, Ismail A, Kuo S-W, Valori CF, Cox L, Sharrack B, Wharton SB, Ince PG, Shaw PJ, Azzouz M (2011) Phosphatase and tensin homologue/protein kinase B pathway linked to motor neuron survival in human superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134(2):506–517
Kanekura K, Hashimoto Y, Kita Y, Sasabe J, Aiso S, Nishimoto I, Matsuoka M (2005) A Rac1/phosphatidylinositol 3-kinase/Akt3 anti-apoptotic pathway, triggered by AlsinLF, the product of the ALS2 gene, antagonizes Cu/Zn-superoxide dismutase (SOD1) mutant-induced motoneuronal cell death. J Biol Chem 280(6):4532–4543
Nawa M, Kanekura K, Hashimoto Y, Aiso S, Matsuoka M (2008) A novel Akt/PKB-interacting protein promotes cell adhesion and inhibits familial amyotrophic lateral sclerosis-linked mutant SOD1-induced neuronal death via inhibition of PP2A-mediated dephosphorylation of Akt/PKB. Cell Signal 20(3):493–505
Nawa M, Kage-Nakadai E, Aiso S, Okamoto K, Mitani S, Matsuoka M (2012) Reduced expression of BTBD10, an Akt activator, leads to motor neuron death. Cell Death Differ 19(8):1398–1407
Brodbeck D, Cron P, Hemmings BA (1999) A human protein kinase Bgamma with regulatory phosphorylation sites in the activation loop and in the C-terminal hydrophobic domain. J Biol Chem 274(14):9133–9136
Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA (1997) Role of translocation in the activation and function of protein kinase B. J Biol Chem 272(50):31515–31524
Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15(23):6541–6551
Peviani M, Kurosaki M, Terao M, Lidonnici D, Gensano F, Battaglia E, Tortarolo M, Piva R, Bendotti C (2012) Lentiviral vectors carrying enhancer elements of Hb9 promoter drive selective transgene expression in mouse spinal cord motor neurons. J Neurosci Methods 205(1):139–147
Belsh JM (1999) Diagnostic challenges in ALS. Neurology 53(8 Suppl 5):S26–S30, discussion S35-26
Tortarolo M, Crossthwaite AJ, Conforti L, Spencer JP, Williams RJ, Bendotti C, Rattray M (2004) Expression of SOD1 G93A or wild-type SOD1 in primary cultures of astrocytes down-regulates the glutamate transporter GLT-1: lack of involvement of oxidative stress. J Neurochem 88(2):481–493
Pizzasegola C, Caron I, Daleno C, Ronchi A, Minoia C, Carri MT, Bendotti C (2009) Treatment with lithium carbonate does not improve disease progression in two different strains of SOD1 mutant mice. Amyotroph Lateral Scler 10(4):221–228
Tortarolo M, Grignaschi G, Calvaresi N, Zennaro E, Spaltro G, Colovic M, Fracasso C, Guiso G, Elger B, Schneider H, Seilheimer B, Caccia S, Bendotti C (2006) Glutamate AMPA receptors change in motor neurons of SOD1G93A transgenic mice and their inhibition by a noncompetitive antagonist ameliorates the progression of amytrophic lateral sclerosis-like disease. J Neurosci Res 83(1):134–146
Tortarolo M, Veglianese P, Calvaresi N, Botturi A, Rossi C, Giorgini A, Migheli A, Bendotti C (2003) Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol Cell Neurosci 23(2):180–192
Bendotti C, Atzori C, Piva R, Tortarolo M, Strong MJ, DeBiasi S, Migheli A (2004) Activated p38MAPK is a novel component of the intracellular inclusions found in human amyotrophic lateral sclerosis and mutant SOD1 transgenic mice. J Neuropathol Exp Neurol 63(2):113–119
Dewil M, dela Cruz VF, Van Den Bosch L, Robberecht W (2007) Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1(G93A)-induced motor neuron death. Neurobiol Dis 26(2):332–341
Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH (2003) Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 301(5634):839–842
Nagano I, Ilieva H, Shiote M, Murakami T, Yokoyama M, Shoji M, Abe K (2005) Therapeutic benefit of intrathecal injection of insulin-like growth factor-1 in a mouse model of Amyotrophic Lateral Sclerosis. J Neurol Sci 235(1–2):61–68
Manabe Y, Nagano I, Gazi MS, Murakami T, Shiote M, Shoji M, Kitagawa H, Setoguchi Y, Abe K (2002) Adenovirus-mediated gene transfer of glial cell line-derived neurotrophic factor prevents motor neuron loss of transgenic model mice for amyotrophic lateral sclerosis. Apoptosis 7(4):329–334
Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, Carmeliet P (2005) Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci 8(1):85–92
Wu DC, Re DB, Nagai M, Ischiropoulos H, Przedborski S (2006) The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci U S A 103(32):12132–12137
Azzouz M, Hottinger A, Paterna JC, Zurn AD, Aebischer P, Bueler H (2000) Increased motoneuron survival and improved neuromuscular function in transgenic ALS mice after intraspinal injection of an adeno-associated virus encoding Bcl-2. Hum Mol Genet 9(5):803–811
Kim AH, Khursigara G, Sun X, Franke TF, Chao MV (2001) Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol 21(3):893–901
Zhang R, Luo D, Miao R, Bai L, Ge Q, Sessa WC, Min W (2005) Hsp90-Akt phosphorylates ASK1 and inhibits ASK1-mediated apoptosis. Oncogene 24(24):3954–3963
Veglianese P, Lo Coco D, Bao Cutrona M, Magnoni R, Pennacchini D, Pozzi B, Gowing G, Julien JP, Tortarolo M, Bendotti C (2006) Activation of the p38MAPK cascade is associated with upregulation of TNF alpha receptors in the spinal motor neurons of mouse models of familial ALS. Mol Cell Neurosci 31(2):218–231
Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H, Noguchi T, Matsuzawa A, Takeda K, Ichijo H (2008) ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev 22(11):1451–1464
Um JW, Im E, Park J, Oh Y, Min B, Lee HJ, Yoon JB, Chung KC (2010) ASK1 negatively regulates the 26 S proteasome. J Biol Chem 285(47):36434–36446
Urushitani M, Kurisu J, Tsukita K, Takahashi R (2002) Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem 83(5):1030–1042
Puttaparthi K, Wojcik C, Rajendran B, DeMartino GN, Elliott JL (2003) Aggregate formation in the spinal cord of mutant SOD1 transgenic mice is reversible and mediated by proteasomes. J Neurochem 87(4):851–860
Kabashi E, Agar JN, Hong Y, Taylor DM, Minotti S, Figlewicz DA, Durham HD (2008) Proteasomes remain intact, but show early focal alteration in their composition in a mouse model of amyotrophic lateral sclerosis. J Neurochem 105(6):2353–2366
Cheroni C, Peviani M, Cascio P, Debiasi S, Monti C, Bendotti C (2005) Accumulation of human SOD1 and ubiquitinated deposits in the spinal cord of SOD1G93A mice during motor neuron disease progression correlates with a decrease of proteasome. Neurobiol Dis 18(3):509–522
Cheroni C, Marino M, Tortarolo M, Veglianese P, De Biasi S, Fontana E, Zuccarello LV, Maynard CJ, Dantuma NP, Bendotti C (2009) Functional alterations of the ubiquitin–proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum Mol Genet 18(1):82–96
Bendotti C, Marino M, Cheroni C, Fontana E, Crippa V, Poletti A, De Biasi S (2012) Dysfunction of constitutive and inducible ubiquitin–proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Prog Neurobiol 97(2):101–126
Palomo V, Perez DI, Gil C, Martinez A (2011) The potential role of glycogen synthase kinase 3 inhibitors as amyotrophic lateral sclerosis pharmacological therapy. Curr Med Chem 18(20):3028–3034
Ahn SW, Kim JE, Park KS, Choi WJ, Hong YH, Kim SM, Kim SH, Lee KW, Sung JJ (2012) The neuroprotective effect of the GSK-3beta inhibitor and influence on the extrinsic apoptosis in the ALS transgenic mice. J Neurol Sci 320(1–2):1–5
Suzuki M, McHugh J, Tork C, Shelley B, Klein SM, Aebischer P, Svendsen CN (2007) GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS One 2(1):e689
Rouaux C, Panteleeva I, Rene F, Gonzalez de Aguilar JL, Echaniz-Laguna A, Dupuis L, Menger Y, Boutillier AL, Loeffler JP (2007) Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci 27(21):5535–5545
Parone PA, Da Cruz S, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, Tseng E, Cleveland DW (2013) Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci 33(11):4657–4671
Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD (2004) Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 185(2):232–240
Taes I, Timmers M, Hersmus N, Bento-Abreu A, Van Den Bosch L, Van Damme P, Auwerx J, Robberecht W (2013) Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum Mol Genet 22(9):1783–1790. doi:10.1093/hmg/ddt028
Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S, Belia S, Wannenes F, Nicoletti C, Del Prete Z, Rosenthal N, Molinaro M, Protasi F, Fano G, Sandri M, Musaro A (2008) Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab 8(5):425–436
Wong M, Martin LJ (2010) Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum Mol Genet 19(11):2284–2302
Capitanio D, Vasso M, Ratti A, Grignaschi G, Volta M, Moriggi M, Daleno C, Bendotti C, Silani V, Gelfi C (2012) Molecular signatures of amyotrophic lateral sclerosis disease progression in hind and forelimb muscles of an SOD1(G93A) mouse model. Antioxid Redox Signal 17(10):1333–1350
Leger B, Vergani L, Soraru G, Hespel P, Derave W, Gobelet C, D'Ascenzio C, Angelini C, Russell AP (2006) Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J 20(3):583–585
Wang LJ, Lu YY, Muramatsu S, Ikeguchi K, Fujimoto K, Okada T, Mizukami H, Matsushita T, Hanazono Y, Kume A, Nagatsu T, Ozawa K, Nakano I (2002) Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J Neurosci 22(16):6920–6928
Guillot S, Azzouz M, Deglon N, Zurn A, Aebischer P (2004) Local GDNF expression mediated by lentiviral vector protects facial nerve motoneurons but not spinal motoneurons in SOD1(G93A) transgenic mice. Neurobiol Dis 16(1):139–149
Acknowledgments
We wish to acknowledge the financial support of Istituto Superiore di Sanità, project no. 526D/6: “Projects on rare diseases funded within the bilateral agreement Italy (Istituto Superiore di Sanità) and USA (NIH, Office for Rare Disesases) on joint research and development of public health actions,” the FP6 STREP EU grant (no. 12702) and Italian ALS association (AISLA).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peviani, M., Tortarolo, M., Battaglia, E. et al. Specific Induction of Akt3 in Spinal Cord Motor Neurons is Neuroprotective in a Mouse Model of Familial Amyotrophic Lateral Sclerosis. Mol Neurobiol 49, 136–148 (2014). https://doi.org/10.1007/s12035-013-8507-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8507-6