
Abstract
Spinal muscular atrophy (SMA) is a devastating and often fatal neurodegenerative disease that affects spinal motor neurons and leads to progressive muscle wasting and paralysis. The survival of motor neuron (SMN) gene is mutated or deleted in most forms of SMA, which results in a critical reduction in SMN protein. Motor neurons appear particularly vulnerable to reduced SMN protein levels. Therefore, understanding the functional role of SMN in protecting motor neurons from degeneration is an essential prerequisite for the design of effective therapies for SMA. To this end, there is increasing evidence indicating a key regulatory antiapoptotic role for the SMN protein that is important in motor neuron survival. The aim of this review is to highlight key findings that support an antiapoptotic role for SMN in modulating cell survival and raise possibilities for new therapeutic approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive infantile neurodegenerative disease that has an incidence of one in 6,000 live births. SMA is characterized by the progressive degeneration of motor neurons of the anterior horn of the spinal cord. Clinically typified by profound muscle weakness, hypotonia, and trunk paralysis, SMA is classified into three main subtypes (I–III) based on age of onset and disease severity [1]. Type I SMA accounts for ≈50 % of SMA cases, manifests within 6 months of birth, and is usually fatal before the age of four. Patients with type II SMA develop muscle weakness before 18 months of age, often develop severe orthopedic and pulmonary complications [2], and may survive into adolescence [3]. Patients with type III SMA, typically display a later onset and suffer from a milder but still debilitating phenotype.
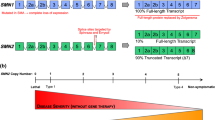
Approximately 95 % of SMA patients have a deletion or mutation in the survival of motor neuron 1 (SMN1) gene [4] that encodes the survival of motor neuron (SMN) protein. A second highly homologous copy of this gene, SMN2, differs by only a single translationally silent base change within exon 7. However, this base change causes aberrant splicing (exon 7 exclusion) in ≈90 % of SMN2 transcripts and when translated, results in truncation, instability, and reduced activity of the SMN protein (Fig. 1). Full-length SMN protein is ubiquitously expressed and occurs in the cytoplasm and nucleus of most cells. Among other functions, SMN plays an important role in the biogenesis of spliceosomal small nuclear ribonuclear proteins (snRNPs) and pre-mRNA splicing. SMN also modulates apoptosis by directly blocking caspase activation and by affecting other key regulators of cell survival such as Bcl-2, p53, ZPR1, and Bcl-xL [5–9]. However, a clear understanding of SMA pathogenesis and the full functions of the SMN protein is still to be determined. In this review, we summarize the evidence that antiapoptotic and prosurvival pathways are affected by reduced SMN protein levels and explain the importance of targeting apoptosis as a potential therapeutic approach for SMA.

Schematic diagram showing the SMN1 and SMN2 genes. Due to a 500-kb inverted duplication on chromosome 5, there exists two almost identical survival of motor neuron genes SMN1 and SMN2. In unaffected individuals, the SMN1 gene is normally spliced and produces a functional SMN protein. In contrast, the SMN1 gene is mutated or deleted in 95 % of SMA patients. A single base change (C → T) affects the splicing of the SMN2 gene causing the majority of SMN2 transcripts to lack exon 7. This translates to a mostly truncated SMN protein (SMN7) missing the amino acids encoded by exon 7. Therefore, SMA patients have much lower levels of full-length functional SMN protein and, consequently, show all the hallmark pathologies of SMA disease. [10]
Genetics of Spinal Muscular Atrophy
In 1995, Lefebvre et al. [10] discovered that mutations in the SMN1 gene caused SMA. The functional SMN1 gene encodes the SMN protein that is necessary for motor neuron development and survival. During human evolution, a second, almost identical, copy of the SMN gene, termed SMN2, arose following a 500-kb inverted duplication of the region containing SMN1 on chromosome 5 [11]. Both the SMN1 and SMN2 genes contain identical promoter sequences and display enhanced neuronal expression [12]. Although the two genes differ by only five nucleotides, a critical translationally silent single C → T nucleotide base change in exon 7 of the SMN2 gene causes exclusion of this exon in 90 % of SMN2 RNA transcripts [13] (Fig. 1). Exclusion of exon 7 occurs due to disruption of a splice modulator site/sequence described as acting as an exon splicing enhancer (ESE) or an exon splicing silencer (ESS) [14, 15]. The C terminus of the SMN protein, which contains the region encoded by exon 7, is essential for survival, and when deleted, the mutant nonfunctional SMN protein (SMNΔ7) fails to reduce cell death following injury [16]. The importance of exon 7 is further exemplified in conditional exon 7 knockout mice that demonstrate similar phenotypes to different SMA mouse models [17].
The Survival of Motor Neuron Protein and Spinal Muscular Atrophy Pathogenesis
The Survival of Motor Neuron Protein
The SMN1 gene encodes a 38-kDa full-length and functional protein. In contrast, while encoding the same protein, alternative splicing of the SMN2 transcript results in the expression of relatively small amounts of full-length SMN protein (10–20 %; Fig. 1). The vast majority of protein encoded by the SMN2 gene is missing the 16 carboxyl end amino acids encoded by exon 7, causing the protein to be less active and unstable [18, 19]. Ubiquitously expressed and developmentally regulated [20], full-length SMN protein is found in both the cytoplasm and nucleus. In the nucleus, SMN protein localizes to structures called Gemini of coiled bodies (gems) that coincide with Cajal bodies in most cell lines but are distinct from Cajal bodies in fetal tissues [21–23]. Cajal bodies and gems colocalize to some degree, and this relationship may be partly mediated by the interaction of SMN with the Cajal body protein coilin [24, 25]. The SMN protein is also present in axons and dendrites [26, 27] and independently localizes to the neuromuscular junction (NMJ) [28].
Spinal Muscular Atrophy Pathogenesis
While not all cell types are equally affected by reduced SMN levels, SMA is characterized by the degeneration of alpha motor neurons of the anterior horn of the spinal cord [29]. The increased vulnerability displayed by motor neurons may be due to a motor neuron-specific SMN2 splicing inefficiency, whereby reduced SMN levels further exacerbates SMN exon 7 exclusion in motor neurons [30]. It is clear that reduced levels of oligomerization-competent intracellular SMN initiate the pathogenic cascade, but the sequence of events in SMA pathogenesis is debatable [31]. The most-studied function of the SMN protein is in forming a complex containing Gemin proteins (Gemins 2–8) and small nuclear ribonucleoproteins (snRNP) assembly [21, 32–38]. The SMN complex specifically binds to and assembles Sm proteins (ribonucleoproteins) onto small nuclear RNAs (snRNAs), generating an active snRNP complex [25, 39–41]. As a component of the spliceosome [42], snRNPs play a role in pre-mRNA splicing that is essential for the expression of mature mRNA [43]. An important question is to what extent is SMN's role in RNA processing fundamental to SMA disease? Theoretically, the SMN protein impacts many mRNAs processed, suggesting defective splicing is playing an important role in the pathophysiology of SMA. Burghes and Beattie [44] suggest that disruption of snRNPs alters the splicing of genes critical to motor neuron function. Recently, Lotti et al. [45] identified an SMN-regulated U12 intron-containing gene, named Stasimon, responsible for normal synaptic transmission and motor neuron function. Restoration of Stasimon in a zebrafish SMA-like model rescued SMN-dependent motor neuron defects [45], suggesting dysregulation of Stasimon plays a key role in the motor neuron-specific pathology of SMA.
Another suggestion is that NMJ formation and axonal function is affected in SMA, leading to the degeneration and eventual cell death of the motor neurons [31]. This is supported by the axonal location and axonal transportation of the SMN protein [27, 46]. To this end, knockdown of SMN expression causes defects in motor neuron axonogenesis, branching, and NMJ formation independent of snRNP biogenesis [47–49]. In addition, anterograde transport of the truncated SMNΔ7 protein does not occur and overexpression of SMNΔ7 results in axonal defects [46].
While SMN appears important to motor neuron function, pathogenic models of SMA suggest a general requirement for the SMN protein in many other tissues. This review will delineate the antiapoptotic functions of the SMN protein and the contribution made by apoptosis to the pathogenesis of SMA. In addition, we will discuss the current and future therapeutic approaches aimed at achieving neuroprotection in SMA.
Apoptosis in Spinal Muscular Atrophy
Apoptosis, or programmed cell death, is a specific and deliberate cellular mechanism that results in nuclear fragmentation, chromatin condensation, and the formation of apoptotic bodies [50, 51]. This phenomenon is required during embryonic development and for the homeostatic maintenance of proliferative tissues [52, 53]. Apoptosis is essential for normal development in the Central Nervous System (CNS) and functions to rapidly remove redundant neurons and nerve cells that fail to establish the appropriate synaptic connections [54]. Apoptosis during development and later in adult life is strictly regulated by a balance of antiapoptotic and proapoptotic signals. Dysregulation of apoptosis due to a disturbance of this balance can be associated with the development of cancer, autoimmune diseases, ischemia-related injuries, and neurodegenerative disorders [51].
There is increasing evidence that neurodegeneration in motor neuron disorders such as SMA and amyotrophic lateral sclerosis (ALS) may also involve apoptosis [55–57]. In genetically confirmed cases of SMA, examination of fetal tissue has revealed that during embryonic development, immature motor neurons undergo a more prolonged period of apoptotic programmed cell death, compared with motor neurons from control human fetal tissue [58, 59]. Furthermore, it is considered that the apoptotic loss of motor neurons in SMA patients after birth has been significantly underestimated due to the rapid elimination of these cells from spinal cord tissue [31]. Elevated levels of proapoptotic genes and apoptosis have also been observed in the CNS of SMA mouse models [60, 61]. Taken together, these human and animal studies provide strong evidence that the excessive loss of motor neurons by apoptosis plays an important role in the early stages of SMA.
The important function of SMN in regulating cell survival and apoptosis is supported in experimental studies showing that SMN levels are proportional to cell death rates following apoptotic stimuli [62]. For example, the SMN protein can protect various cell types against a range of apoptotic stimuli, including growth factor deprivation, PI 3-kinase inhibition, and camptothecin- and staurosporine-induced apoptosis [62–65]. In addition, SMN knockdown in neuronal-like NSC-34 cells results in reduced cell survival and elevated levels of apoptosis [62]. Therefore, irrespective of its other functions, it is clear that SMN can reduce apoptosis in normal cells and in cells induced to undergo apoptosis.
Proposed Antiapoptotic Mechanisms Used by Survival of Motor Neuron
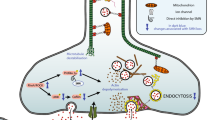
Studies aimed at identifying SMN's specific antiapoptotic mechanisms are limited. Kerr et al. [66] first postulated that SMN may play a crucial role in modulating neuronal-specific apoptotic mechanisms. To date, the most well-defined antiapoptotic mechanism of action of SMN is through inhibition of active caspase-3 subunit formation. Caspase-3 is a key mediator of apoptotic cell death and is usually activated by the apoptosome complex consisting of cytochrome c, caspase-9, and Apaf-1. The activation of procaspase-3 (32 kDa) involves a two-stage process: calpain-mediated removal of the 3-kDa prodomain resulting in a 29-kDa subunit (p29), followed by spontaneous proteolytic cleavage of p29 into large and small subunits (17/12 kDa) by caspase-9 [67–69]. Caspase-3 is also activated by caspase-8 through the extrinsic pathway. This pathway involves the activation of death receptors such as Fas and tumor necrosis factor (TNF), the recruitment of death receptor-associated molecules such as Fas-associated death domain containing protein (FADD), and the subsequent activation of the caspase-8 (reviewed in [51]). The ability of SMN to block calpain-mediated activation of procaspase-3 [65] and thereby reduce caspase-3 activation [62–65, 70] provides strong support for the view that SMN functions in an antiapoptotic caspase-3-dependent manner (Fig. 2). In support of this, human SMA motor neurons derived from induced pluripotent stem cells (iPSCs) exhibit increased levels of caspase subunits and caspase-3 cleavage [71].

Proposed cell signaling pathways linking SMN expression to the regulation of cell survival. Neurotrophic factors commonly signal through the PI 3-kinase pathway, phosphorylating Akt and preventing BAD sequestration of Bcl-2 and Bcl-xL. SMN interacts with Bcl-2 and regulates the expression of Bcl-xL, both of which prevent cytochrome c release from the mitochondria. The SMN protein also interacts with the tumor suppressor protein, p53, and when bound, colocalizes across the nuclear membrane. Under normal conditions, the p53 protein tightly regulates Bax activation, and the interaction between Bcl-2 and SMN synergistically prevents Bax-mediated apoptosis. In SMA patient cells, levels of the p53 modulator, MDM2, are reduced and unable to prevent p53 activation of Bax. Prolactin and erythropoietin signal via the Jak/Stat pathway. Prolactin increases SMN levels by stimulating Stat5, a transcription factor known to increase Bcl-xL levels. Activation of this pathway by prolactin is also known to activate Fyn, a tyrosine kinase involved in the activation of the prosurvival PI 3-kinase pathway and the phosphorylation of the RNA binding protein, Sam68. Phosphorylated Sam68 splices Bcl-x and SMN transcripts, promoting the long Bcl-xL isoform and inclusion of exon 7 in SMN transcripts. End-stage apoptosis begins with the activation of caspase-9, via cytochrome c-mediated activation of Apaf-1 or by activation of cell surface death receptors. Activation of caspase-3 requires cleavage of the 3-kDa procaspase-3 prodomain, a step blocked by SMN. Expression of ZPR1 and NAIP are correlated with SMA severity and SMN protein levels, and both proteins can directly regulate caspase-3 activation. The overall inhibition of caspase-3, directly or indirectly by SMN, prevents cleavage of death substrates and apoptosis. [5, 6, 9, 65, 81, 92, 93, 101, 131, 159]
What makes the involvement of caspase-3 and calpain potentially interesting is their ability to directly cleave full-length SMN protein following injury [66, 72, 73]. Caspases are predicted to cleave SMN at amino acid Asp-252, generating a ≈29-kDa truncated protein that is present in cells undergoing apoptosis in brain tissue [66]. Not surprisingly, mutating the SMN caspase cleavage site results in an SMN protein with enhanced antiapoptotic properties [66]. However, it is unclear whether caspase cleavage of SMN abolishes its intrinsic antiapoptotic properties or if the resulting SMN cleavage products have their own unique proapoptotic functions.
Cell Death and Prosurvival Proteins Affected in Spinal Muscular Atrophy
The Role of the Bcl-2 Protein
As a consequence of its multifunctional roles, the SMN protein impacts a variety of pathways and genes implicated in SMA pathogenesis. Of particular interest are the relationships and interactions between SMN and key proteins that are directly or indirectly involved in regulating cell survival (Table 1). For example, SMN is known to interact with Bcl-2, a well-characterized member of the Bcl-2 family that negatively modulates apoptosis by several mechanisms [74, 75]. The interaction of SMN with Bcl-2 provides a synergistic antiapoptotic action against Bax- and Fas-mediated apoptosis [5, 76]. Moreover, the truncated SMNΔ7 protein is unable to produce a similar antiapoptotic effect, further supporting the importance of exon 7 to SMN protein functionality [5]. Finally, in SMA model mice, the transcription factor WT-1, a key regulator of Bcl-2 expression and apoptosis, is downregulated [77]. The WT-1 protein modulates apoptosis by upregulating Bcl-2 expression and by directly interacting with p53 to inhibit apoptosis [78, 79].
The altered expression of Bcl-2 in SMA is likely to have a significant impact on neuronal development. During murine embryogenesis, Bcl-2 expression peaks at embryonic day 11 and plays an essential role in regulating developmental apoptosis [53]. Interestingly, at 15 weeks of gestation, in the spinal cord tissue of SMA patients, expression of Bcl-2 is downregulated, a factor which is likely to lead to increased total motor neuron cell death [8, 58].
An Interaction with ZPR1
Another protein involved in negatively regulating caspase activation and known to interact and colocalize with the SMN protein is ZPR1 [7]. Like Bcl-2, expression of ZPR1 is reduced in SMA patients [80] and is also thought to play a role in SMA pathogenesis [81]. Similar to SMN, complete knockout of ZPR1 is embryonically lethal, supporting an essential role for ZPR1 in cell survival [81]. Indeed, ZPR1 deficiency causes Cajal body defects, axonal abnormalities, defective embryonic growth, mislocalization of SMN, and increased apoptosis [7, 81, 82]. Interestingly, ZPR1 knockdown can induce caspase-3 activation and play a significant role in motor neuron degeneration in mice [82]. Taken together, these findings suggest that ZPR1 may play an important role in determining the extent of neuronal apoptosis in SMA and thereby contribute to disease pathogenesis.
The Role of p53
The SMN protein also interacts with the proapoptotic p53 protein [6]. The p53 protein is a sequence-specific transcription factor that targets both mitochondrial and death receptor-induced apoptotic pathways, such as Bax, NOXA, and PUMA, resulting in cytochrome c release and Apaf-1/caspase-9 activation [83]. Although p53 is not subject to SMN-dependent regulation [80], it is thought that high levels of SMN are responsible for p53 localization to nuclear bodies [6]. Young et al. [6] determined that the SMN/p53 interaction prevented p53-mediated apoptosis. Not surprisingly, the truncated SMNΔ7 protein was unable to interact with p53 and prevent activation of p53 apoptotic pathways. The primary regulator of p53 is the MDM2 protein, which inhibits p53 transcriptional activity and targets p53 for proteasome degradation. MDM2 is a transcriptional target of p53 and, via an autoregulatory feedback loop, its expression is increased as p53 activity increases [84]. Interestingly, both MDM2 and its regulator, PSME3, are downregulated in SMA model mice [77], possibly secondary to reduced SMN and p53 levels. The importance of MDM2 as a regulator of p53 has been demonstrated in MDM2 knockout mice whereby p53-driven apoptosis causes embryonic lethality [85].
What Role Does the Bcl-xL Protein Play?
The Bcl-x gene encodes several alternatively spliced mRNA's, of which Bcl-xL is the predominant transcript [86, 87]. Unsequestered Bcl-xL targets the mitochondria, inhibiting cytochrome c release and Apaf-1-dependent caspase-9 activation, subsequently blocking apoptosis [88, 89]. Bcl-xL expression is high during embryogenesis [53] and is thought to play an essential role in CNS development, as Bcl-xL-deficient mice exhibit extensive apoptotic cortical and spinal neuronal loss [90]. In SMA, downregulation and irregular expression of Bcl-xL have been documented in human SMA fetal tissue [8] and SMA model mice [91]. However, since the identification of Bcl-2 as an interacting partner of SMN [5], Bcl-xL has been largely overlooked as playing a role in SMA pathogenesis. We recently reported that expression of Bcl-xL and SMN is coregulated, suggesting a common regulatory mechanism [9]. In addition, we reported that a regulator of both Bcl-x and SMN mRNA alternative splicing, the RNA-binding protein Sam-68, is reduced in SH-SY5Y cells when Bcl-xL or SMN is overexpressed [9]. Reduced levels of Sam-68 cause accumulation of both antiapoptotic Bcl-xL and full-length SMN2 transcripts [92, 93]. In SMA, Sam 68 regulation may be altered, resulting in abnormally high protein levels and potentially leading to the downregulation of Bcl-xL and increased exon 7 skipping in SMN2 transcripts.
The Role of the Neuronal Apoptosis Inhibitory Protein Gene
Lying immediately adjacent to the SMN gene, the neuronal apoptosis inhibitory protein (NAIP) gene is located within the 500-kb inverted duplication on chromosome 5. NAIP is the founding member of the human IAP protein family [94] that together inhibit many key caspases and procaspases. Interestingly, numerous studies have correlated NAIP deletions with SMA disease severity [95–100]. These studies show that up to 90 % of type I SMA patients have deletions in the NAIP gene. In contrast, deletions in NAIP are less frequent in types II and III SMA. Therefore, it is likely that NAIP gene expression is directly implicated as a modulator in SMA and absence of this gene exacerbates the SMA phenotype.
The NAIP protein directly inhibits caspase-3 and caspase-7 through the action of its baculovirus inhibitor of apoptosis protein repeat (BIR) [101]. Furthermore, NAIP suppresses apoptosis in neural tissues [102–104] and the loss of endogenous NAIP results in increased neuronal vulnerability [105]. In vivo, hippocampal pyramidal neurons from transgenic mice lacking the NAIP1 gene display an increased sensitivity to kainic acid-induced apoptosis [105]. However, these mice are morphologically normal and do not show any features or characteristics of an SMA phenotype. Attempts to determine the exact effects of NAIP deletions on SMA phenotype have, so far, not been successful, mainly due to the presence of six different NAIP genes [106], making in vivo modeling difficult.
Current Therapeutic Strategies Toward Treating Spinal Muscular Atrophy
Increasing Survival of Motor Neuron Protein Levels
Viral Delivery of Survival of Motor Neuron
The primary goal of current SMA therapeutics is threefold: firstly, to increase SMN protein levels; secondly, to promote the inclusion of exon 7 in SMN2 transcripts; and thirdly, to promote motor neuron survival. Considered to be the most promising of the current SMA therapeutic approaches, viral delivery of SMN presents an efficient method for systemic delivery of SMN protein. Demonstrating the feasibility of this approach for the first time, Azzouz et al. used a lentiviral vector to retrogradely increase SMN expression in motor neurons in an SMA mouse model [107]. Following this initial study, improvements in gene delivery efficiency have led to the rescue of a severe mouse model of SMA using an adeno-associated viral vector (AAV9), which can infect peripheral tissue as well as cross the blood–brain barrier [108]. However, it is still not clear what effects viral delivery will have on already diseased human motor neurons and if any immunological responses will occur.
Exon 7 Inclusion of Survival of Motor Neuron
An alternative approach to SMN1 gene replacement is the use of antisense oligonucleotides to alter SMN2 splicing. The antisense oligonucleotide approach targets pre-mRNA processing, reducing exon 7 exclusion in SMN2 mRNA and increasing full-length SMN transcripts and protein levels. Antisense oligonucleotide technology has previously been used to alter gene expression in different forms of cancer (reviewed in [109]) and has been used successfully to restore dystrophin expression in Duchenne muscular dystrophy patients [110, 111]. Applying this methodology to treating SMA has proven successful both in vitro [112] and in vivo using SMA mouse models [113]. However, oligonucleotide delivery requires a direct route of administration to the CNS, such as intrathecal or intracerebroventricular injections, and will require additional optimization for an efficient delivery to peripheral organs.
Neuroprotection and Antiapoptotic Approaches
Survival of Motor Neuron-independent Therapies
While current strategies have focused solely on the importance of increasing SMN levels, the use of neuroprotective therapeutics as either a separate or complementary treatment for SMA also needs to be considered. For example, neurotrophic factors have demonstrated therapeutic benefits clinically and in animal models of neurodegenerative diseases, such as ALS, Alzheimer's disease, Parkinson's disease, and Huntington's disease (reviewed in [114]).
Cardiotrophin
Cardiotrophin-1 (CT-1) regulates apoptosis by blocking the proapoptotic functions of p53 and Bax, activating the Akt prosurvival signaling pathway and promoting Bcl-2 expression [115]. CT-1 delivery has proven to be neuroprotective in in vitro models of cerebral ischemia [116], while intramuscular delivery of an adenovirus-expressing CT-1 is able to delay motor impairment and muscle atrophy in an ALS mouse model [117]. With respect to SMA, Lesbordes et al. were the first to demonstrate the effective use of CT-1 treatment in an SMA mouse model [118]. In this study, overexpression of CT-1 in SMA mice resulted in increased survival, reduced NMJ disorganization, and diminished axonal degeneration [118]. Importantly, CT-1 is capable of reducing neuronal apoptosis by blocking caspase-3 and caspase-8 activation [119]. These findings suggest that there is potential for CT-1 to be used to reduce motor neuron apoptosis and prolong survival in SMA patients.
Insulinlike Growth Factor
Similarly, insulinlike growth factor (IGF-1) activates several prosurvival signaling pathways including PI 3-kinase/Akt and Erk1/2. In addition, IGF-1 enhances motor neuron axonal growth [120] and is thought to act in a retrograde manner to exert a neuroprotective effect on motor neurons. Expression of IGF-1 reduces muscle wastage in a mouse model of Duchenne muscular dystrophy (mdx) and causes significant improvements in life span and motor function in animals models of spinal bulbar muscular atrophy [121] and ALS [122].
In a severe mouse model of SMA, transgenic expression of IGF-1 in skeletal muscle tissue improved median survival and also resulted in improved overall motor function [123]. While significant improvements were observed in this study, they remain modest in comparison to recent studies using SMN gene replacement or correction of SMN2 splicing using antisense oligonucleotide delivery [124–126]. In contrast, in a SMA type III mouse model, CNS-mediated delivery of IGF-1 did not improve overall survival or motor function but did reduce motor neuron loss [127]. These results suggest the neurotrophic and antiapoptotic effects of IGF-1 therapy may be more beneficial in severe forms of the disease, where motor neuron loss is greater. Current therapeutic strategies are also using IGF-1 in combination with SMN-dependent approaches [128], demonstrating the importance of achieving neuroprotection and gene replacement for maximal therapeutic benefit in SMA.
Bcl-xL and Bcl-2
Both Bcl-2 and Bcl-xL expressions are reduced in SMA fetus spinal cords [8], potentially contributing to the irregular neuronal morphology and increased levels of apoptosis observed during SMA development [59, 129]. With this in mind, Tsai et al. [91] first postulated that Bcl-xL may compensate for a deficiency in SMN levels, showing that in transgenic SMA type III mice, increased Bcl-xL expression resulted in improved viability and reduced muscle atrophy. Recently, Bcl-xL has also been shown to phenotypically rescue mouse motor neurons exhibiting all the effects of reduced SMN levels [130]. Extending on these studies, we have demonstrated a relationship between Bcl-xL and SMN expressions, whereby coexpression of both proteins provided additive protection from apoptosis [9]. From these studies, we postulate that Bcl-xL is an important regulator of SMN expression and may be involved in mediating the antiapoptotic effects of SMN.
In addition to Bcl-xL, there also exists a relationship between the Bcl-2 and SMN proteins [5, 76]. Coexpression of Bcl-2 and SMN causes a synergistic reduction in Bax-mediated apoptosis [5]. Hence, it is not surprising that levels of proapoptotic Bax protein are elevated in the spinal cords of SMA mice and that inhibiting Bax in SMA mice results in improved motor neuron numbers and median survival [60]. Therefore, targeting apoptosis in SMA appears to be a viable treatment approach and the use of Bcl-xL and/or Bcl-2 in future SMA therapies should be considered.
Platelet-derived Growth Factor Prolactin and Erythropoietin
Recently, both platelet-derived growth factor (PDGF) and prolactin have been shown to elevate SMN protein levels through different pathways [131, 132]. PDGF was found to inhibit GSK-3, an important regulator of p53 activity, via activation of the Akt signaling pathway. In comparison, prolactin receptor activation stimulates the Janus kinase 2 pathway (JAK2), resulting in STAT5-mediated transcriptional activation of SMN and other antiapoptotic proteins [131, 133]. In this study, prolactin treatment of a severe mouse model of SMA resulted in elevated full-length SMN protein levels in the CNS, improved motor function, and enhanced survival [131]. Signaling via the Jak2/Stat5 pathway, erythropoietin (EPO) is another growth factor that could potentially show therapeutic potential in SMA. EPO is able to cross the blood–brain barrier [134] and several studies have demonstrated its effectiveness to ameliorate or reduce neuronal injury in cell culture and/or animal models of stroke, epilepsy, spinal cord injury, and ALS [135–138]. Interestingly, both PDGF- and prolactin-stimulated pathways result in SMN upregulation and the regulation of key survival proteins. This suggests that molecular regulation of SMN can be tightly controlled, independent of a splicing function, via different cell signaling pathways.
Stem Cells to Treat and Model Spinal Muscular Atrophy
Embryonic Stem Cells
Human embryonic stem cells (hESCs) were first isolated in 1998 and have generated much interest in their use to develop in vitro disease models and for stem cell transplantation [139]. Pluripotent hESCs are derived from the “inner cell mass” of blastocyst stage embryos. While embryonic stem cells are relatively easy to obtain and differentiate, ethical challenges relating to their source need to be overcome if they are to be considered as a mainstream therapy. Recently, embryonic stem cell-derived neural stem cells were successfully transplanted into SMA model mice [140]. In this study, intrathecal transplantation of ESC-derived neural stem cells demonstrated appropriate migration patterns, differentiated into motor neurons, and increased overall survival [140]. In SMA, stem cell transplantation of donor-derived hESCs may replenish motor neuron numbers but will not target reduced SMN protein levels in peripheral tissues. Therefore, for this approach to be feasible, it will need to be used in conjunction with other SMA therapies.
Induced Pluripotent Stem Cells
Reprograming of human fibroblasts presents another potential source of pluripotent stem cells and bypasses the ethical issues surrounding obtaining hESC's. While at present this method does not offer a therapeutic avenue, reprograming of SMA fibroblasts into human SMA motor neurons has potentially opened the door to targeted drug screening and improved SMA therapeutics [141]. SMA-derived motor neurons show axonal deficits and increased levels of apoptosis but importantly respond to SMN-dependent therapies. In addition, SMA-derived motor neurons display increased caspase-3 activation and selectively blocking caspase-3 rescues these neurons from an apoptotic death [71]. Utilization of iPSCs to model SMA can potentially provide a reliable and accurate platform to identify and assess therapies aimed at increasing SMN levels and targeting apoptosis in SMA.
Clinical Trials
Completed and Potential Clinical Trials in Spinal Muscular Atrophy
Currently, there are no approved treatments to prolong the survival of SMA patients. Clinically, numerous compounds have been trialed to increase SMN transcript and protein levels, but to date, no therapeutic candidate has been adopted as a treatment for SMA. The first candidates identified for treating SMA were the histone deacetylaste (HDAC) inhibitors, which were found to increase SMN2 expression [142]. Preclinical studies using HDAC inhibitors result in increased full-length SMN2 transcripts in vitro [143–145] and increased median survival in vivo [146]. However, the increased survival observed in HDAC-treated SMA mice could be due to an increased expression of several antiapoptotic genes as a result of the treatment [147]. Clinical trials of two HDAC inhibitors, phenylbutyrate and valproic acid, failed to show any significant clinical benefits in SMA patients [148–150]. Similarly, hydroxyurea enhances SMN2 expression in SMA patient cells [151] but has little or no clinical effects on SMA patients [152]. Currently, direct replacement of SMN using a scAAV9-expressing SMN appears the most promising therapeutic approach. Preclinical studies demonstrate dramatic improvements in systemic SMN expression and median survival in SMA mouse models [124, 125]. Furthermore, this approach results in sustained and systemic expression in nonhuman primates [153]. As a result of these studies and recent approval from the NIH Recombinant DNA Advisory Committee, phase I clinical trials using AAV9 to treat SMA are anticipated to commence in 2013.
Neuroprotective and antiapoptotic therapeutics could potentially target and reduce the early motor neuron loss that occurs in SMA patients. To date, clinical trials in SMA patients using the neuroprotective gabapentin have demonstrated negative or minimal results [154]. Neurotrophic factors have also been used in preclinical studies in numerous neurodegenerative diseases with success. However, in clinical trials of ALS, IGF-1, BDNF, and CNTF have failed to show any clinical benefit [155–158]. From these results, it appears that solely targeting apoptosis in SMA is not the answer. However, to combat the motor neuron loss due to elevated apoptosis occurring early in the disease, the addition of neurotrophic and/or antiapoptotic factors alongside SMN replacement may provide additional clinical benefits.
Conclusions
SMN is a multifunctional protein that interacts with and mediates the expression of numerous other proteins involved in DNA repair, calcium handling, cell cycle, and apoptosis and which major function is the maintenance of motor neuron survival. Importantly, in SMA, the expression of critical modulators of cell viability is significantly affected (Table 1) that is likely to affect the disease phenotype. Expression of these genes in SMA may be affected through direct or indirect regulatory roles of the SMN protein and/or dysfunction of the snRNP complex, thereby causing altered splicing of these important genes [44]. The diverse range of proteins affected in SMA strongly supports a multifunctional role for the SMN protein. Further elucidation of SMN functions and downstream effects of SMN deficiency is likely to lead to a better understanding of the disease mechanisms that underlie impaired motor neuron survival in SMA.
It is important that SMN-independent approaches to promote neuronal survival should not be ruled out as they may complement therapies aimed at increasing SMN protein levels. In particular, neuroprotective and antiapoptotic proteins have shown promising results in promoting neuronal survival, rescuing axonal defects and increasing endogenous SMN protein levels. As summarized in this review, a variety of proteins involved in survival/apoptosis are affected in SMA mouse models and patient tissues. A more complete understanding of the signaling pathways involving SMN is required to substantiate the key cellular mechanisms involved in SMA pathogenesis. However, the role of apoptosis in SMA pathogenesis is well-established and targeting the early-stage apoptosis that occurs in SMA may be paramount to attempting to completely alleviate the disease.
References
Munsat TL, Davies KE (1992) International SMA consortium meeting. (26–28 June 1992, Bonn, Germany). Neuromuscul Disord 2(5–6):423–428
Bertini E et al (2005) 134th ENMC International workshop: outcome measures and treatment of spinal muscular atrophy, 11–13 february, 2005 Naarden, The Netherlands. Neuromuscul Disord 15(11):802–816
Russman BS et al (1996) Function changes in spinal muscular atrophy II and III. The DCN/SMA Group. Neurology 47(4):973–976
Wirth B (2000) An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat 15(3):228–237
Iwahashi H et al (1997) Synergistic anti-apoptotic activity between Bcl-2 and SMN implicated in spinal muscular atrophy. Nature 390(6658):413–417
Young PJ et al (2002) A direct interaction between the survival motor neuron protein and p53 and its relationship to spinal muscular atrophy. J Biol Chem 277(4):2852–2859
Gangwani L et al (2001) Spinal muscular atrophy disrupts the interaction of ZPR1 with the SMN protein. Nat Cell Biol 3(4):376–383
Soler-Botija C et al (2003) Downregulation of Bcl-2 proteins in type I spinal muscular atrophy motor neurons during fetal development. J Neuropathol Exp Neurol 62(4):420–426
Anderton RS et al (2012) Co-regulation of survival of motor neuron and Bcl-xL expression: Implications for neuroprotection in spinal muscular atrophy. Neuroscience 220:228–236
Lefebvre S et al (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80(1):155–165
Rochette CF, Gilbert N, Simard LR (2001) SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum Genet 108(3):255–266
Boda B et al (2004) Survival motor neuron SMN1 and SMN2 gene promoters: identical sequences and differential expression in neurons and non-neuronal cells. Eur J Hum Genet 12(9):729–737
Lorson CL et al (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA 96(11):6307–6311
Cartegni L, Krainer AR (2002) Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 30(4):377–384
Kashima T, Manley JL (2003) A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet 34(4):460–463
Wang J, Dreyfuss G (2001) Characterization of functional domains of the SMN protein in vivo. J Biol Chem 276(48):45387–45393
Frugier T et al (2000) Nuclear targeting defect of SMN lacking the C-terminus in a mouse model of spinal muscular atrophy. Hum Mol Genet 9(5):849–858
Pellizzoni L, Charroux B, Dreyfuss G (1999) SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc Natl Acad Sci USA 96(20):11167–11172
Burnett BG et al (2009) Regulation of SMN protein stability. Mol Cell Biol 29(5):1107–1115
La Bella V et al (1998) Survival motor neuron (SMN) protein in rat is expressed as different molecular forms and is developmentally regulated. Eur J Neurosci 10(9):2913–2923
Liu Q, Dreyfuss G (1996) A novel nuclear structure containing the survival of motor neurons protein. EMBO J 15(14):3555–3565
Carvalho T et al (1999) The spinal muscular atrophy disease gene product, SMN: A link between snRNP biogenesis and the Cajal (coiled) body. J Cell Biol 147(4):715–728
Young PJ et al (2001) Nuclear gems and Cajal (coiled) bodies in fetal tissues: nucleolar distribution of the spinal muscular atrophy protein, SMN. Exp Cell Res 265(2):252–261
Hebert MD et al (2001) Coilin forms the bridge between Cajal bodies and SMN, the spinal muscular atrophy protein. Genes Dev 15(20):2720–2729
Gubitz AK, Feng W, Dreyfuss G (2004) The SMN complex. Exp Cell Res 296(1):51–56
Francis JW et al (1998) Heterogeneity of subcellular localization and electrophoretic mobility of survival motor neuron (SMN) protein in mammalian neural cells and tissues. Proc Natl Acad Sci USA 95(11):6492–6497
Pagliardini S et al (2000) Subcellular localization and axonal transport of the survival motor neuron (SMN) protein in the developing rat spinal cord. Hum Mol Genet 9(1):47–56
Broccolini A, Engel WK, Askanas V (1999) Localization of survival motor neuron protein in human apoptotic-like and regenerating muscle fibers, and neuromuscular junctions. Neuroreport 10(8):1637–1641
Battaglia G et al (1997) Expression of the SMN gene, the spinal muscular atrophy determining gene, in the mammalian central nervous system. Hum Mol Genet 6(11):1961–1971
Ruggiu M et al (2012) A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol Cell Biol 32(1):126–138
Simic G (2008) Pathogenesis of proximal autosomal recessive spinal muscular atrophy. Acta Neuropathol 116(3):223–234
Liu Q et al (1997) The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 90(6):1013–1021
Charroux B et al (1999) Gemin3: a novel DEAD box protein that interacts with SMN, the spinal muscular atrophy gene product, and is a component of gems. J Cell Biol 147(6):1181–1194
Charroux B et al (2000) Gemin4. A novel component of the SMN complex that is found in both gems and nucleoli. J Cell Biol 148(6):1177–1186
Baccon J et al (2002) Identification and characterization of Gemin7, a novel component of the survival of motor neuron complex. J Biol Chem 277(35):31957–31962
Gubitz AK et al (2002) Gemin5, a novel WD repeat protein component of the SMN complex that binds Sm proteins. J Biol Chem 277(7):5631–5636
Pellizzoni L, Yong J, Dreyfuss G (2002) Essential role for the SMN complex in the specificity of snRNP assembly. Science 298(5599):1775–1779
Carissimi C et al (2006) Gemin8 is a novel component of the survival motor neuron complex and functions in small nuclear ribonucleoprotein assembly. J Biol Chem 281(12):8126–8134
Raker VA et al (1999) Spliceosomal U snRNP core assembly: Sm proteins assemble onto an Sm site RNA nonanucleotide in a specific and thermodynamically stable manner. Mol Cell Biol 19(10):6554–6565
Pellizzoni L (2007) Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep 8(4):340–345
Kolb SJ, Battle DJ, Dreyfuss G (2007) Molecular functions of the SMN complex. J Child Neurol 22(8):990–994
Will CL, Luhrmann R (2001) Spliceosomal UsnRNP biogenesis, structure and function. Curr Opin Cell Biol 13(3):290–301
Kolb SJ, Sutton S, Schoenberg DR (2010) RNA processing defects associated with diseases of the motor neuron. Muscle Nerve 41(1):5–17
Burghes AH, Beattie CE (2009) Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 10(8):597–609
Lotti F et al (2012) An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151(2):440–454
Zhang HL et al (2003) Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J Neurosci 23(16):6627–6637
McWhorter ML et al (2003) Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol 162(5):919–931
Carrel TL et al (2006) Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J Neurosci 26(43):11014–11022
Fallini C et al (2011) The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(A) mRNA in primary motor neuron axons. J Neurosci 31(10):3914–3925
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4):239–257
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516
Mattson MP (2000) Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1(2):120–129
Lindsten T, Zong WX, Thompson CB (2005) Defining the role of the Bcl-2 family of proteins in the nervous system. Neuroscientist 11(1):10–15
Oppenheim RW (1991) Cell death during development of the nervous system. Annu Rev Neurosci 14:453–501
Desjardins P, Ledoux S (1998) The role of apoptosis in neurodegenerative diseases. Metab Brain Dis 13(2):79–96
Martin LJ (1999) Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol 58(5):459–471
Cleveland DW, Rothstein JD (2001) From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2(11):806–819
Simic G et al (2000) Ultrastructural analysis and TUNEL demonstrate motor neuron apoptosis in Werdnig–Hoffmann disease. J Neuropathol Exp Neurol 59(5):398–407
Soler-Botija C et al (2002) Neuronal death is enhanced and begins during foetal development in type I spinal muscular atrophy spinal cord. Brain 125(Pt 7):1624–1634
Tsai MS et al (2006) Abolishing Bax-dependent apoptosis shows beneficial effects on spinal muscular atrophy model mice. Mol Ther 13(6):1149–1155
Tsai MS et al (2006) Abolishing Trp53-dependent apoptosis does not benefit spinal muscular atrophy model mice. Eur J Hum Genet 14(3):372–375
Parker GC et al (2008) Survival motor neuron protein regulates apoptosis in an in vitro model of spinal muscular atrophy. Neurotox Res 13(1):39–48
Vyas S et al (2002) Involvement of survival motor neuron (SMN) protein in cell death. Hum Mol Genet 11(22):2751–2764
Wang W et al (2005) Increased susceptibility of spinal muscular atrophy fibroblasts to camptothecin-induced cell death. Mol Genet Metab 85(1):38–45
Anderton RS et al (2011) Survival of motor neuron protein over-expression prevents calpain-mediated cleavage and activation of procaspase-3 in differentiated human SH-SY5Y cells. Neuroscience 181:226–233
Kerr DA et al (2000) Survival motor neuron protein modulates neuron-specific apoptosis. Proc Natl Acad Sci USA 97(24):13312–13317
Han Z et al (1997) A sequential two-step mechanism for the production of the mature p17:p12 form of caspase-3 in vitro. J Biol Chem 272(20):13432–13436
Deveraux QL et al (1998) IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 17(8):2215–2223
Meergans T et al (2000) The short prodomain influences caspase-3 activation in HeLa cells. Biochem J 349(Pt 1):135–140
Trulzsch B et al (2007) Knockdown of SMN by RNA interference induces apoptosis in differentiated P19 neural stem cells. Brain Res 1183:1–9
Sareen D et al (2012) Inhibition of apoptosis blocks human motor neuron cell death in a stem cell model of spinal muscular atrophy. PLoS One 7(6):e39113
Walker MP et al (2008) SMN complex localizes to the sarcomeric Z-disc and is a proteolytic target of calpain. Hum Mol Genet 17(21):3399–3410
Fuentes JL, Strayer MS, Matera AG (2010) Molecular determinants of survival motor neuron (SMN) protein cleavage by the calcium-activated protease, calpain. PLoS One 5(12):e15769
Gross A, McDonnell JM, Korsmeyer SJ (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13(15):1899–1911
Burlacu A (2003) Regulation of apoptosis by Bcl-2 family proteins. J Cell Mol Med 7(3):249–257
Sato K et al (2000) Regions essential for the interaction between Bcl-2 and SMN, the spinal muscular atrophy disease gene product. Cell Death Differ 7(4):374–383
Anderson K et al (2003) Protein expression changes in spinal muscular atrophy revealed with a novel antibody array technology. Brain 126(Pt 9):2052–2064
Maheswaran S et al (1995) The WT1 gene product stabilizes p53 and inhibits p53-mediated apoptosis. Genes Dev 9(17):2143–2156
Mayo MW et al (1999) WT1 modulates apoptosis by transcriptionally upregulating the bcl-2 proto-oncogene. EMBO J 18(14):3990–4003
Helmken C et al (2003) Evidence for a modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet 114(1):11–21
Gangwani L, Flavell RA, Davis RJ (2005) ZPR1 is essential for survival and is required for localization of the survival motor neurons (SMN) protein to Cajal bodies. Mol Cell Biol 25(7):2744–2756
Doran B et al (2006) Deficiency of the zinc finger protein ZPR1 causes neurodegeneration. Proc Natl Acad Sci USA 103(19):7471–7475
Ryan KM, Phillips AC, Vousden KH (2001) Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol 13(3):332–337
Alarcon-Vargas D, Ronai Z (2002) p53-Mdm2—the affair that never ends. Carcinogenesis 23(4):541–547
de Rozieres S et al (2000) The loss of mdm2 induces p53-mediated apoptosis. Oncogene 19(13):1691–1697
Boise LH et al (1993) bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74(4):597–608
Gonzalez-Garcia M et al (1994) bcl-XL is the major bcl-x mRNA form expressed during murine development and its product localizes to mitochondria. Development 120(10):3033–3042
Kim CN et al (1997) Overexpression of Bcl-X(L) inhibits Ara-C-induced mitochondrial loss of cytochrome c and other perturbations that activate the molecular cascade of apoptosis. Cancer Res 57(15):3115–3120
Hu Y et al (1998) Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc Natl Acad Sci USA 95(8):4386–4391
Motoyama N et al (1995) Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 267(5203):1506–1510
Tsai LK et al (2008) Restoring Bcl-x(L) levels benefits a mouse model of spinal muscular atrophy. Neurobiol Dis 31(3):361–367
Paronetto MP et al (2007) The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol 176(7):929–939
Pedrotti S et al (2010) The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J 29(7):1235–1247
Roy N et al (1995) The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 80(1):167–178
Samilchuk E et al (1996) Deletion analysis of the SMN and NAIP genes in Kuwaiti patients with spinal muscular atrophy. Hum Genet 98(5):524–527
Capon F et al (1996) Deletion analysis of SMN and NAIP genes in spinal muscular atrophy Italian families. Muscle Nerve 19(3):378–380
Chang JG et al (1997) Molecular analysis of survival motor neuron (SMN) and neuronal apoptosis inhibitory protein (NAIP) genes of spinal muscular atrophy patients and their parents. Hum Genet 100(5–6):577–581
Tsai CH et al (2001) Molecular analysis of SMN, NAIP and P44 genes of SMA patients and their families. J Neurol Sci 190(1–2):35–40
Kesari A et al (2005) Study of survival of motor neuron (SMN) and neuronal apoptosis inhibitory protein (NAIP) gene deletions in SMA patients. J Neurol 252(6):667–671
Watihayati MS et al (2009) Combination of SMN2 copy number and NAIP deletion predicts disease severity in spinal muscular atrophy. Brain Dev 31(1):42–45
Maier JK et al (2002) The neuronal apoptosis inhibitory protein is a direct inhibitor of caspases 3 and 7. J Neurosci 22(6):2035–2043
Hutchison JS et al (2001) Neuronal apoptosis inhibitory protein expression after traumatic brain injury in the mouse. J Neurotrauma 18(12):1333–1347
Liston P et al (1996) Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 379(6563):349–353
Perrelet D et al (2000) IAP family proteins delay motoneuron cell death in vivo. Eur J Neurosci 12(6):2059–2067
Holcik M et al (2000) The hippocampal neurons of neuronal apoptosis inhibitory protein 1 (NAIP1)-deleted mice display increased vulnerability to kainic acid-induced injury. Proc Natl Acad Sci USA 97(5):2286–2290
Gotz R et al (2000) The neuronal apoptosis inhibitory protein suppresses neuronal differentiation and apoptosis in PC12 cells. Hum Mol Genet 9(17):2479–2489
Azzouz M et al (2004) Lentivector-mediated SMN replacement in a mouse model of spinal muscular atrophy. J Clin Invest 114(12):1726–1731
Foust KD et al (2010) Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol 28(3):271–274
Stahel RA, Zangemeister-Wittke U (2003) Antisense oligonucleotides for cancer therapy—an overview. Lung Cancer 41(Suppl 1):S81–S88
van Deutekom JC et al (2007) Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med 357(26):2677–2686
Cirak S et al (2011) Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378(9791):595–605
Hua Y et al (2007) Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol 5(4):e73
Passini MA et al (2011) Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med 3(72):72ra18
Lim ST, Airavaara M, Harvey BK (2010) Viral vectors for neurotrophic factor delivery: a gene therapy approach for neurodegenerative diseases of the CNS. Pharmacol Res 61(1):14–26
Ruixing Y, Dezhai Y, Jiaquan L (2004) Effects of cardiotrophin-1 on hemodynamics and cardiomyocyte apoptosis in rats with acute myocardial infarction. J Med Invest 51(1–2):29–37
Wen TC et al (2005) Cardiotrophin-1 protects cortical neuronal cells against free radical-induced injuries in vitro. Neurosci Lett 387(1):38–42
Bordet T et al (2001) Protective effects of cardiotrophin-1 adenoviral gene transfer on neuromuscular degeneration in transgenic ALS mice. Hum Mol Genet 10(18):1925–1933
Lesbordes JC et al (2003) Therapeutic benefits of cardiotrophin-1 gene transfer in a mouse model of spinal muscular atrophy. Hum Mol Genet 12(11):1233–1239
Peng H et al (2010) Caspase inhibition by cardiotrophin-1 prevents neuronal death in vivo and in vitro. J Neurosci Res 88(5):1041–1051
Ozdinler PH, Macklis JD (2006) IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat Neurosci 9(11):1371–1381
Palazzolo I et al (2009) Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 63(3):316–328
Kaspar BK et al (2003) Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 301(5634):839–842
Bosch-Marce M et al (2011) Increased IGF-1 in muscle modulates the phenotype of severe SMA mice. Hum Mol Genet 20(9):1844–1853
Passini MA et al (2010) CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J Clin Invest 120(4):1253–1264
Dominguez E et al (2011) Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet 20(4):681–693
Porensky PN et al (2012) A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet 21(7):1625–1638
Tsai LK et al (2012) IGF-1 delivery to CNS attenuates motor neuron cell death but does not improve motor function in type III SMA mice. Neurobiol Dis 45(1):272–279
Shababi M, Glascock J, Lorson CL (2011) Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum Gene Ther 22(2):135–144
Simic G et al (2008) Abnormal motoneuron migration, differentiation, and axon outgrowth in spinal muscular atrophy. Acta Neuropathol 115(3):313–326
Garcera A et al (2011) A new model to study spinal muscular atrophy: neurite degeneration and cell death is counteracted by BCL-X(L) Overexpression in motoneurons. Neurobiol Dis 42(3):415–426
Farooq F et al (2011) Prolactin increases SMN expression and survival in a mouse model of severe spinal muscular atrophy via the STAT5 pathway. J Clin Invest 121(8):3042–3050
Makhortova NR et al (2011) A screen for regulators of survival of motor neuron protein levels. Nat Chem Biol 7(8):544–552
Ting CH et al (2007) Stat5 constitutive activation rescues defects in spinal muscular atrophy. Hum Mol Genet 16(5):499–514
Brines ML et al (2000) Erythropoietin crosses the blood–brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA 97(19):10526–10531
Minnerup J et al (2009) The efficacy of erythropoietin and its analogues in animal stroke models: a meta-analysis. Stroke 40(9):3113–3120
Kondo A et al (2009) Erythropoietin exerts anti-epileptic effects with the suppression of aberrant new cell formation in the dentate gyrus and upregulation of neuropeptide Y in seizure model of rats. Brain Res 1296:127–136
Leist M et al (2004) Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 305(5681):239–242
Grunfeld JF et al (2007) Erythropoietin delays disease onset in an amyotrophic lateral sclerosis model. Exp Neurol 204(1):260–263
Thomson JA et al (1998) Embryonic stem cell lines derived from human blastocysts. Science 282(5391):1145–1147
Corti S et al (2010) Embryonic stem cell-derived neural stem cells improve spinal muscular atrophy phenotype in mice. Brain 133(Pt 2):465–481
Ebert AD et al (2009) Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457(7227):277–280
Chang JG et al (2001) Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci USA 98(17):9808–9813
Brichta L et al (2003) Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet 12(19):2481–2489
Andreassi C et al (2004) Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet 12(1):59–65
Sumner CJ et al (2003) Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol 54(5):647–654
Narver HL et al (2008) Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann Neurol 64(4):465–470
Grierson AJ, Shaw CE, Miller CC (2001) Androgen induced cell death in SHSY5Y neuroblastoma cells expressing wild-type and spinal bulbar muscular atrophy mutant androgen receptors. Biochim Biophys Acta 1536(1):13–20
Mercuri E et al (2007) Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology 68(1):51–55
Swoboda KJ et al (2010) SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS One 5(8):e12140
Kissel JT et al (2011) SMA CARNIVAL TRIAL PART II: a prospective, single-armed trial of L-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS One 6(7):e21296
Grzeschik SM et al (2005) Hydroxyurea enhances SMN2 gene expression in spinal muscular atrophy cells. Ann Neurol 58(2):194–202
Chen TH et al (2010) Randomized, double-blind, placebo-controlled trial of hydroxyurea in spinal muscular atrophy. Neurology 75(24):2190–2197
Bevan AK et al (2011) Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther 19(11):1971–1980
Miller RG et al (2001) A placebo-controlled trial of gabapentin in spinal muscular atrophy. J Neurol Sci 191(1–2):127–131
ALS CNTF Treatment Study Group (1996) A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. Neurology 46:1244–1249
Kasarkis E (1999) A controlled trial of recombinant methionyl human BDNF in 1097 ALS: the BDNF Study Group (Phase III). Neurology 52(7):1427–33
Beck M et al (2005) Autonomic dysfunction in ALS: a preliminary study on the effects of intrathecal BDNF. Amyotroph Lateral Scler Other Motor Neuron Disord 6(2):100–103
Sorenson EJ et al (2008) Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology 71(22):1770–1775
Clevenger CV, Medaglia MV (1994) The protein tyrosine kinase P59fyn is associated with prolactin (PRL) receptor and is activated by PRL stimulation of T-lymphocytes. Mol Endocrinol 8(6):674–681
Acknowledgments
These studies were supported by the Neuromuscular Foundation and Muscular Dystrophy Association of Western Australia.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Anderton, R.S., Meloni, B.P., Mastaglia, F.L. et al. Spinal Muscular Atrophy and the Antiapoptotic Role of Survival of Motor Neuron (SMN) Protein. Mol Neurobiol 47, 821–832 (2013). https://doi.org/10.1007/s12035-013-8399-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8399-5