
Abstract
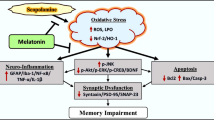
Alzheimer’s disease (AD) is the most prevalent type of dementia in elderly people. There are decreased melatonin levels in the serum of AD patients, and melatonin supplements are able to reverse AD pathology and memory deficits in many animal experiments and clinical trials. However, the underlying mechanism regarding how melatonin rescues the AD-like memory/synaptic disorder remains unknown. Here, we use the Morris water maze, step-down inhibitory avoidance task, in vivo long-term potentiation recording, and Golgi staining and report that intraperitoneal injection of melatonin (1 mg/kg/day) for 14 days in rats effectively reverses the memory and synaptic impairment in scopolamine-induced amnesia, a well-recognized dementia animal model. Using real-time polymerase chain reaction and western blotting experiments, we further determined that melatonin rescues the EPACs/miR-124/Egr1 signal pathway, which is important in learning and memory, as reported recently. Our studies provide a novel underlying epigenetic mechanism for melatonin to attenuate the synaptic disorder and could benefit drug discovery in neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Melatonin is the major hormone secreted by the pineal gland and is crucial for the regulation of the circadian rhythm. It plays an important role in antioxidative impairments, protection of nuclear and mitochondrial DNA, interactions with the immune system, and other biological functions [1]. Melatonin is important in synaptic plasticity and learning/memory [2]. Melatonin decreases in the elderly people, particularly in patients with Alzheimer’s disease (AD). AD is one of the most common neurodegenerative diseases, characterized by progressive memory decline [3]. Inhibition of melatonin biosynthesis induces AD-like spatial memory deficits and pathological changes in rats, and supplementation with exogenous melatonin reverses these changes [2]. Melatonin administration increases dendrite maturation and complexity in new neurons [4] and stimulates dendritogenesis in hippocampal organotypic cultures [5]. Melatonin also preserves dendritic spines in hippocampal CA1 pyramidal neurons upon exposure to global cerebral ischemia [6] and preserves basilar dendritic outgrowth of pyramidal cortical cells exposed to toluene vapors [7]. In addition, supplemental melatonin alleviates the memory deficits induced by streptozotocin, calyculin A, and Aβ [8–10]. Thus, melatonin might be useful for the treatment of neuropsychological diseases.
Memory deficits are the major clinical syndrome of AD, which is accompanied by neuronal degeneration in the brain, particularly in the cholinergic system. Scopolamine is a nonselective muscarinic receptor antagonist and impairs learning and memory by blocking cholinergic signaling. It has been used as a pharmacological tool to model AD-related cognitive decline by addressing the aspect of cholinergic system dysfunction. Scopolamine-treated rats display reduced alternation and exploratory behavior, and impaired spatial learning/memory. Many drugs that are used for dementia therapy have been screened using this model. Although it has been reported that melatonin can arrest cholinergic activity and reverse the amnesia induced by scopolamine [11], direct evidence for a role of melatonin in synaptic disorders and its underlying mechanism are still missing.
MicroRNA (miRNA) is a short ribonucleic acid molecule that is nonencoding RNA but is widely distributed in eukaryotic cells. miRNA plays an important role in the normal function of synapses, and dysfunctional miRNA is associated with synaptic disorders [12]. In a whole genome expression screen, there are many miRNAs identified that could be regulated by melatonin in cancer [13]. Most of these have been reported to be crucial for synaptic plasticity, but a direct link of melatonin on miRNA expression in the brain has not been shown.
Here, we demonstrate that melatonin can attenuate scopolamine-induced synaptic disorder in rats. We also found that melatonin can activate the EPACs/miR-124/Egr1 signaling pathway and rescue the expression of synaptic proteins and the impairment of dendritic spines.
Materials and Methods
Reagents
The primary antibodies employed in this study and their properties are listed in Table 1. Scopolamine and melatonin were purchased from Sigma Chemical Co. (St. Louis, MO, USA).
Animals and Drug Administration
Male Sprague Dawley rats (grade II, male, 200–250 g, 4 months old) were supplied by the Experimental Animal Center of Tongji Medical College. All animal experiments were performed according to the “Policies on the Use of Animals and Humans in Neuroscience Research.” All rats were housed in an air-conditioned room (22 ±2 °C, 12 h light and 12 h dark; lights on at 06:00 am) with free access to food and water.
Scopolamine (sco) and melatonin (mel) were dissolved in 3 % dimethylsulfoxide before injection. Forty rats were randomly divided into four groups (con, sco, s+m, and mel; n = 10 for each group). The rats were treated with scopolamine (1 mg/kg/day) or saline in the same volume with or without a simultaneous supplement of melatonin (1 mg/kg/day) for 14 consecutive days through intraperitoneal injection. The scopolamine injection was carried out 30 min before behavior training, and the melatonin injection was carried out at 6:00 pm. Animals were sacrificed 24 h after the final injection.
Morris Water Maze Test
Morris water maze test was used to measure the spatial learning and memory ability of rats [14, 15]. Briefly, the rats were trained for six consecutive days to find a transparent platform hidden 1 cm under water through using a stationary array of cues outside the pool. A digital tracking device connected to a computer was used to record the swimming pathway and escape latency. On the seventh day, the swimming pathway and escape latency of rats to reach the hidden platform were examined. On the ninth day, the percent time spent in the target quadrant and the number of platform quadrant crosses were tested with removed platform.
Step-Down Inhibitory Avoidance Task
A 30 × 30 × 30-cm electronic avoidance-response chamber consists of Plexiglas on its three sides and black plastic on the fourth side. A series of parallel stainless steel bars (0.5 cm in diameter, spacing 1 cm apart) were equipped at the bottom of the chamber. An insulated platform, 5 cm high and 5 cm in diameter, was placed at a corner to provide a shelter for the rats. Before tests, the rats were trained three times from 2:00 pm to 5:00 pm. During training sessions, the rats were placed on the platform and would receive a 0.5-s, 1-Hz, 36-V DC electronic foot shock immediately after stepping down. The rats were then tested twice, 2 and 24 h after the training. In test sessions, step-down latencies and errors (placing their all four paws onto the grid) within 3 min were used as measurements of retention.
Golgi Staining
The rats were anesthetized by chloral hydrate and perfused with 400 mL normal saline containing 0.5 % sodium nitrite, followed by 400 mL 4 % formaldehyde solution. The brains were further perfused with 400 mL dying solution made of 5 % chloral hydrate, 5 % potassium dichromate, and 4 % formaldehyde. The brains were then incubated in dying solution for 3 days in the dark and transferred to silver solution containing 1 % silver nitrate for 3 days in the dark; 30-μm brain sections of hippocampal tissue were cut using a vibrate microtome (Leica, Wetzlar, Germany).
Western Blotting
Hippocampi removed from brain were homogenized in a buffer containing Tris–Cl (pH 7.6) 10 mmol/L, Na3VO4 1 mmol/L, NaF 50 mmol/L, benzamidine 1 mmol/L, edetic acid 1 mmol/L, and PMSF 1 mmol/L. Three volumes of the homogenized tissue were added to one volume of an extracting buffer containing Tris–Cl (pH 7.6) 200 mmol/L, 8 % sodium dodecyl sulfate (SDS), and 40 % glycerol, and extracts were boiled in a water bath for 10 min. The lysates were sonicated briefly and centrifuged at 12,000 × g for 5 min. Protein concentration of the supernatants was measured by the bicinchoninic acid Protein Assay Kit (Pierce, Rockford, IL, USA). The proteins were separated by SDS–polyacrylamide gel electrophoresis (10 % gel) and transferred to a nitrocellulose membrane. After blocking in 3 % nonfat milk for 1 h at 25 °C, the membranes were incubated with primary antibodies at 4 °C overnight. The blots were then incubated with anti-mouse or anti-rabbit IgG conjugated to IRDye™ (800CW) for 1 h at 25 °C and visualized with the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA).
Immunohistochemistry
The rats were anesthetized and sacrificed by overdose of chloral hydrate and immediately perfused with 400 mL normal saline, and then with 400 mL 4 % paraformaldehyde solution. The brains were dissected and postfixed for another 24 h at 4 °C. The fixed brains were sliced coronally at 25 μm using a vibrate microtome (Leica). For a primary antibody, three to five consecutive sections from each brain were used. The sections were further incubated with biotin-labeled secondary antibodies for 1 h at 37 °C and visualized with 3,3′-diaminobenzidine (Sigma Chemical Co.) as brown in color. The images were observed using a microscope (Olympus BX60, Tokyo, Japan) and quantitatively analyzed by Image-Pro Plus software (Media Cybernetics, Carlsbad, CA, USA).
Long-Term Potentiation Measurement
The rats were anesthetized using urethane (1.2 g/kg) and placed on the stereotaxic instrument (Narishige, Tokyo, Japan). The body temperature of the rats was maintained at 37 °C in a bath circulator. After the skull was exposed, a small hole was made at the appropriate coordinates to enable vertical penetration through the stimulating and recording electrodes. The stimulating electrode was placed in the area of perforant path at coordinates of anteroposterior (AP) −6.9 to −7.1 mm, mediolateral (ML) ±4.4 to ±4.6 mm, and dorsoventral (DV) −3.4 to −3.6 mm according to the rat brain atlas, and the recording electrode was placed in CA3 at coordinates of AP −3.4 to −3.6 mm, ML ±3.4 to ±3.6 mm, and DV −3.2 to −3.4 mm. For each measurement, a stable baseline (±10 % change) for at least 20 min was required before high-frequency stimulation (HFS). Long-term potentiation (LTP) was elicited using HFS consisting of four trains of 50 pulses delivered at 200 Hz with a 2-s intertrain interval. LTP was analyzed by the measurement of excitatory postsynaptic potential and population spike [16].
Real-Time Quantitative PCR
Total RNA was isolated using TRIzol reagents according to the instructions (Invitrogen, Carlsbad, CA, USA) and reverse-transcribed to cDNA using reverse transcription reagents kit (Takara, Dalian, China). Poly(A) was added to microRNAs and reverse-transcribed to cDNA using All-in-One reverse transcription kit (GeneCopoeia, MD, USA). Fifty nanograms of cDNA was used for real-time PCR. Primers for EPAC1, EPAC2, RagA, and β-actin were listed in Table 2. Primer for rno-miR-124 and U6 were purchased from GeneCopoeia. The PCR cycle was as follows: 95 °C/30 s, 40 cycles of 95 °C/5 s, 60 °C/30 s, and 72 °C/30 s, and the melt-curve analysis was performed following each experiment. The amplification and analysis were performed using a StepOnePlus Real-Time PCR Detection System (Life Technologies, NY, USA). Samples were compared using the relative CT method.
Statistical Analysis
All results were expressed as means ± SE and were analyzed using SPSS 16.0 statistical software (SPSS, Chicago, IL, USA) by one-way ANOVA followed by a least significant difference post hoc test. A level of P < 0.05 was accepted as statistically significant.
Results
Melatonin Arrests Scopolamine-Induced Memory Deficits
To explore whether melatonin could reverse scopolamine-induced spatial memory impairments, we used the Morris water maze. We first evaluated learning ability by training the rats with a hidden platform. We found that scopolamine-treated rats display a prolonged latency to find the platform and melatonin supplements shorten the latency (Fig. 1b, c). On the seventh day, the scopolamine-treated rats took a tortuous swimming path to the platform but rats given a melatonin supplement took a linear path to the platform (Fig. 1a). We removed the platform on the ninth day and allowed the rats to swim freely. We observed fewer crossing times and less time spent in the target quadrant in the scopolamine-treated rats (Fig. 1d, e), indicating that scopolamine induced severe memory deficits, as previously reported. However, simultaneous melatonin supplement effectively restored the memory deficits (Fig. 1d, e).

Melatonin arrests scopolamine-induced memory deficits. a The path to find the platform on the seventh day in Morris water maze. b The escape latency to find the hidden platform from the first day to the sixth day. c The escape latency to find the hidden platform on the seventh day. d The time spent in the target quadrant on the ninth day. e The number to cross the target quadrant on the ninth day. f The step-down latency to land on the bottom of the chamber, 2 and 24 h after training, respectively. g The number to land on the bottom of the chamber, 2 and 24 h after training, respectively. con control group, sco scopolamine-treated group, s + m scopolamine plus melatonin-treated group, mel melatonin-treated group. *P < 0.05, **P < 0.01, compared with con; #P < 0.05, ##P < 0.01, compared with sco group
We also used the step-down inhibitory avoidance task to examine whether melatonin could reverse the scopolamine-induced fear-motivated memory impairment. We found that scopolamine injection dramatically decreased the step-down latency and increased the number of errors at 2 and 24 h after training, suggesting severe deficits in both short-term memory and long-term memory. In addition, we found that supplemental melatonin arrests the latency and reduces the number of errors, indicating a recovery of memory capacity in these rats (Fig. 1f, g). These data strongly suggested that melatonin supplementation could restore the memory deficits induced by scopolamine.
Melatonin Attenuates Scopolamine-Induced Synaptic Disorder
Synaptic plasticity is the basis of learning and memory. Therefore, we examined synaptic transmission using LTP recordings as described previously [16]. We found that in scopolamine-treated rats, the amplitude and slope of field excitatory postsynaptic potential (fEPSP) increased approximately 1.2- and 1.3-fold, respectively; this was much lower than the increase in vehicle-treated rats. Concomitant supplement of melatonin restored the amplitude and slope of fEPSP (Fig. 2). These data suggested that melatonin could effectively reverse the synaptic transmission deficits induced by scopolamine.

Melatonin reverses scopolamine-induced synaptic transmission deficits. a The representative traces of the evoked potential before (pre, dashed line) and after (post, solid line) high-frequency stimulation (HFS). b Normalized population spike (PS) amplitude. c Normalized excitatory postsynaptic potential (EPSP) slope
We also examined alteration of dendritic spines, the most important postsynaptic compartment that is crucial for synaptic strength [17]. Using a well-established Golgi staining method, we found that scopolamine injection decreased the density of spines and the percentage of mushroom-type spines in CA1 neurons but melatonin supplementation recovered the dendritic spine deficits (Fig. 3a–c). These data suggested that melatonin may attenuate the spine impairment induced by scopolamine.

Melatonin attenuates scopolamine-induced synaptic disorder. a–c Golgi staining was used to evaluate the changes of dendritic spines. Representative photomicrographs (a) were chosen from three independent experiments, bar = 1 μm, and the amplified images were chosen as indicated by black rectangles. Quantification of dendritic spine density (b) and the ratio of mushroom-type spine (c) were performed by NIH ImageJ software. d Immunohistochemistry of presynaptic synaptophysin. e Western blot of postsynaptic PSD95 and quantification of synaptophysin and PSD95. *P < 0.05, **P < 0.01, compared with con; #P < 0.05, ##P < 0.01, compared with sco group
Normal synaptic function relies on the stable expression of synaptic proteins, such as PSD95, in the postsynapse and synaptophysin in the presynapse. Therefore, we evaluated the expression of PSD95 and synaptophysin by using western blotting and immunohistochemistry. We found that scopolamine dramatically suppresses PSD95 expression and synaptophysin staining in the hippocampus, and melatonin completely reverses these effects (Fig. 3d, e). These data suggested that melatonin supplementation could reverse scopolamine-induced synaptic disorder.
Melatonin Activates the EPACs/miR-124/Egr1 Pathway to Rescue Scopolamine-Induced Memory Deficits
In a genomic array analysis in the scopolamine-injected hippocampus, the mRNA levels of Ras-like GTPase superfamily proteins, including cAMP-regulated guanine nucleotide exchange factor I (also known as EPAC1) and Ras-related GTP-binding protein (RagA), changed significantly [18]. Because signals mediated by these proteins are crucial for memory formation and synaptic plasticity, it suggested the possible involvement of these proteins in scopolamine-induced memory deficits. Melatonin also affects the expression of cAMP-related proteins [19]; therefore, we proposed that melatonin might activate cAMP-related proteins and rescue the memory/synaptic disorder induced by scopolamine.
To test this hypothesis, we first employed real-time PCR to examine the mRNA levels of EPACs (EPAC1 and EPAC2) and RagA, which were altered in the previous reports [18]. We found that EPAC1 and EPAC2 decreased to ~0.61- and ~0.28-fold, respectively, in scopolamine-treated rats compared to vehicle-treated rats. After melatonin supplementation, EPAC1 and EPAC2 recovered to ~0.86- and ~0.92-fold, respectively. Interestingly, melatonin alone did not affect the EPAC1 and EPAC2 mRNA levels. We also found similar alterations in EPAC protein expression by using immunoblot analyses (Fig. 4c, d). In addition, RagA mRNA decreased to ~0.72-fold after scopolamine was administered but there was no rescue with the addition of melatonin. These data suggested that EPACs, but not RagA, might be involved in the memory prevention effects of melatonin.

a Melatonin activates the EPACs/miR-124/Egr1 pathway. Relative quantity of EPAC1, EPAC2, and RagA by real-time PCR. b Relative quantity of miR-124 by real-time PCR. c, d Protein levels of EPAC1, EPAC2, and Egr1 between groups were measured by western blot (c) and quantitative analysis (d). *P < 0.05, **P < 0.01, compared with con; #P < 0.05, ##P < 0.01, compared with sco group
In our recent report, we identified that aberrant regulation of miR-124 and Egr1 translation mediates the learning and memory deficits that result from a lack of EPACs [20]. We further explored whether this signaling pathway is involved in the effects of melatonin on scopolamine-induced memory deficits. Using real-time PCR, we found that miR-124 levels increased significantly in scopolamine-treated rats and supplemental melatonin completely suppressed the increase (Fig. 4b). In further experiments, we confirmed that the substrate of miR-124, Egr1 (an immediate early gene), decreased dramatically after scopolamine treatment. Again, supplemental melatonin successfully restored the downregulated Egr1 protein level (Fig. 4c, d). Thus, our results strongly suggest that melatonin attenuates scopolamine-induced memory/synaptic disorder by rescuing the EPACs/miR-124/Egr1 pathway.
Discussion
Understanding the mechanisms that underlie memory deficits will benefit drug discovery for many neurological diseases. Scopolamine-induced memory impairment is a well-established, short duration amnesia model that represents dementia and can be used for drug screening [21]. Many studies including two microarray analyses have been undertaken to determine the signaling pathways mediating memory dysfunction after scopolamine treatment [18, 22]. On the basis of these data, many important molecules have been suggested to be affected by scopolamine in the hippocampus, including some cAMP-related proteins such as EPACs, RagA, as well as the immediate response gene Egr1. Here, we report that scopolamine decreased the level of EPACs and suppressed Egr1 expression, which in turn caused memory impairment.
As a new alternative cAMP target, EPACs are crucial for many cAMP actions including insulin secretion, cardiac contraction, and vascular permeability. EPACs are widely distributed in the brain, including the hippocampus, and their role in memory has been recently revealed by genetic deletion mutant studies or pharmacological regulation [23]. EPAC null mice display severe deficits in synaptic plasticity, spatial memory, and social interactions. Injection of an EPAC activator to the hippocampus improves contextual fear memory retrieval ([24]; Ouyang et al. [25]). EPAC-mediated signaling enhances the formation of long-term memory in the hippocampus independent of protein kinase A [26], suggesting it has a critical role in memory formation. Using electrophysiological recordings, it has been shown that EPAC activation enhanced LTP [27] whereas EPAC−/− neurons exhibit LTP impairments [20]. In another study, EPACs induced long-term depression in hippocampal CA1 excitatory synapses in a p38MAPK-, Ca2+-, and protein synthesis-dependent manner [28]. EPACs are also enriched in the synaptic compartment, indicating a role in synaptic plasticity. The EPAC activator 8-pCPT-2′-O-Me-cAMP [ESCA (1)] enhances neurotransmitter release in excitatory central synapses [29]. In addition, EPAC2 plays an important role in synaptic remodeling and spine structure [30]. The well-known downstream effector of EPACs is Rap1 [31]. In our recent publication, we demonstrated that Rap1 acts as the downstream factor of EPACs and physically restricts miR-124 transcription by inhibiting its upstream regulatory element [20]. This further extends our understanding of EPACs signaling in memory formation and epigenetic regulation. In HEK-293 cells stably expressing the M3 muscarinic acetylcholine receptor, EPAC activation is essential for multiple cellular functions mediated by Ras-related GTPase R-Ras activation, implying an intrinsic link of cholinergic function with EPACs physiologically [32]. In this study, we found that although EPACs and RagA are downregulated in the scopolamine-treated hippocampus, only EPACs are restored by melatonin. This result suggests that loss of function of EPACs plays an important role in cholinergic system dysfunction-induced memory deficits.
miRNAs are small noncoding RNAs that act as posttranscriptional regulators of gene expression and are important for synapse development and plasticity [12]. miR-124 is a brain-enriched miRNA that promotes neuronal differentiation [33]. In EPACs null mutant, miR-124 is upregulated and silences Egr1 expression. We also found that in scopolamine-treated rats, EPACs/miR-124/Egr1 signaling was damaged but it could be retrieved by melatonin. The role of miRNAs in memory, synapse, and circadian rhythm has been well elucidated. miR-124 is also abundant in the pineal gland and targets Mat2a, an enzyme for the Asmt-catalyzed O-methylation of N-acetylserotonin to form melatonin [34]. A previous report suggested melatonin exposure of a human breast cell line caused alterations of multiple miRNAs but without miR-124 [13]. We found that melatonin could downregulate miR-124 level by promoting EPAC expression, indicating a novel regulatory approach by this hormone in the nervous system. We propose this discrepancy may due to a different expression profile of miR-124 in different cell types.
As a free-radical scavenger, melatonin has been suggested to be neuroprotective in the central nervous system. It reduces reactive oxygen species production induced by chronical β-amyloid injection in the mice hippocampus [35], restores mitochondrial dysfunction and neuronal death induced by toxic PrP (106–126) peptide [36], and attenuates gray and white matter damage in a mouse model of transient focal cerebral ischemia [37]. Thus, melatonin has been recommended in the therapy of multiple neural diseases, including Alzheimer’s disease and Parkinson’s disease. These diseases are characterized by memory impairments, and melatonin supplements benefit memory recovery. For example, decreased melatonin in the serum and cerebrospinal fluid was found in AD patients and the cognitive function of these patients was improved after melatonin supplementation [38, 39]. In this study, we demonstrate that melatonin restores synaptic plasticity, as well as memory, which are damaged by scopolamine. We firstly reported that melatonin supplement rescued spinopathy induced by scopolamine, consistent with a pervious study regarding melatonin in dendrite formation and complexity in the hilus region of organotypic hippocampal slice cultures [5]. Melatonin also increased the expression PSD95, a member of the PSD subfamily of the membrane-associated guanylate kinase proteins that interact with N-methyl-d-aspartate and 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl) propanoic acid receptors to regulate their membrane localization and neuronal signaling in the dendritic spine that contains postsynaptic compartment. Combined with the rescue effect of LTP, we hypothesize that melatonin administration will be a useful approach to reverse the synaptic disorder in neurological diseases. It is interesting that melatonin almost completely restored the levels of EPAC1, EPAC2, and Egr1, and of PSD 95 and synaptophysin and the dendritic spines but only partially rescues the disorder of synaptic plasticity induced by scopolamine. We presume that other mechanisms that are important to the activity-dependent synaptic plasticity, such as activity-dependent vesicle release in the presynaptic loci or the traffic of glutamate receptors in the postsynaptic compartment [40, 41], may be involved in the effect of melatonin, because the expression of those synaptic proteins represents the normal synaptic structure, which is the basis for synaptic transmission. A further separated study will be performed for this issue.
In conclusion, we found that melatonin supplementation reverses EPACs/miR-124/Egr1 signaling and rescues the synaptic disorder and memory deficit induced by scopolamine. This will provide a new therapeutic approach for the treatment of neurological diseases with memory impairments.
References
Chang CF, Huang HJ, Lee HC, Hung KC, Wu RT, Lin AM (2012) Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and alpha-synuclein aggregation. J Pineal Res 52:312–321. doi:10.1111/j.1600-079X.2011.00945.x
Zhu LQ, Wang SH, Ling ZQ, Wang DL, Wang JZ (2004) Effect of inhibiting melatonin biosynthesis on spatial memory retention and tau phosphorylation in rat. J Pineal Res 37:71–77. doi:10.1111/j.1600-079X.2004.00136.xJPI136 [pii]
Zhou JN, Liu RY, Kamphorst W, Hofman MA, Swaab DF (2003) Early neuropathological Alzheimer's changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J Pineal Res 35:125–130
Ramirez-Rodriguez G, Ortiz-Lopez L, Dominguez-Alonso A, Benitez-King GA, Kempermann G (2010) Chronic treatment with melatonin stimulates dendrite maturation and complexity in adult hippocampal neurogenesis of mice. J Pineal Res 50:29–37. doi:10.1111/j.1600-079X.2010.00802.x
Dominguez-Alonso A, Ramirez-Rodriguez G, Benitez-King G (2012) Melatonin increases dendritogenesis in the hilus of hippocampal organotypic cultures. J Pineal Res 52:427–436. doi:10.1111/j.1600-079X.2011.00957.x
Gonzalez-Burgos I, Letechipia-Vallejo G, Lopez-Loeza E, Morali G, Cervantes M (2007) Long-term study of dendritic spines from hippocampal CA1 pyramidal cells, after neuroprotective melatonin treatment following global cerebral ischemia in rats. Neurosci Lett 423:162–166. doi:10.1016/j.neulet.2007.06.050
Pascual R, Bustamante C (2010) Melatonin promotes distal dendritic ramifications in layer II/III cortical pyramidal cells of rats exposed to toluene vapors. Brain Res 1355:214–220. doi:10.1016/j.brainres.2010.07.086
Saxena G, Bharti S, Kamat PK, Sharma S, Nath C (2009) Melatonin alleviates memory deficits and neuronal degeneration induced by intracerebroventricular administration of streptozotocin in rats. Pharmacol Biochem Behav 94:397–403. doi:10.1016/j.pbb.2009.09.022
Shen YX, Wei W, Yang J, Liu C, Dong C, Xu SY (2001) Improvement of melatonin to the learning and memory impairment induced by amyloid beta-peptide 25–35 in elder rats. Acta Pharmacol Sin 22:797–803
Yang X et al (2010) Melatonin ameliorates Alzheimer-like pathological changes and spatial memory retention impairment induced by calyculin A. J Psychopharmacol 25:1118–1125. doi:10.1177/0269881110367723
Agrawal R, Tyagi E, Shukla R, Nath C (2008) Effect of insulin and melatonin on acetylcholinesterase activity in the brain of amnesic mice. Behav Brain Res 189:381–386. doi:10.1016/j.bbr.2008.01.015
Schratt G (2009) microRNAs at the synapse. Nat Rev Neurosci 10:842–849. doi:10.1038/nrn2763
Lee SE, Kim SJ, Youn JP, Hwang SY, Park CS, Park YS (2011) MicroRNA and gene expression analysis of melatonin-exposed human breast cancer cell lines indicating involvement of the anticancer effect. J Pineal Res 51:345–352. doi:10.1111/j.1600-079X.2011.00896.x
Morris R (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11:47–60
Yin YY et al (2010) Acetyl-l-carnitine attenuates okadaic acid induced tau hyperphosphorylation and spatial memory impairment in rats. J Alzheimers Dis 19:735–746. doi:10.3233/JAD-2010-1272
Zhu LQ et al (2007) Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J Neurosci 27:12211–12220. doi:10.1523/JNEUROSCI.3321-07.2007
Segal M (2005) Dendritic spines and long-term plasticity. Nat Rev Neurosci 6:277–284. doi:10.1038/nrn1649
Hsieh MT, Hsieh CL, Lin LW, Wu CR, Huang GS (2003) Differential gene expression of scopolamine-treated rat hippocampus-application of cDNA microarray technology. Life Sci 73:1007–1016
Roy D, Belsham DD (2002) Melatonin receptor activation regulates GnRH gene expression and secretion in GT1-7 GnRH neurons. Signal transduction mechanisms. J Biol Chem 277:251–258. doi:10.1074/jbc.M108890200 M108890200 [pii]
Yang Y et al (2012) EPAC null mutation impairs learning and social interactions via aberrant regulation of miR-124 and Zif268 translation. Neuron 73:774–788. doi:10.1016/j.neuron.2012.02.003
Verloes R, Scotto AM, Gobert J, Wulfert E (1988) Effects of nootropic drugs in a scopolamine-induced amnesia model in mice. Psychopharmacol (Berl) 95:226–230
Brouillette J, Young D, During MJ, Quirion R (2007) Hippocampal gene expression profiling reveals the possible involvement of Homer1 and GABA(B) receptors in scopolamine-induced amnesia. J Neurochem 102:1978–1989. doi:10.1111/j.1471-4159.2007.04666.x
Laurent AC, Breckler M, Berthouze M, Lezoualc'h F (2012) Role of Epac in brain and heart. Biochem Soc Trans 40:51–57. doi:10.1042/BST20110642
Ostroveanu A, van der Zee EA, Eisel UL, Schmidt M, Nijholt IM (2009) Exchange protein activated by cyclic AMP 2 (Epac2) plays a specific and time-limited role in memory retrieval. Hippocampus 20:1018–1026. doi:10.1002/hipo.20700
Ouyang M, Zhang L, Zhu JJ, Schwede F, SA T (2008) Epac signaling is required for hippocampus-dependent memory retrieval. Proc Natl Acad Sci USA 105:11993–11997
Ma N, Abel T, Hernandez PJ (2009) Exchange protein activated by cAMP enhances long-term memory formation independent of protein kinase A. Learn Mem 16:367–370. doi:10.1101/lm.1231009
Gelinas JN, Banko JL, Peters MM, Klann E, Weeber EJ, Nguyen PV (2008) Activation of exchange protein activated by cyclic-AMP enhances long-lasting synaptic potentiation in the hippocampus. Learn Mem 15:403–411. doi:10.1101/lm.830008
Ster J et al (2009) Epac mediates PACAP-dependent long-term depression in the hippocampus. J Physiol 587:101–113. doi:10.1113/jphysiol.2008.157461
Gekel I, Neher E (2008) Application of an Epac activator enhances neurotransmitter release at excitatory central synapses. J Neurosci 28:7991–8002. doi:10.1523/JNEUROSCI.0268-08.2008
Woolfrey KM et al (2009) Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nat Neurosci 12:1275–1284. doi:10.1038/nn.2386
Enserink JM et al (2004) The cAMP-Epac-Rap1 pathway regulates cell spreading and cell adhesion to laminin-5 through the alpha3beta1 integrin but not the alpha6beta4 integrin. J Biol Chem 279:44889–44896. doi:10.1074/jbc.M404599200 M404599200 [pii]
Lopez De Jesus M et al (2006) Cyclic AMP-dependent and Epac-mediated activation of R-Ras by G protein-coupled receptors leads to phospholipase D stimulation. J Biol Chem 281:21837–21847. doi:10.1074/jbc.M604156200
Makeyev EV, Zhang J, Carrasco MA, Maniatis T (2007) The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell 27:435–448. doi:10.1016/j.molcel.2007.07.015
Clokie SJ, Lau P, Kim HH, Coon SL, Klein D (2012) Micro RNAs in the pineal gland: mir-483 regulates melatonin synthesis by targeting arylalkylamine N-acetyltransferase. J Biol Chem. doi:10.1074/jbc.M112.356733
Masilamoni JG et al (2008) The neuroprotective role of melatonin against amyloid beta peptide injected mice. Free Radic Res 42:661–673. doi:10.1080/10715760802277388
Jeong JK, Moon MH, Lee YJ, Seol JW, Park SY (2012) Melatonin-induced autophagy protects against human prion protein-mediated neurotoxicity. J Pineal Res. doi:10.1111/j.1600-079X.2012.00980.x
Lee EJ et al (2005) Melatonin attenuates gray and white matter damage in a mouse model of transient focal cerebral ischemia. J Pineal Res 38:42–52. doi:10.1111/j.1600-079X.2004.00173.x
Asayama K, Yamadera H, Ito T, Suzuki H, Kudo Y, Endo S (2003) Double blind study of melatonin effects on the sleep-wake rhythm, cognitive and non-cognitive functions in Alzheimer type dementia. J Nihon Med Sch 70:334–341
Uchida K, Okamoto N, Ohara K, Morita Y (1996) Daily rhythm of serum melatonin in patients with dementia of the degenerate type. Brain Res 717:154–159
Crozier RA, Bi C, Han YR, Plummer MR (2008) BDNF modulation of NMDA receptors is activity dependent. J Neurophysiol 100:3264–3274. doi:10.1152/jn.90418.2008
Evans GJ, Cousin MA (2007) Activity-dependent control of slow synaptic vesicle endocytosis by cyclin-dependent kinase 5. J Neurosci 27:401–411. doi:10.1523/JNEUROSCI.3809-06.2007
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (30971478, 91132725, 31201011), the New Century Excellent Talent of Education Ministry (NCET-10-0421), the Ministry of Science and Technology of China (2011DFG33250), the New Investigator Research Grant of Alzheimer’s Association (NIRG-11-205737), and the Fundamental Research Funds for the Central Universities, HUST, (nos. 0118510011 and 0118510019).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Xiong Wang and Zhi-Hao Wang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Wang, X., Wang, ZH., Wu, YY. et al. Melatonin Attenuates Scopolamine-Induced Memory/Synaptic Disorder by Rescuing EPACs/miR-124/Egr1 Pathway. Mol Neurobiol 47, 373–381 (2013). https://doi.org/10.1007/s12035-012-8355-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-012-8355-9