
Abstract
All sensory receptors adapt. As the mean level of light or sound or odor is altered, the sensitivity of the receptor is adjusted to permit the cell to function over as wide a range of ambient stimulation as possible. In a rod photoreceptor, adaptation to maintained background light produces a decrease (or “sag”) in the response to the prolonged illumination, as well as an acceleration in response decay time and a Weber–Fechner-like decrease in sensitivity. Earlier work on salamander indicated that adaptation is controlled by the intracellular concentration of Ca2+. Three Ca2+-dependent mechanisms were subsequently identified, namely, regulation of guanylyl cyclase, modulation of activated rhodopsin lifetime, and alteration of channel opening probability, with the contribution of the cyclase thought to be the most important. Later experiments on mouse that exploit the powerful techniques of molecular genetics have shown that cyclase does indeed play a significant role in mammalian rods, but that much of adaptation remains even when regulation of cyclase and both of the other proposed pathways have been genetically deleted. The identity of the missing mechanism or mechanisms is unclear, but recent speculation has focused on direct modulation of spontaneous and light-activated phosphodiesterase.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As John Maynard Keynes so justly observed, “The difficulty lies not in the new ideas, but in escaping from the old ones.” The mechanism of vertebrate photoreceptor light adaptation has been so thoroughly described and reviewed [see for example 1–6] that it would seem no longer of current interest. The discovery that adaptation is mediated primarily if not exclusively by a change in intracellular Ca2+ concentration [7–9] and the identification of the Ca2+-dependence of guanylyl cyclase led to the proposal that most [10] if not all [11] of adaptation is produced by a Ca2+-dependent acceleration of the guanylyl cyclase and a speeding up of the turnover of cyclic guanosine monophosphate (cGMP), the second messenger of photoreceptor transduction.
Much of this earlier research was done on amphibian photoreceptors, which are large and relatively easy to manipulate. More recently, physiologists have increasingly turned to mouse because of powerful techniques for manipulating the mouse genome. Nearly all of the proteins used in photoreceptor transduction are unique to the photoreceptors; they can be knocked out, over- or under-expressed, and mutated or otherwise altered, and the worst that will happen is that the mouse becomes blind. But blind mice feed and reproduce and can be quite active, as the farmer's wife found to her chagrin. When results emerged from these newer studies, it became clear that adaptation cannot be entirely the result of modulation of guanylyl cyclase, as the amphibian experiments seemed to have indicated, because much of adaptation in mouse rods survives the genetic ablation of Ca2+ control of the cyclase [12–14]. Moreover, the adaptation that remains cannot be explained by any alternative mechanism of Ca2+-dependent modulation so far described [14], indicating that there is at least one major component of light adaptation in rods that remains to be elucidated.
In this review, I briefly summarize the experiments on amphibian rods that led to our earlier understanding of adaptation and then focus on the newer results from mouse that have brought those former interpretations into question. The challenge for the future will be to escape from the old ideas that have so long dominated our thinking and to arrive at some new understanding based on more recent studies. This will require not only new interpretation but also considerable experimentation to reveal novel mechanisms that modulate photoreceptor transduction during exposure to steady light.
What is Background Adaptation?
When the outer segment of a rod is exposed to light (see Fig. 1), the activated rhodopsin molecules (Rh*) interact with the heterotrimeric G protein transducin on the rod disk membrane to produce activated transducin alpha subunit bound to GTP (TαGTP). The TαGTP then binds to the inhibitory gamma subunit of the enzyme phosphodiesterase 6 (PDEγ), which displaces the inhibitory subunits from the PDE6 catalytic α and β subunits, allowing the catalytic subunits to hydrolyze cGMP. In darkness, the concentration of cGMP is relatively elevated, and the cGMP maintains channels in the rod plasma membrane in an open state that is conductive to cations, principally Na+ and Ca2+. These cations enter the rod in darkness and produce a tonic depolarization. As the concentration of cGMP in the rod is decreased by activated PDE6 in the light, the probability of opening of cGMP-gated channels in the rod plasma membrane is decreased, the channels close, and the rod membrane potential hyperpolarizes. The recovery of the rod following illumination requires inactivation of Rh* by phosphorylation by rhodopsin kinase and binding of arrestin, inactivation of PDE6 by the hydrolysis of TαGTP to TαGDP assisted by a complex of GTPase-activating proteins (the GAP complex), and resynthesis of cGMP by guanylyl cyclase [for recent reviews of the biochemistry of transduction, see 4, 5, 15–18].

Mechanism of transduction in vertebrate rod photoreceptor. a Rod morphology. The outer segment contains disk membranes which have most of the proteins necessary for transduction and adaptation. The inner segment is the metabolic part of the cell. b Major proteins and mechanisms. Light (hν) activates rhodopsin in the disks of a rod outer segment, forming its excited Rh* conformation. The heterotrimeric G-protein transducin binds to Rh* and produces activated transducin alpha subunit bound to GTP (TαGTP). The TαGTP then binds to the γ inhibitory subunit of the phosphodiesterase 6 (PDEγ), which displaces the PDEγ from the catalytic α and β subunits and activates the enzyme. The catalytic subunits of PDE6 hydrolyze guanosine 3',5'-cyclic monophosphate (cGMP), and this activity decreases the cGMP concentration and causes the cGMP-gated Na+/Ca2+ channels to close. Ca2+ entering through the rod plasma membrane is extruded by means of a Na+/Ca2+–K+ exchanger, and the entering Na+ is extruded by a Na+/K+ ATPase in the inner segment (not shown). The GTPase-activating protein (GAP) complex is a group of three proteins, including RGS9, which accelerate the hydrolysis of GTP by Tα. GTP guanosine triphosphate, GDP guanosine diphosphate, GMP guanosine monophosphate, P i inorganic phosphate
When a rod is exposed to steady light, the closure of the channels produces a decrease in inward current. In Fig. 2a, I show responses of a mouse rod to prolonged illumination, recorded by placing a suction electrode over the rod outer segment (see Fig. 1a). The suction electrode registers the current entering the outer segment through the cGMP-gated channels and shows how the current first decreases and then increases during maintained illumination. If the light is sufficiently bright, as in Fig. 2a, all of the channels are initially closed, and the circulating current briefly goes to zero. The rod then adapts to the steady illumination, and some channels open back up, producing first a rapid (Fig. 2a, inset) and then a more prolonged sag in current. The rapid and slow decays are both exponential, with time constants in mouse rods of a few hundred milliseconds and a few tens of seconds [14, 19]. Depending upon the intensity, as much as a third of the dark circulating current can recover in the presence of steady light, permitting the rod to continue to respond even in bright illumination.

Adaptation of WT mouse rods. a Data traces are suction-electrode recordings of superimposed means of currents of seven WT rods each exposed three times to steps of light 10, 30, 60, and 120 s in duration, of intensity 440 photons μm-2 s-1, and beginning at t = 0. Inset, same data at faster time resolution showing only the first 4 s. b Flash responses recorded with suction electrodes in background light. Shown are means from five presentations each of ten WT rods to 20-ms flashes beginning at t = 0; intensity was kept at 453 photons μm-2 for flashes in backgrounds of the following intensities (in photons μm-2 s-1): 0, 12, 38, 118, 438, and 1354. Responses have been normalized cell by cell to the peak amplitude of the response and averaged. The most slowly decaying response is with no background; decay time course progressively accelerated as background intensity was increased. c Weber–Fechner plot. Sensitivity (S F in pA photons μm-2) was calculated in darkness and in the presence of background light as the peak amplitude of the response in the linear range divided by the flash intensity. The graph gives the mean sensitivity divided by the sensitivity in the absence of a background (\( S_F^D \)) as a function of background intensity (I B), averaged from ten WT rods. The solid line is the best-fitting Weber–Fechner function for WT rods given by Eq. (1), with I 0 = 77 photons μm-2 s-1
If flashes are given in the presence of the background light, the decay time of the response can be seen to be accelerated. In Fig. 2b, I have plotted normalized responses to the same 20-ms flash intensity in the absence of a background (most slowly decaying response in the figure) and in the presence of gradually increasing background intensities. The decay of the flash response in its linear range can be characterized by a single exponential time constant of recovery, τ REC, which in the absence of a background has a value for mouse rods of about 200 ms [see for example 20, 21]. This time constant decreases in bright background light by at least a factor of 2 to about 100 ms; backgrounds also produce a similar decrease in the limiting time constant of response decay, usually symbolized as τ D [14, 22]. The acceleration of the time course of decay was first recognized to be an important feature of light adaptation by Fuortes and Hodgkin almost 50 years ago [23], and it is, in part, responsible for the well-known behavioral increase in flicker-fusion frequency in background light. Rods decay slowly in darkness with a long integration time in order to sum as many photons as possible. In brighter light when photons are abundant, the integration time decreases so that the visual system can more readily detect change and motion.
A third consequence of exposure to background light is a regulation of flash sensitivity, which allows rods to avoid saturation and to continue to respond within the dynamic range of rod vision. The flash sensitivity follows a well-known relationship, which I have plotted in Fig. 2c. The data points in that figure are the mean sensitivity of wild-type (WT) mouse rods, measured as the amplitude of the response divided by the intensity of the flash and normalized to the sensitivity in darkness. The sensitivity has been plotted as a function of the intensity of the background light. The curve fitted to the data is called the Weber–Fechner relation,
where S F is the flash sensitivity of the rod in background light, \( S_F^D \) is the flash sensitivity of the dark-adapted rod, I B is the intensity of the background, and I 0 is a constant. The sensitivity in the brightest background deviates from the curve because in light this bright the PDE6 is hydrolyzing cGMP so rapidly that the rod can maintain very few of the channels in an open state. Even brighter backgrounds produce complete saturation of rod vision, such that nearly all of the channels are continuously closed and no detectable response to superimposed flashes can be recorded. Saturation is an important feature of duplex visual systems, which permits cone vision to replace rod vision in bright ambient illumination.
The changes in steady inward current, response decay time, and sensitivity illustrated in Fig. 2 all occur in relatively dim light. The brightest steady light in Fig. 2c produced fewer than 2,000 activated rhodopsin molecules per second, amounting to less than 2 × 105 Rh* after 100 s of exposure. Since a WT mouse rod contains on the order of 2.5 × 107 rhodopsin molecules [24], the brightest light in Fig. 2 would have bleached less than 1% of the total amount of visual pigment; thus, background adaptation is unlikely to be affected by a reduction in the concentration of visual pigment produced by bleaching. Bright light has been shown to produce translocation of both transducin and arrestin between the rod inner and outer segments [see 25], which could conceivably also contribute to changes in photoreceptor sensitivity [26], but the light intensity required to produce significant migration of these proteins is much brighter than the background lights used in Fig. 2 [see for example 27], indicating that translocation of arrestin and transducin does not contribute to background adaptation in a mammalian rod.
Adaptation is Produced by Ca2+-dependent Modulation of Transduction
Adaptation to background light in a rod requires a second messenger. In Fig. 2c, the best-fitting value of the constant I 0 is 77 photons μm-2 s-1, which is equivalent to 35–40 Rh* per second. Changes in sensitivity occur rather quickly after turning on the background: Much of the sensitivity change of a mouse rod for dim background light is complete in the first 2–3 s (unpublished observations). Now, from Eq. (1), we can see that I 0 is the intensity of the background that reduces the sensitivity of the rod by half. It is therefore possible to reduce sensitivity by a factor of 2 from the excitation of something like 100 rhodopsins, and since a mouse rod has about 750 disks, light exciting single rhodopsin molecules in only a fraction of the disks can produce a considerable reduction in sensitivity. This effect could only occur if a second messenger diffused within the rod so that an Rh* in one disk altered the sensitivity in neighboring disks. Measurements in amphibian receptors indicate that an adaptatory signal can spread several micrometers up and down the rod outer segment [28].
Initial experiments with the Ca chelator BAPTA suggested that the second messenger of adaptation may be Ca2+ [29], and this suggestion was more thoroughly explored with a novel technique of rapidly perfusing the rod outer segment with a solution having low Ca2+ and all of the external Na+ replaced with guanidinium [7–9]. Because guanidinium can carry current through the cGMP-gated channels but cannot support the Na+/K+–Ca2+ exchange mechanism responsible for extruding Ca2+ from the rod outer segment (see Fig. 1b), perfusion with this solution makes possible the recording of light responses from the rod with minimal entry or exit of calcium, thus keeping the Ca2+ concentration nearly constant. When changes in Ca2+ are minimized in this way, every aspect of light adaptation in an amphibian rod is prevented: The “sag” in current to maintained background light no longer occurs, response decay is no longer accelerated, and sensitivity declines with background intensity not according to the Weber–Fechner relationship as in Fig. 2c, but by a theoretical curve of simple summation of responses to absorbed photons without any active adjustment of sensitivity.
In the same issue of Nature in which these experiments were first described, Koch and Stryer [30] reported a highly co-operative dependence of the rod enzyme guanylyl cyclase on Ca2+ concentration, confirming the less quantitative observations of Lolley and Racz [31]. Guanylyl cyclase is a membrane-bound enzyme in the rod outer segment that synthesizes the second messenger cGMP (see Fig. 1). Koch and Stryer showed that the cyclase is activated by low Ca2+, and since the closing of the channels in light reduces Ca2+ influx and decreases the free-Ca2+ concentration [see for example 32], the activity of the cyclase increases during illumination, producing negative feedback: Light activates PDE6 and decreases the cGMP concentration, which closes the channels, decreases Ca2+, and activates the cyclase, thus helping to restore the concentration of cGMP initially reduced by light. The calcium dependence of the cyclase is mediated by low-molecular-weight, calcium-binding proteins called guanylyl-cyclase-activating proteins, or GCAPs [see 33], of which the mouse rod has two, GCAP1 and GCAP2, which both may contribute to cyclase regulation [34, 35].
Additional experimentation revealed two further Ca2+-dependent mechanisms of modulation: one produced by the Ca2+-binding protein S-modulin or recoverin, which controls the rate of the rhodopsin kinase [36, 37] and modulates the lifetime of Rh* [38], and a second mechanism of Ca2+-dependent modulation of the cyclic nucleotide-gated channels by the Ca2+-binding protein calmodulin [39]. Both of these further mechanisms were proposed also to contribute to adaptation in background light. As Ca2+ concentration declines in the light, recoverin was hypothesized to fall off the rhodopsin kinase, accelerating phosphorylation of rhodopsin and decreasing the lifetime of Rh*. In addition, the decrease in Ca2+ produces a calmodulin-dependent increase in the probability of opening of the cGMP-gated channels [40–42], compensating, to some extent, for the decrease in channel opening caused by the decrease in cGMP in the light.
Which of these three Ca2+-dependent pathways is the most important for regulating response waveform and sensitivity in background light? The first attempt to estimate the relative significance of these mechanisms was made by Yiannis Koutalos, King-Wai Yau, and their collaborators [43, 44], who used a truncated rod outer segment preparation from cells isolated from salamander retina. The truncated preparation made it possible for the experimenters to record light responses from rods while internally perfusing the rod cytoplasm. By cleverly manipulating the concentration of nucleotides, Koutalos et al. were able to estimate separately the Ca2+ dependence of the guanylyl cyclase, the channels, and light-dependent phosphodiesterase activity. They showed that the modulation of the channels made a minimal contribution to regulation of sensitivity and that the predominant mechanism particularly at dim to medium intensity backgrounds was regulation of guanylyl cyclase; in bright background light, a significant contribution was also made by Ca2+-dependent regulation of phosphodiesterase activity, but Koutalos and collaborators [45] could not distinguish between an effect of Ca2+ on the lifetime of Rh* mediated by Ca2+-dependent binding of recoverin and a direct effect of Ca2+ on the PDE6 enzyme itself.
The relative importance of the various proposed mechanisms for light adaptation was also investigated by Nikonov et al. [11], who used careful measurements of rod responses (again from salamander) and extensive theoretical calculation to conclude that nearly all of background adaptation can be explained by an acceleration of the turnover of cGMP produced by activation of the PDE6, together with the Ca2+-dependent regulation of the cyclase which increases the light-evoked current and prevents the rod from saturating. As the rate of the PDE6 is increased in background light, a larger change in PDE6 rate would be required to produce a significant change in cGMP level, and this by itself would contribute to the change in sensitivity, as Koutalos and collaborators had also realized [45d]. The acceleration of turnover of cGMP could also increase the rate of decay of the flash response as in Fig. 2b [see also Appendix of 2]. Nikonov and collaborators hypothesized that the contributions of regulation of rhodopsin lifetime and channel open probability were minor and of much less significance.
Mouse Mutant Lines and Genetic Deletion of Ca2+ Regulation of Guanylyl Cyclase
The experiments that I have described up to this point from salamander rods led to nearly universal agreement that most of adaptation was produced by Ca2+-dependent regulation of cyclase and an increase in the steady rate of the PDE6 with accelerated turnover of cGMP. Universal agreement can mean that everyone is right; it can also mean that everyone is wrong. New approaches often lead to new conclusions, and this generalization is nowhere more relevant than for our understanding of vertebrate photoreceptor light adaptation.
What happened next is that mice became available with targeted deletions of the Ca+-dependent mechanisms thought to be responsible for regulation of sensitivity. The first of these lines to be produced was one in which both of the two GCAP proteins in mouse rods were deleted [12]. Since deletion of the GCAPs removes all Ca2+-dependent regulation of the cyclase [12], rods from these GCAPs−/− animals should no longer be able to keep their photocurrents from saturating even in relatively dim light, and all the regulatory mechanisms of adaptation should be eliminated, much as in the salamander experiments of rods exposed to low Ca2+/zero Na+ solution.
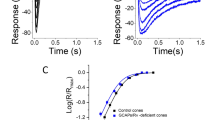
In Fig. 3, I show, for convenience, recordings made from GCAPs−/− rods by Mike Woodruff in my laboratory, taken from the paper of Chen et al. [14], but similar observations were made in part from earlier studies by Mendez et al. [12] and by Burns et al. [13]. The recordings in Fig. 3 are presented in the same format as that for WT rods in Fig. 2. Fig. 3a shows that GCAPs−/− rods exposed to steady light lack the rapid component of response decay of the inset of Fig. 2a, but there is still a prominent slow decay again with a time constant of tens of seconds, which restores as much as a third of the circulating current in steady light. Thus, even in the absence of Ca2+-dependent cyclase regulation, the channels can somehow reopen during long exposures to steady light. When the background light is turned off, there are prominent overshoots during which the current becomes transiently larger than in darkness before the illumination [see also 13].

Adaptation of GCAPs−/− mouse rods [data replotted from 14]. Both genes for GCAP proteins were deleted from genome. a Data traces are suction-electrode recordings of superimposed means of currents from three presentations each of five GCAPs−/− rods to steps of light of intensity 38 photons μm-2 s-1 beginning at t = 0 for the following durations: 10, 30, 60, and 120 s. b Flash responses recorded with suction electrodes in background light. Shown are means from five presentations each of seven GCAPs−/− rods to 20-ms flashes beginning at t = 0; intensity was kept at 17 photons μm-2 for flashes in backgrounds of the following intensities (in photons μm-2 s-1): 0, 4, 13, 38, and 118. Responses have been normalized cell by cell to the peak amplitude of the response and averaged. The most slowly decaying response is with no background; decay time course progressively accelerated as background intensity was increased. c Weber–Fechner plot as in Fig. 2c. The graph gives the mean relative sensitivity as a function of background intensity, averaged from 10 WT rods (filled circles), 14 rods lacking the protein recoverin (filled squares), 13 rods from which the Ca2+–calmodulin binding site of the cGMP-gated channel had been deleted (filled triangles), and 5 GCAPs−/− rods (open circles). The solid line is the best-fitting Weber–Fechner function for WT rods given by Eq. (1), with I 0 = 77 photons μm-2 s-1. The black dashed and dotted curves give theoretical predictions of change of sensitivity produced by simple saturation (dashed) or in the absence of cyclase and PDE feedback (dotted)
In Fig. 3b, I show normalized responses to 20-ms flashes of the same intensity in the absence of a background light (most slowly decaying response) and in the presence of gradually increasing background intensities, completely analogous to the records of WT rods in Fig. 2b. The decay of the response is uniformly slower than in WT rods because the rate of the cyclase is slower in the absence of the GCAP proteins. The recovery rate can again be characterized by a single exponential time constant, τ REC, which, as for WT rods, is modulated over about a factor of 2: It is about 500 ms in the absence of a background and about 250 ms in the brightest background in which responses were recorded.
Finally, I show measurements of sensitivity as a function of background intensity in Fig. 3c, which replicate the earlier work of Mendez et al. [12]. The filled circles are from WT rods as in Fig. 2c, and the open circles show the mean sensitivity of GCAPs−/− rods. The continuous line is again the Weber–Fechner relation, and it is clear that genetic deletion of the GCAPs alters the relationship between sensitivity and background illumination so that GCAPs−/− rods do not regulate their sensitivity as effectively as WT rods. On the other hand, much of light adaptation remains intact. We can tell that this is so by comparing the open circles to the curve of large dashes, which is the prediction of a simple expression that assumes that all of adaptation has been eliminated and that the rod is simply summing responses of single photons with no active mechanism of sensitivity regulation [12]. The open circles should also be compared to the dotted curve of Chen et al. [14], from a model developed by Dan Tranchina based on earlier models of Hamer and collaborators [44] and of Soo et al. [46]. The Tranchina model successfully predicts Weber–Fechner-like adaptation for WT rods [14], but the dotted curve in Fig. 3c gives the prediction for the case in which all feedback modulation of either cyclase or phosphodiesterase has been eliminated so that there is no active regulation of sensitivity during exposure to steady illumination. This model does however incorporate the small speeding up of response kinetics produced by the steady-state increase in the rate of the phosphodiesterase [2, 11]. Comparison of the data points to either the dashed or dotted curves shows that the sensitivity of the GCAPs−/− rods is many orders of magnitude greater than expected had all of the feedback mechanisms of adaptation been eliminated.
These surprising results demonstrate that elimination of Ca2+ control of the cyclase still preserves a substantial amount of adaptation: Responses continue to sag in steady light, decay time of the flash response continues to accelerate, and sensitivity regulation—though certainly affected by elimination of the GCAPs—is much less altered than anticipated. Can the adaptation that survives deletion of the GCAPs be produced by one of the two other proposed Ca2+-dependent mechanisms? Makino and collaborators [47] recorded from mouse rods in which modulation of Rh* lifetime had been removed by genetic deletion of recoverin, and they found that adaptation was little affected. Responses to steady light continued to show the prominent sag of WT responses, and sensitivity as a function of background intensity was indistinguishable from WT and closely followed the predictions of Eq. (1). We have replicated their experiments with similar results [Fig. 3c, filled squares, from 14]. I note in addition that Krispel et al. [20] reported that there is no effect of a four-fold change in expression level of rhodopsin kinase on sensitivity, and we have now extended their observations to a 70-fold change in kinase expression, again with sensitivity unaltered [unpublished observations and 38]. These results also argue that changes in kinase activity are unlikely to play a significant role in regulation of rhodopsin lifetime. Finally, the deletion of the Ca2+–calmodulin binding site from the cGMP-gated channel has no effect on light adaptation [filled triangles in Fig. 3c, from 14], confirming the earlier claims of Koutalos et al. and of Nikonov et al. that channel modulation makes little or no contribution to light adaptation in rods.
The overshoots of the GCAPs−/− rods in Fig. 3a are of particular significance [see also 13]. They cannot be caused by modulation of rhodopsin lifetime by any conceivable mechanism because there is no possible way in which a change in Rh* lifetime could cause the current at light offset to become even larger than it was before the light was turned on. The production of activated Rh* by light causes a stimulation of the PDE6 (see Fig. 1b), which reduces the cGMP concentration. Altering the lifetime of activated Rh* could modulate the size of the cGMP decrease, but it couldn't cause the cGMP to increase above the resting level in darkness. Burns et al. [13] made the interesting suggestion that the overshoots of the GCAPs−/− rods might be produced by channel modulation. When however we recorded from rods that were GCAPs−/− and also lacked the Ca2+–calmodulin binding site of the cGMP-gated channel [14] so that both cyclase and channel modulation had been eliminated, responses to steady light were very similar to those in Fig. 3c, and the overshoots not only did not disappear, they actually became even larger. These results taken together show that there is at least one important component of light adaptation that cannot be explained by modulation of the cyclase, of Rh* lifetime, or of opening probability of the cGMP-gated channels, but must be produced by some process not as yet elucidated.
What is Missing?
Is there only a single additional mechanism? Is it controlled by Ca2+? The truth is that we do not know. Even though experiments in salamander retina seem to show that all of light adaptation requires a change in intracellular Ca2+ concentration and we have always assumed that adaptation in amphibians and mammals is likely to be the same, we cannot be certain that mammalian rods behave similarly until experiments with low Ca2+/zero Na+ perfusion are done on the much more difficult preparation of isolated mouse rods. Moreover at present, we have no biochemical evidence for mechanisms of further modulation of the cascade by either Ca2+-dependent or Ca2+-independent pathways. In spite of these many uncertainties, I will review recent speculation, which has centered on a role for direct modulation of the PDE6.
I begin with the results of Fig. 3a, showing that currents in steady light still reopen almost to the same extent in GCAP−/− rods as in WT rods, even in the absence of Ca2+ control of the cyclase. This behavior cannot be produced by modulation of Rh* lifetime, at least not entirely, since it is followed by pronounced overshoots in which the current becomes even larger after exposure to the light than in darkness. As I explained earlier, regulation of Rh* lifetime could not produce such an effect. It cannot be produced by channel modulation [14]. That leaves only modulation of the lifetime of TαGTP or of the PDE6 itself as possible mechanisms of the current sag of the GCAPs−/− rods.
Consider, however, the long time constant of return of current to the baseline during decay of the overshoot. This long decay would make it surprising for the overshoot to be produced by modulation of decay of light-activated TαGTP or PDE6, since the TαGTP would be expected to be hydrolyzed and the light-activated PDE6 to subside to its dark level, with a time course much more rapid than the several seconds it takes for the overshoot to return to baseline. This is because the decay of TαGTP and light-activated PDE6 cannot have time constants slower than the time constants of response recovery τ REC and of the dominant time constant of response decay τ D, which are both on the order of 200 ms in darkness and even smaller during [22] or after [48] exposure to bright light. The decay of the overshoot is more likely to be produced by a slow time-course modulation of the rate of decay of spontaneously activated PDE6 [14]. Modulation of the decay of spontaneously activated PDE6 would therefore seem to be a likely mechanism for at least part of the sag of the current in GCAPs−/− rods.
The results of Fig. 3b also seem to support a role of modulation of PDE6, but for a rather different reason. Recent evidence indicates that the rate-limiting step of the recovery of the light response is the GAP-assisted hydrolysis of TαGTP and the decay of light-activated PDE6 [20]. Although this result has been recently disputed [49], these objections seem to have been effectively refuted [50]. One seldom-mentioned observation relevant to this controversy is the acceleration of the falling phase of the light response of rods in which the gamma subunit of the PDE6 has been over-expressed [21]. It is difficult to imagine how over-expression of PDEγ could accelerate response decay unless the decline of light-activated PDE6 activity was responsible for the decline of the light response. The decay of Rh* is likely to be much more rapid than the decay of PDE6, with a time constant certainly less than 55 ms [38] and perhaps even shorter [50].
If the decay of the flash response is set by the decay of light-activated PDE6, then the acceleration of the decay of the flash response in background light (as in Figs. 2b and 3b) is very likely to be caused by acceleration of light-activated PDE6 decay. This conclusion was first argued by Krispel et al. [48], who exposed a mouse rod to a long-duration bright light bleaching about 2% of the rhodopsin. They then observed that, when the light was turned off, the circulating current recovered rapidly in only a few seconds, but responses continued to decay with an accelerated time course much like the light-adapted flash responses in Fig. 2b, and the rate of response decay only slowly returned to the dark-adapted rate over a time course of several minutes. Already suspecting that the limiting time constant for response recovery was set by PDE6 decay, they attributed the changes in τ REC (as well as the changes in the dominant time constant of response decay τ D) during recovery from bright light exposure to modulation of PDE6 decay.
Woodruff et al. [22] then showed that Krispel's results of recovery after bright light exposure can be extended to background adaptation. They demonstrated for the first time [and in contrast to previous results from salamander, see 51] that both τ REC and τ D are accelerated in steady background light. Assuming that the rate constants for the decay of the response are limited by the decay of light-activated PDE6, Woodruff et al. concluded that acceleration of τ REC and τ D in background light can only be the result of the modulation of decay of light-activated PDE. They also showed that a single amino-acid mutation of the PDEγ subunit (T35A) could prevent the acceleration of response decay, also implicating a central role of the PDE6 in modulation of response recovery.
Further indications for a role of PDE6 modulation have come from modeling studies. The experiments of Soo et al. [46] were most successfully modeled if light was hypothesized to produce a slow increase in the decay of light-activated PDE, though no distinction could be made between modulation of Rh* decay and direct modulation of the PDE itself. A model like the one Soo et al. used was also exploited by Daniel Tranchina in the study of Chen et al. [14]. Tranchina showed that both WT and GCAPs−/− mouse rod response waveform and sensitivity could be modeled by assuming that light directly regulates the decay of PDE6. Unlike Soo et al. [46], however, Tranchina found that modulation of only light-activated PDE6 could not explain the responses of WT and GCAPs−/− rods; the decay of both spontaneous and light-activated PDE6 had to be accelerated to produce model calculations that replicated actual recordings.
Where are We Now?
The most likely explanation of light adaptation in mammalian rods, in my view, is that it is produced by GCAP and Ca2+-dependent regulation of guanylyl cyclase, together with modulation of both the spontaneous and light-activated decay of PDE6. There must also be some contribution of increased hydrolysis and turnover of cGMP [2, 11], but Tranchina's model calculations suggest that this contribution in mouse is likely to be small. Regulation of Rh* lifetime and channel modulation are unlikely to play any significant role.
The mechanism of modulation of cyclase is well established, but we know next to nothing about direct modulation of the PDE6. Since the rate-limiting step for the decay of light-activated PDE6 is the GAP-assisted hydrolysis of TαGTP [20], the simplest mechanism for modulating decay of PDE6 is alteration of some reaction at the level of the GAP complex. Spontaneously activated PDE6 could also be modulated by such a mechanism, at least in theory, but experiments by Rieke and Baylor [52] argue that spontaneous activation of PDE in salamander occurs by a process that does not involve transducin but is rather the result of some change in molecular conformation of the PDE enzyme itself. Similar experiments have not been done on mammalian rods, and at present, there is no information for mouse about the molecular origin of spontaneous activation of PDE. We must therefore be open to the possibility that spontaneous and light-activated PDEs are modulated by different mechanisms.
The greatest difficulty that we presently face is that we have no biochemical information indicating how the PDE might be modulated. Once definite possibilities are delimited, new mice can be made to test them; at present, however, we have next to nothing to go on. It is frustrating but also quite exhilarating to realize how little we still know about adaptation in photoreceptors, whose G-protein cascade is nevertheless the best studied of any in the body. Photoreceptors are not alone in this regard: Adaptation in olfactory receptor cells was once thought to be almost entirely the result of channel modulation [53], but recent results indicate that channel modulation plays little if any role [54]. The origin of olfactory adaptation is still unclear, though recent evidence indicates that modulation of phosphodiesterase may make a contribution here too [55]. The complexity of sensory adaptation has puzzled us for many years, and there is every indication that many more years will pass before we understand this intriguing process in its entirety.
References
Pugh EN Jr, Nikonov S, Lamb TD (1999) Molecular mechanisms of vertebrate photoreceptor light adaptation. Curr Opin Neurobiol 9:410–418
Fain GL, Matthews HR, Cornwall MC, Koutalos Y (2001) Adaptation in vertebrate photoreceptors. Physiol Rev 81:117–151
Arshavsky VY, Lamb TD, Pugh EN Jr (2002) G proteins and phototransduction. Annu Rev Physiol 64:153–187
Fain GL (2003) Sensory transduction. Sinauer, Inc., Sunderland, MA
Burns M, Arshavsky V (2005) Beyond counting photons: trials and trends in vertebrate visual transduction. Neuron 48:387–401
Burns ME, Pugh EN Jr (2011) Lessons from photoreceptors: turning off g-protein signaling in living cells. Physiology (Bethesda) 25:72–84
Matthews HR, Murphy RL, Fain GL, Lamb TD (1988) Photoreceptor light adaptation is mediated by cytoplasmic calcium concentration. Nature 334:67–69
Nakatani K, Yau KW (1988) Calcium and light adaptation in retinal rods and cones. Nature 334:69–71
Fain GL, Lamb TD, Matthews HR, Murphy RL (1989) Cytoplasmic calcium as the messenger for light adaptation in salamander rods. J Physiol (Lond) 416:215–243
Koutalos Y, Yau KW (1996) Regulation of sensitivity in vertebrate rod photoreceptors by calcium. Trends Neurosci 19:73–81
Nikonov S, Lamb TD, Pugh EN Jr (2000) The role of steady phosphodiesterase activity in the kinetics and sensitivity of the light-adapted salamander rod photoresponse. J Gen Physiol 116:795–824
Mendez A, Burns ME, Sokal I, Dizhoor AM, Baehr W, Palczewski K, Baylor DA, Chen J (2001) Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc Natl Acad Sci U S A 98:9948–9953
Burns ME, Mendez A, Chen J, Baylor DA (2002) Dynamics of cyclic GMP synthesis in retinal rods. Neuron 36:81–91
Chen J, Woodruff ML, Wang T, Concepcion F, Tranchina D, Fain GL (2010) Channel modulation and the mechanism of light adaptation in mouse rods. J Neurosci 30:16232–16240
Ridge KD, Palczewski K (2007) Visual rhodopsin sees the light: structure and mechanism of G protein signaling. J Biol Chem 282:9297–9301
Hofmann KP, Scheerer P, Hildebrand PW, Choe HW, Park JH, Heck M, Ernst OP (2009) A G protein-coupled receptor at work: the rhodopsin model. Trends Biochem Sci 34:540–552
Koch KW, Duda T, Sharma RK (2010) Ca(2+)-modulated vision-linked ROS-GC guanylate cyclase transduction machinery. Mol Cell Biochem 334:105–115
Smith SO (2010) Structure and activation of the visual pigment rhodopsin. Annu Rev Biophys 39:309–328
Calvert PD, Govardovskii VI, Arshavsky VY, Makino CL (2002) Two temporal phases of light adaptation in retinal rods. J Gen Physiol 119:129–145
Krispel CM, Chen D, Melling N, Chen YJ, Martemyanov KA, Quillinan N, Arshavsky VY, Wensel TG, Chen CK, Burns ME (2006) RGS expression rate-limits recovery of rod photoresponses. Neuron 51:409–416
Tsang SH, Woodruff ML, Chen CK, Yamashita CY, Cilluffo MC, Rao AL, Farber DB, Fain GL (2006) GAP-independent termination of photoreceptor light response by excess gamma subunit of the c-GMP-phosphodiesterase. J Neurosci 26:4472–4480
Woodruff ML, Janisch KM, Peshenko IV, Dizhoor AM, Tsang SH, Fain GL (2008) Modulation of phosphodiesterase6 turnoff during background illumination in mouse rod photoreceptors. J Neurosci 28:2064–2074
Fuortes MG, Hodgkin AL (1964) Changes in time scale and sensitivity in the ommatidia of limulus. J Physiol 172:239–263
Fan J, Woodruff ML, Cilluffo MC, Crouch RK, Fain GL (2005) Opsin activation of transduction in the rods of dark-reared Rpe65 knockout mice. J Physiol 568:83–95
Fain GL (2006) Why photoreceptors die (and why they don't). Bioessays 28:344–354
Sokolov M, Lyubarsky AL, Strissel KJ, Savchenko AB, Govardovskii VI, Pugh EN Jr, Arshavsky VY (2002) Massive light-driven translocation of transducin between the two major compartments of rod cells: a novel mechanism of light adaptation. Neuron 34:95–106
Lobanova ES, Finkelstein S, Song H, Tsang SH, Chen CK, Sokolov M, Skiba NP, Arshavsky VY (2007) Transducin translocation in rods is triggered by saturation of the GTPase-activating complex. J Neurosci 27:1151–1160
Lamb TD, McNaughton PA, Yau KW (1981) Spatial spread of activation and background desensitization in toad rod outer segments. J Physiol 319:463–496
Matthews HR, Torre V, Lamb TD (1985) Effects on the photoresponse of calcium buffers and cyclic GMP incorporated into the cytoplasm of retinal rods. Nature 313:582–585
Koch KW, Stryer L (1988) Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature 334:64–66
Lolley RN, Racz E (1982) Calcium modulation of cyclic GMP synthesis in rat visual cells. Vision Res 22:1481–1486
Woodruff ML, Sampath AP, Matthews HR, Krasnoperova NV, Lem J, Fain GL (2002) Measurement of cytoplasmic calcium concentration in the rods of wild- type and transducin knock-out mice. J Physiol 542:843–854
Palczewski K, Sokal I, Baehr W (2004) Guanylate cyclase-activating proteins: structure, function, and diversity. Biochem Biophys Res Commun 322:1123–1130
Makino CL, Peshenko IV, Wen XH, Olshevskaya EV, Barrett R, Dizhoor AM (2008) A role for GCAP2 in regulating the photoresponse. Guanylyl cyclase activation and rod electrophysiology in GUCA1B knock-out mice. J Biol Chem 283:29135–29143
Howes KA, Pennesi ME, Sokal I, Church-Kopish J, Schmidt B, Margolis D, Frederick JM, Rieke F, Palczewski K, Wu SM, Detwiler PB, Baehr W (2002) GCAP1 rescues rod photoreceptor response in GCAP1/GCAP2 knockout mice. EMBO J 21:1545–1554
Kawamura S (1993) Rhodopsin phosphorylation as a mechanism of cyclic GMP phosphodiesterase regulation by S-modulin. Nature 362:855–857
Chen CK, Inglese J, Lefkowitz RJ, Hurley JB (1995) Ca(2+)-dependent interaction of recoverin with rhodopsin kinase. J Biol Chem 270:18060–18066
Chen CK, Woodruff ML, Chen FS, Chen D, Fain GL (2010) Background light produces a recoverin-dependent modulation of activated-rhodopsin lifetime in mouse rods. J Neurosci 30:1213–1220
Hsu YT, Molday RS (1993) Modulation of the cGMP-gated channel of rod photoreceptor cells by calmodulin [published erratum appears in Nature 1993 Sep 16;365(6443):279] [see comments]. Nature 361:76–79
Chen TY, Illing M, Molday LL, Hsu YT, Yau KW, Molday RS (1994) Subunit 2 (or beta) of retinal rod cGMP-gated cation channel is a component of the 240-kDa channel-associated protein and mediates Ca(2+)– calmodulin modulation. Proc Natl Acad Sci U S A 91:11757–11761
Grunwald ME, Yu WP, Yu HH, Yau KW (1998) Identification of a domain on the beta-subunit of the rod cGMP-gated cation channel that mediates inhibition by calcium–calmodulin. J Biol Chem 273:9148–9157
Grunwald ME, Yau KW (2000) Modulation of rod cGMP-gated cation channel by calmodulin. Methods Enzymol 315:817–828
Nakatani K, Koutalos Y, Yau KW (1995) Ca2+ modulation of the cGMP-gated channel of bullfrog retinal rod photoreceptors. J Physiol (Lond) 484:69–76
Hamer RD, Nicholas SC, Tranchina D, Lamb TD, Jarvinen JL (2005) Toward a unified model of vertebrate rod phototransduction. Vis Neurosci 22:417–436
Koutalos Y, Nakatani K, Yau KW (1995) The cGMP-phosphodiesterase and its contribution to sensitivity regulation in retinal rods. J Gen Physiol 106:891–921
Soo FS, Detwiler PB, Rieke F (2008) Light adaptation in salamander L-cone photoreceptors. J Neurosci 28:1331–1342
Makino CL, Dodd RL, Chen J, Burns ME, Roca A, Simon MI, Baylor DA (2004) Recoverin regulates light-dependent phosphodiesterase activity in retinal rods. J Gen Physiol 123:729–741
Krispel CM, Chen CK, Simon MI, Burns ME (2003) Novel form of adaptation in mouse retinal rods speeds recovery of phototransduction. J Gen Physiol 122:703–712
Doan T, Azevedo AW, Hurley JB, Rieke F (2009) Arrestin competition influences the kinetics and variability of the single-photon responses of mammalian rod photoreceptors. J Neurosci 29:11867–11879
Gross OP, Burns ME (2010) Control of rhodopsin's active lifetime by arrestin-1 expression in mammalian rods. J Neurosci 30:3450–3457
Pepperberg DR, Cornwall MC, Kahlert M, Hofmann KP, Jin J, Jones GJ, Ripps H (1992) Light-dependent delay in the falling phase of the retinal rod photoresponse. Vis Neurosci 8:9–18
Rieke F, Baylor D (1996) Molecular origin of continuous dark noise in rod photoreceptors. Biophys J 71:2553–2572
Kurahashi T, Menini A (1997) Mechanism of odorant adaptation in the olfactory receptor cell. Nature 385:725–729
Song Y, Cygnar KD, Sagdullaev B, Valley M, Hirsh S, Stephan A, Reisert J, Zhao H (2008) Olfactory CNG channel desensitization by Ca2+/CaM via the B1b subunit affects response termination but not sensitivity to recurring stimulation. Neuron 58:374–386
Cygnar KD, Zhao H (2009) Phosphodiesterase 1C is dispensable for rapid response termination of olfactory sensory neurons. Nat Neurosci 12:454–462
Acknowledgements
I am grateful to Ray Goldstein, the Cambridge University Department of Applied Mathematics and Theoretical Physics, and Churchill College, Cambridge, for their hospitality during the writing of this review, and I would like to thank Alapakkam P. Sampath for his very helpful comments on the manuscript and Margery Fain for her assistance with Figure 1. Research in the author's laboratory is supported by NIH R01 EY01844.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fain, G.L. Adaptation of Mammalian Photoreceptors to Background Light: Putative Role for Direct Modulation of Phosphodiesterase. Mol Neurobiol 44, 374–382 (2011). https://doi.org/10.1007/s12035-011-8205-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-011-8205-1