
Abstract
Injury to the central nervous system (CNS) initiates a cascade of responses that is inhibitory to the regeneration of neurons and full recovery. At the site of injury, glial cells conspire with an inhibitory biochemical milieu to construct both physical and chemical barriers that prevent the outgrowth of axons to or beyond the lesion site. These inhibitors include factors derived from myelin, repulsive guidance cues, and chondroitin sulfate proteoglycans. Each bind receptors on the axon surface to initiating intracellular signaling cascades that ultimately result in cytoskeletal reorganization and growth cone collapse. Here, we present an overview of the molecules, receptors, and signaling pathways that inhibit CNS regeneration, with a particular focus on the intracellular signaling machinery that may function as convergent targets for multiple inhibitory ligands.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Following injury to the adult mammalian central nervous system (CNS), axons do not spontaneously regenerate, and functional connections between neurons are not re-established. This failure to regenerate can be partly attributed to an unfavorable CNS environment [1]. The glial scar is a physical barrier that consists primarily of reactive astrocytes, which plays an important role in blocking axonal regeneration past the site of injury [2]. In addition, factors from the glial scar and from myelin debris create an inhibitory biochemical barrier that actively signals to the surface of regenerating axons to induce growth cone collapse and inhibit axon extension [3]. Several proteins that inhibit neurite outgrowth have been identified, including myelin-associated glycoprotein (MAG), nogo-A, oligodendrocyte-myelin glycoprotein (OMgp), netrin-1, semaphorin4D, ephrinB3, and chondroitin sulfate proteoglycans (CSPGs). Several of these proteins are upregulated by cells that form the glial scar [4], and the remainder are expressed by oligodendrocytes, which form the insulating myelin membrane. Although these inhibitory ligands engage multiple distinct receptors on the neuronal cell surface, they signal through overlapping pathways. This review will overview the inhibitory ligands and receptors through which they signal (Fig. 1), and then will focus on recent advances in our understanding of the intracellular signaling machinery activated in response to inhibitors of CNS regeneration.

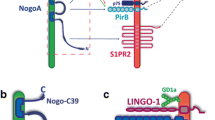
The inhibitory ligands of neural regeneration and their receptors. Multiple inhibitory ligands prevent axonal repair in the central nervous system. Significant receptor redundancy exists as several ligands bind to multiple receptors; however, individual ligands can also bind to multiple receptors. This is highlighted by myelin-associated glycoprotein (MAG), which has been shown to interact with the gangliosides (GT1b and GD1a), the Nogo receptors (NgR1 and 2), and PirB. Also shown is the binding of the various Nogoligands (Nogo66 and Amino Nogo, top left) with the tripartite Nogo receptor complex (NgR1, LINGO-1, and p75), PirB, and a third yet-to-be-identified receptor. Oligodendrocyte-myelin glycoprotein (OMgp) has similar binding partners and is shown in the top left. Netrin-1, EphrinB3, Sema4D, and Sema3A are shown to the right with their respective receptors
Ligands Associated with CNS Injury
The Glial Scar and CSPGs
Damage to the CNS initiates a cascade of events that leads to the formation of a glial scar. Following injury, this scar beneficially stabilizes fragile CNS tissue, protects surrounding healthy tissue from inflammatory damage, and aids in the repair of the blood–brain barrier [3, 5]. However, the scar is also a critical barrier to regeneration; it forms a physical and biochemical barrier preventing the elongation of axons into damaged tissue [6]. The glial scar contains astrocytes, oligodendrocyte precursor cells, microglia, macrophages, and an extracellular matrix of secreted basal membrane components [2, 6].
Astrocytes undergo reactive gliosis and are the major contributor to the inhibitory nature of the scar [7]. Following injury, these astrocytes form tight junctions to create a physical barrier between injured and healthy CNS tissue. In addition, they secrete chondroitin sulfate proteoglycans (CSPGs), a class of extracellular matrix molecules comprising a protein core and numerous variable glycosaminoglycan (GAG) side chains [8]. The family of CSPGs includes more than a half dozen members, including aggrecan, brevican, neurocan, versican, NG2, and phosphacan, that have been demonstrated to be upregulated following certain types of CNS injury [9]. Collectively, they are potent inhibitors of neurite outgrowth in vitro from both adult and embryonic neurons, but the relative contribution of each member has not been fully characterized [8]. Interestingly, the neurite outgrowth inhibitory activity of CSPGs has been associated with both the core protein [10, 11] and the chondroitin sulfated GAG side chains [11–13]. Treatment with chondroitinase ABC to remove these side chains has demonstrated some promise in promoting plasticity and axon regeneration following CNS injury [9, 14].
Myelin-Associated Inhibitors
A number of inhibitors of neurite outgrowth are associated with myelin, including MAG, Nogo-A, and OMgp. MAG, also known as sialic acid binding Ig-like lectin (SIGLEC-4), is a glycoprotein member of the immunoglobulin (Ig)-like superfamily [15]. It is a 100-kDa type I transmembrane glycoprotein selectively localized to the periaxonal membrane of both peripheral nervous system Schwann cells and CNS oligodendrocytes. Its extracellular domain consists of five sequence-related glycosylated Ig-like domains, while the cytoplasmic domain is subject to developmentally regulated alternative splicing [16, 17]. MAG expression correlates with the onset of myelination and continues at high levels into adulthood; it may play multiple roles during the development and maintenance of myelinated axons [18]. More than a decade ago, two groups independently identified MAG as a potent inhibitor of neurite outgrowth in vitro [19, 20]. MAG not only inhibits neurite outgrowth of various types of mature neurons but also promotes axon outgrowth at early developmental stages (discussed below) [20]. In MAG knockout mice, CNS compact myelin formation is relatively normal but it is significantly delayed and misguided in a small proportion of cells [21–23]. Furthermore, in aging MAG knockout mice, alterations in periaxonal oligodendrocyte processes are consistent with oligodendrogliopathy [24]. While myelin from MAG knockout mice is a less inhibitory substrate for neurite outgrowth of neonatal cerebellar or P5 DRG neurons in vitro [25], regeneration of retinal ganglion cells or corticospinal tract neurons is not enhanced in MAG knockout mice following optic nerve or spinal cord lesions, respectively [26]. Therefore, MAG does not account for a significant proportion of in vivo myelin inhibition or there is a non-additive redundancy between different myelin-associated inhibitors.
The most widely studied myelin-associated inhibitor is Nogo-A. Nogo-A was initially described as biochemical fractions of myelin approximately 250 and 35 kDa (NI-35/250) in size that were non-permissive substrates for neurite outgrowth [27]. Subsequently, Schwab and colleagues generated a function-blocking antibody (IN-1) against NI-35/250 that could alleviate the inhibition of neurite outgrowth by CNS white matter both in vitro [28] and in vivo [29–31]. More than a decade following the discovery of NI-35/250, NI-250 was identified as Nogo-A, also known as reticulon 4A [32–34]. Nogo-A is encoded by the nogo/RTN-4 gene. This locus also generates two other isoforms, Nogo-B and Nogo-C, by alternative splicing [35]. The Nogo-A transcript is expressed in adult CNS tissue, including oligodendrocytes and neurons in the brain and spinal cord [33, 36, 37]. Nogo-A protein is predominantly expressed by oligodendrocytes and localizes to the outer and inner myelin loops [35, 37]. Nogo-B and Nogo-C are also expressed by neurons although Nogo-C appears to be the predominate species on northern blots [33, 37]. Given the neuronal pattern of Nogo-A, the protein has been proposed to play a role in neuronal development and maturity beyond its role as a myelin-associated inhibitor.
The three Nogo splice variants contain a common C-terminal domain with an inhibitory 66 amino acid lumenal/extracellular domain found between two hydrophobic regions. This domain is termed Nogo-66 [33]. In addition, the amino terminus of Nogo-A is also inhibitory [32, 38]. Of the three Nogo splice variants, Nogo-A is considered to be the major player in myelin inhibition because it is expressed in oligodendrocytes, the IN-1 antibody recognizes Nogo-A, and the inhibitory activity residing in the amino terminal region is unique to Nogo-A [38]. However, Nogo-A is a reticulon, and the majority of the protein is found associated with the endoplasmic reticulum intracellularly. The potential functions of Nogo-A and other isoforms beyond their role at myelin membranes are not clear. Rapoport and colleagues have proposed that Nogo-A is critical for the formation of ER tubules. However, the restricted expression pattern and mild phenotype of several different strains of Nogo-A mutant mice are inconsistent with the protein such a central role in ER morphology [39].
Three groups independently generated mutant mice in which the rtn4 locus was perturbed. [40–42]. The inhibitory activity of myelin prepared from all three strains of mutant mice is significantly diminished as demonstrated with in vitro neurite outgrowth assays. Two of the three strains [41, 42] demonstrated some limited corticospinal tract (CST) regeneration following a dorsal hemisection injury to the spinal cord but no improvement was detectable in the third line [40]. Follow-up studies have revealed that strain background, age, and the nature of the Nogo mutant allele all influence the regenerative capacity of the corticospinal tract [43, 44]. Instructively, when the two strains of Nogo-A mutant with the greatest and least anatomical plasticity and functional recovery following spinal cord injury were examined following a more precise lesion to the corticospinal tract, intact uninjured CST exhibited greater local sprouting in both lines [45]. Thus, each of these mutant Nogo strains possess some degree of enhanced outgrowth or sprouting following CNS injury. Collectively, these studies have largely dispelled concerns regarding artifactual tract tracing in the Nogo regeneration studies but the specific differences between the strains that account for the different initial reports of recovery, or lack thereof, after spinal cord injury have not been fully elucidated [46]. More recently, a deletion mutant line that is null for all Nogo isoforms was generated [47]. CST regeneration is not significantly enhanced in this strain following experimental spinal cord injury; however, the targeting vectors used to generate the strain are identical to those used for the previously generated Nogo-C and Nogo-A, B targeted mutants that showed no regeneration [40].
Oligodendrocyte-myelin glycoprotein (OMgp) is another myelin-associated inhibitor [48, 49]. OMgp is a membrane glycoprotein anchored to the plasma membrane though a glycosylphosphatidylinositol (GPI) lipid moeity [48, 50]. This 433 amino acid protein contains a series of leucine-rich repeats (LRRs). LRRs are present in a spectrum of proteins, but these domains are generally considered to provide an interface for interacting with other proteins [51]. Expression of OMgp correlates with the onset of myelination but can also be detected on pre-myelinated axons as well as on the surface of oligodendrocytes [50, 52]. Mikol and colleagues [50] demonstrated that OMgp does not execute a direct role in myelination itself but mediates the adhesion of membranes at the nodes of Ranvier between either two oligodendrocytes or between an oligodendrocyte and axon. Further, OMgp plays a role in inhibition of axonal sprouting at the nodes of Ranvier [53]. Similar to Nogo-A, OMgp appears to constrain neurite outgrowth and can induce growth cone collapse [48, 50]. Myelin from OMgp knockout mice is less inhibitory for neurite outgrowth and there are improvements in the numbers of ascending sensory axons and 5-HT-positive descending axons following spinal cord injury in OMgp knockout mice; however, this effect is dependent on the strain background [54].
Other Myelin-Associated Inhibitory Ligands: Netrins, Semaphorins, and Ephrins
Netrin-1 is a secreted protein that acts as a long-range chemotropic axon guidance cue during neuronal development [55]. Netrins function both as a chemoattractant and chemorepellent, depending on the types of receptors expressed by the responding axon [56]. Netrin was first described as a chemoattractant expressed by the floor plate of the developing neural tube [57]. Subsequent studies have demonstrated that netrin-1 is also expressed by neurons and oligodendrocytes in the adult brain and spinal cord [58]. Netrin-1 is associated with the extracellular matrix in the periaxonal myelin allowing for close contact between axon and oligodendrocyte [58]. Consistent with this expression pattern, Low and colleagues reported an inhibitory role of netrin-1 on axonal growth [59] and demonstrated that neutralization of netrin-1 in myelin with soluble Unc5 receptor increases neurite outgrowth from spinal neurons. Furthermore, following an in vivo dorsal column lesion, axonal penetration into a graft of netrin-1-secreting fibroblasts is significantly diminished compared to a control graft. Thus, netrin-1 is another category of inhibitor to CNS regeneration associated with myelin.
A second class of axon guidance cues implicated in inhibiting axon regeneration is the semaphorins. Semaphorins are potent mediators of growth cone collapse and axon repulsion [60]. Subsequent studies have demonstrated that semaphorins are upregulated following CNS injury and play roles in limiting regeneration [61, 62]. Expression of both secreted and transmembrane semaphorins have been reported within the glial scar and in preparations of myelin membranes. Sema4D, also called CD100, is a transmembrane semaphorin expressed by myelinating oligodendrocytes and is upregulated following CNS injury [63, 64]. In a stripe assay, Sema4D repels post-natal sensory and cerebellar granule cells. Therefore, it may contribute to failed regeneration in vivo. Sema3A also contributes to axonal inhibition. Increased expression of Sema3A appears mainly within scar tissue following damage to the spinal cord [65, 66]. A selective Sema3A inhibitor created from fungal extract, SM-216289, enhances repair and functional recovery in a spinal cord transection model [67].
Members of the ephrin family of guidance molecules have also been implicated as inhibitors of CNS regeneration [68]. Ephrin-B3 is expressed by post-natal myelinating oligodendrocytes and is dramatically enriched in spinal cord white matter. Purified recombinant Ephrin-B3 inhibits growth of EphA4-positive cortical neurons and myelin from ephrin-B3 knockout mice is significantly less inhibitory than myelin from wild-type mice [69].
Receptors: NgRs, PirB, and Gangliosides
The relative contribution of individual receptor components to inhibitory signaling in response to CSPGs and repulsive guidance cues is not well defined. Receptor components for CSPGs have yet to be identified. Netrin-1, semaphorin4D, and ephrinB3 are likely to signal through their respective receptors, DCC/UNC5 [59], CD72 [64], and EphA4 [69], but the specific roles of these receptors in regeneration in vivo have yet to be fully addressed. Receptor partners for the classic MAIs have been more widely studied and these include members of the Nogo receptor (NgR) family, Paired immunoglobulin-like receptor B (PirB) as well as gangliosides and integrins.
NgR
Despite their differing structural properties, MAG, Nogo-66, and OMgp all bind to a common receptor, Nogo-66 receptor 1 (NgR1) [70–72]. Two similar proteins are also present in the mammalian genome, NgR2 and NgR3 [73]. NgR3 fails to bind to myelin-associated outgrowth inhibitors but NgR2 is a high affinity receptor for MAG [74]. NgR family members are attached to the plasma membrane by GPI anchor and lack an intracellular signaling domain. NgR1 has been proposed to function together with p75 neurotrophin receptor (p75NTR) and LRR and Ig domain-containing Nogo receptor-interacting protein-1 (LINGO-1) to form a tripartite Nogo receptor complex [70, 75, 76]. The expression of p75NTR decreases postnatally, and TROY (tumor necrosis factor receptor superfamily, member 19) has been suggested as a molecular substitute for p75NTR in some adult axons [77, 78] (Fig. 1). Blockade of NgR1 function with the soluble extracellular domain of the protein (NgREcto) or with a small peptide antagonist (NEP1-40) enhances neurite outgrowth on inhibitory substrates in vitro and axon regeneration in vivo [79–84]. Neurons from NgR1 knockout mice are resistant to myelin-dependent growth cone collapse. Following spinal cord injury, regeneration of the raphespinal and rubrospinal axon regeneration is enhanced in NgR1 knockout mice; however, corticospinal tract regeneration remains poor [85, 86]. Subsequent studies have suggested that the physiologic role of NgR1 may be in sensitizing neurons to more acute myelin responses [87]. NgR1 knockout mice have an extended critical period for ocular dominance plasticity [88], demonstrate mild differences in dendritic spine morphology and activity-dependent synaptic strength under specific conditions [89], and have enhanced plasticity in a variety of injury models [45].
PirB
More recently, a second receptor for Nogo-66, MAG, and OMgp was identified as Paired immunoglobulin-like receptor B (PirB) [90]. PirB is a major histocompatiblity complex class I (MHC1) receptor that is expressed in subsets of neurons within the brain. Ocular dominance plasticity is enhanced in mice carrying a loss of function PirB allele suggesting that it may function to stabilize neural circuits [91]. Its identification as a receptor mediating myelin-associated inhibition is particularly intriguing in light of the recent findings implicating NgR1 in synaptic plasticity [45, 89]. Cerebellar granule neurons from the PirB mutant mice or wild-type neurons treated with a PirB function-blocking antibody are less inhibited by Nogo-66 or total myelin in an in vitro neurite outgrowth assay. When the PirB function-blocking antibody was used to treat NgR1-null neurons, myelin inhibition was completely relieved. The in vivo contribution of PirB to regeneration has yet to be reported.
Gangliosides and NgR2
MAG has several additional binding partners that have been implicated in its neurite outgrowth inhibitory activity. It binds the gangliosides GT1b and GD1a [92] and the NgR1 homolog NgR2. NgR2 acts selectively to mediate MAG inhibitory responses [74]. Gangliosides play an essential role in mediating MAG inhibition [93, 94]; however, the extent of this role appears to depend on neuronal cell type [95, 96]. The signaling mechanism utilized by these receptors remains unclear. It is known that GT1b interacts with NgR1 [97] and p75NTR [98], and that both NgR1 and NgR2 bind MAG in a sialic acid-dependent manner [74]. Gangliosides may be components of the Nogo receptor complex. However, studies using biochemical [95] or genetic [96] methods to ablate NgR1 or p75NTR receptors indicate that gangliosides are able to mediate MAG inhibition even in the absence of the Nogo receptor complex in some types of neurons. Thus, gangliosides may bind yet another receptor to mediate MAG inhibition.
Integrin receptors also appear to play a role in signaling by myelin-associated inhibitors. Amino-Nogo inhibits cell adhesion and axon outgrowth as a function of extracellular matrix composition. It reduces focal adhesion kinase (FAK) activity in response to fibronectin and its inhibition can be partially overcome by β1-integrin activation [99]. MAG also appears to signal through beta1-integrin and FAK to induce growth cone turning [100].
Intracellular Signaling Events Inhibitory to CNS Regeneration
Intracellularly, inhibitors of axonal regeneration signal through Ca2+-dependent activation of protein kinase C (PKC) and epidermal growth factor receptor (EGFR), and activation of RhoA GTPase and its downstream effectors to affect cytoskeletal rearrangements. Further, loss of PTEN (phosphatase and tensin homolog) promotes robust retinal ganglion cell survival and regeneration in an optic nerve injury model. This likely occurs by relieving inhibition of mTOR (mammalian target of rapamycin)-dependent protein synthesis. Responses to myelin-associated inhibitors are further subject to modulation by cAMP-dependent signaling. These signals act in a concerted fashion to induce growth cone collapse and/or inhibition of axon extension during regeneration.
Ca2+-Dependent Activation of PKC and EGFR
Calcium-dependent signaling in response to MAIs was first noted by Kater and colleagues [101]. Follow-up studies suggest that MAG induces a rapid rise in intracellular Ca2+ which is dependent on the co-expression and interaction of p75NTR and NgR1 [75] and that a reduction of extracellular Ca2+ abolishes growth cone turning in response to a MAG gradient [102]. MAG-induced calcium elevation in neurons is blocked by inhibitors of phospholipase C (PLC), the heterotrimeric GTP-binding protein Gi, and the IP3 receptor [103]. The conventional PKCs α, β1, and β2 are activated in a PLC-dependent manner by MAG. Neurite outgrowth inhibition and growth cone collapse in response to MAG, Nogo-A, and myelin substrates are mitigated by PKC inhibitors [103]. Similar results were obtained by Sivasankaran et al., who showed that both myelin and CSPGs activate the conventional PKCs α and β, as well as that overexpression of dominant-negative forms of these PKCs overcomes myelin- and CSPG-dependent neurite outgrowth inhibition [104]. Furthermore, intrathecal infusion of Gö6976 (a selective inhibitor of PKCα and PKCβ1) to adult rats with spinal cord dorsal hemisection stimulates regeneration of the dorsal column but not of the corticospinal tract. Taken together, the results of Hasegawa et al. [103] and Sivasankaran et al. [104] strongly implicate PKC in the signaling pathways mediated by myelin-associated inhibitors and CSPGs (see Fig. 2).

Signaling pathways mediated by myelin-associated inhibitors and CSPGs. Inhibitors of axonal regeneration signal through Ca+-dependent activation of PKC, the activation of the epidermal growth factor receptor (EGFR), and the activation of RhoA. RhoA inhibits outgrowth through the remodeling the cytoskeleton. ROCK, CRMP-2, and Cofilin are all downstream of RhoA and have significant impacts on microtubule and actin polymerization. Calcium-dependent activation of PKC also impacts the RhoA pathway. The sequential cleavage of p75 by α- then γ-secretases generates an intracellular domain of 25 kDa that activates RhoA-mediated inhibition of neurite outgrowth. Finally, the developmental regulation of cAMP and PKA plays a role in the inhibition of neuronal outgrowth. A decrease in cAMP levels is coincident with a developmental switch from outgrowth promotion to inhibition in response to MAG
From a small molecule screen using a neurite outgrowth assay on myelin substrates, Koprivica et al. discovered that inhibition of the epidermal growth factor receptor (EGFR) counters outgrowth inhibition. Nogo-66 and OMgp trigger rapid EGFR phosphorylation in neurons, requiring expression of full-length NgR1. However, direct binding or a physical association between EGFR and NgR1 or p75NTR could not be detected suggesting that EGFR is trans-activated in response to myelin inhibitors. Further, calcium chelators can prevent Nogo-66-dependent EGFR phosphorylation, indicating that transactivation likely occurs via a calcium-dependent signal. Interestingly, CSPGs elevate cytoplasmic calcium levels in DRG neurons [105] and signal in a calcium-dependent manner to rapidly phosphorylate EGFR [106]. Furthermore, downstream of EGFR, CSPGs activate the MAPK signaling pathway through phosphorylation of MEK-1/2 and ERK [67]. These findings imply that myelin-associated inhibitors and CSPGs share components of the intracellular signaling pathways blocking axonal regeneration in the CNS.
Activation of RhoA GTPase and its Downstream Effectors
Inhibitors of axonal regeneration must signal to the growth cone cytoskeleton if they are to cause growth cone collapse and inhibition of axonal extension. The Rho family of small GTPases are well characterized as transducers of extracellular cues to the growth cone actin cytoskeleton. These GTPases, which include RhoA, Rac1, and Cdc42, cycle between an active GTP-bound state and an inactive GDP-bound state. Nucleotide state is controlled by several inactivating GTPase-activating proteins (GAPs), activating guanine nucleotide exchange factors (GEFs), and guanine nucleotide dissociation inhibitors (GDI) that maintains GTPases in the inactive GDP-bound state [107]. While Cdc42 and Rac1 activation are associated with growth cone attraction via filopodial and lamellipodial formation, RhoA activation is associated with growth cone collapse and neurite retraction [108]. RhoA has been implicated as an essential component of the myelin inhibitory signaling pathway during regeneration of CNS axons [109–111]. Retinal ganglion neurons grown on an inhibitory substrate of MAG or myelin and in the presence of C3 transferase—an enzyme which selectively ADP-ribosylates RhoA and thereby inactivates it—exhibit greater neurite outgrowth. In vivo regeneration of optic nerve axons past a crush lesion site as well as corticospinal tract regeneration and BBB locomotor score are improved by C3 transferase treatment [110, 112]. Biochemical work by Niederost et al. [109] indicates that a rapid and transient increase in RhoA-GTP, as well as a concomitant but persistent decrease in Rac1-GTP levels, occurs following treatment with either MAG, Amino-Nogo, or Nogo-66 in cerebellar granule neurons. The increase in active RhoA can be blocked by C3 transferase treatment.
Signaling events upstream of RhoA activation involve the regulated PKC-dependent cleavage of the p75NTR intracellular domain (ICD). Upon binding MAG, the p75NTR component of the Nogo receptor complex undergoes sequential proteolytic cleavage by α- and then γ-secretases generating a final ICD fragment of about 25 kDa [113]. It is γ-secretase-mediated cleavage that is dependent on PKC activity. Pharmacological inhibition of either α-secretase, γ-secretase, or PKC prevents formation of the ICD fragment, the activation of RhoA, and reverses MAG inhibition of neurite outgrowth. Overexpression of a cleavage-resistant form of p75NTR can prevent MAG-induced inhibition, while the overexpression of the ICD fragment alone can mimic inhibition. Yamashita and Tohyama suggest that MAG and Nogo stimulation strengthens an interaction between Rho-GDI and p75NTR allowing for RhoA activation [114]. Thus, there are two possible mechanisms for signaling through the p75NTR component of the Nogo receptor complex to activate RhoA.
Results from Sivasankaran et al. also support a central role for PKC in MAI-dependent activation of RhoA. Application of the PKC inhibitor Gö6976 prevents RhoA activation caused by either MAG, Nogo-66, and CSPGs, indicating a convergence of signaling pathways activated by inhibitory ligands binding to different receptors [104]. However, some of these findings are not supported by Hasegawa et al., who reports that pharmacological inhibition of PKC does not impair the RhoA-GTP increase caused by MAG or Nogo-66 treatment [103].
Intriguingly, some inhibitors of axonal regeneration that do not signal through NgR1 do signal through RhoA. Treatment with either C3 transferase or the pharmacological rho-associated kinase (ROCK) inhibitors Y-27632 or HA1077 enhances neurite outgrowth on CSPG [115–117]. Therefore, together with evidence implicating activation of the PKC and EGFR pathways in response to CSPGs [104, 106], it appears that CSPGs activate a set of signaling pathways in common with MAIs. Netrin-1 and ephrinB3 also may signal to inhibit regeneration via RhoA given that their receptors, DCC and ephA4, respectively, can signal through RhoA [118, 119].
Downstream of RhoA activation, ROCK is an important relay point for signal transduction to cytoskeletal regulators. The pharmacological ROCK inhibitors Y-27632 and HA1077, as well a dominant-negative form of ROCK, protect neurons from myelin-induced growth cone collapse and neurite outgrowth inhibition [111, 112, 117, 120]. Nogo-66 treatment activates ROCKII activity, which in turn increases phosphorylation of the actomyosin regulator myosin light chain II (MLCII) [120]. In agreement with this finding, Kubo et al. showed that neurite outgrowth inhibition, growth cone collapse, and the reduction in F-actin density caused by repulsive guidance molecule (RGMa) require MLCII phosphorylation. Furthermore, phosphorylation of MLCII increases after spinal cord injury [121]. Thus, MLCII likely acts downstream of RhoA and ROCK as an effector for cytoskeletal remodeling in response to regeneration inhibitors (Fig. 2).
In addition to MLCII, cofilin is a major regulatory protein of the actin depolymerizing factor/cofilin family, which has also been shown to mediate inhibitory responses to the cytoskeleton [122]. Cofilin regulates actin dynamics by increasing F-actin depolymerization and by severing F-actin. It cycles between active (dephosphorylated) and inactive (phosphorylated) states, as regulated by two kinases, LIM Kinase (LIMK) and TES Kinase (TESK), and two phosphatases, Slingshot (SSH) and Chronophin (CIN) [123]. Hsieh et al. demonstrated that cofilin phosphorylation is regulated by Nogo-66, in a manner that is dependent on LIMK1 and SSH1. In turn, Nogo-66-mediated LIMK1 and SSH1 regulation was shown to be dependent on ROCK. Myelin-dependent growth cone collapse and neurite outgrowth inhibition were blocked by introduction of dominant-negative LIMK1, indicating that an active ROCK–LIMK1–Cofilin–SSH signaling pathway impedes neurite outgrowth through modulation of the actin cytoskeleton [122].
The collapsin-response mediator protein (CRMP) family of cytosolic phosphoproteins has also been implicated in the signaling pathways mediated by myelin-associated inhibitors [124, 125]. CRMP-2 promotes microtubule assembly during axon extension [126] and is phosphorylated in response to Nogo-66 and MAG in a ROCK-dependent manner [124]. Furthermore, mutation of threonine 555 of CRMP-2, a ROCK phosphorylation site, counteracts MAG inhibition of neurite outgrowth in cerebellar neurons [124]. These findings raise the possibility that MAIs act not only via the actin cytoskeleton to reduce outgrowth but may also directly regulate microtubule assembly.
Alabed et al. identified CRMP4b as a protein which physically and functionally interacts with RhoA to mediate neurite outgrowth inhibition in a screen for proteins which interact with constitutively active RhoA in response to Nogo-66 treatment. The interaction between RhoA and CRMP4b is dependent on Nogo-66 stimulation, but independent of the nucleotide binding state of RhoA, indicating that upstream signaling events may differ from those leading to RhoA activation. In fact, the interaction depends on the phosphorylation status of CRMP4, with phosphorylation of CRMP4b preventing the interaction with RhoA. Disruption of the RhoA-CRMP4b interaction attenuates neurite outgrowth inhibition by both myelin and aggrecan. Overexpression of CRMP4b promotes an actin-based filopodial phenotype in growth cones and neurites, suggesting that CRMP4b may participate in outgrowth inhibition by modulating actin dynamics [125]. Furthermore, all members of the CRMP family bind tubulin [126], and CRMP4b is known to interact with intersectin, an endocytic–exocytic adaptor protein [127], raising the possibility that CRMP4b may act through microtubule- or endocytosis-dependent mechanisms. Alternatively, the interaction between RhoA and CRMP4b may be necessary for RhoA-dependent activation of ROCK and its downstream effectors.
Developmental Regulation of cAMP and its Modulation of Myelin Inhibition
A clearer picture of the signaling pathways acutely activated following CNS injury is emerging. However, equally important is the differential interpretation of inhibitory signals by neurons at different developmental stages and the underlying molecular signals responsible for these changes. For example, the response to MAG undergoes a developmental switch from neurite outgrowth promotion to inhibition [20]. In rodents, this occurs around post-natal day 3 or 4 for DRG neurons. The basis for this switch is a developmentally regulated decrease in endogenous levels of cAMP [128].
Several lines of evidence indicate that modulation of cAMP or downstream signaling events can alter the response of neurons to myelin-associated inhibitors. First, in older neurons, which have comparatively lower cAMP levels, elevation of cAMP using an analog such as db-cAMP blocks myelin or MAG inhibition of neurite outgrowth [128]. Further, when applied prior to myelin exposure, the neurotrophins BDNF and GDNF, but not NGF, can elevate cAMP by preventing its degradation, thereby blocking myelin inhibition [129, 130]. Finally, the ability of DRG neurons to regenerate their axon innervating the CNS following a pre-conditioning lesion of the peripheral branch is cAMP and PKA dependent, and can be mimicked by a cAMP analog [131, 132].
cAMP elevation activates the transcription factor cAMP response element binding protein (CREB) that is required to overcome myelin inhibition [133]. This likely initiates long-lasting changes through transcription of a set of genes responsible for cAMP-dependent blockade of myelin inhibition. For example, transcription of Arginase I, a key enzyme in polyamine synthesis, is upregulated, and this results in increased putrescine levels [134]. Both exogenous putrescine and overexpression of Arginase I overcome myelin inhibition in older neurons. It is not clear how polyamines may contribute to block myelin inhibition, but possible mechanisms include initiation of a second round of transcription, modulation of NMDA and AMPA receptor channels, or the promotion of microtubule assembly [134].
The cytokine interleukin-6 (IL-6) is also upregulated in neurons in response to high cAMP levels or a conditioning lesion [135]. Both in vitro and in vivo, IL-6 overcomes the inhibitory effects of myelin on DRG neurons. However, while IL-6 is sufficient to mimic the effects of cAMP elevation or the pre-conditioning lesion, it is not necessary for either response [135, 136].
The Intrinsic Ability of Adult CNS Neurons to Regenerate Following Injury
While inhibitory signaling pathways continue to be elucidated, numerous studies are also assessing ways to enhance the intrinsic ability of adult CNS neurons to regenerate following injury. The reduced intrinsic ability of neurons to re-extend axons following injury and an insufficient ability of non-injured axons to branch and form new synapses constitute a significant limitation to functional recovery. In this regard, strategies altering the activity of several signaling components critical for neuronal development have proven effective in enhancing regeneration.
The optic nerve injury system has been used extensively to study intrinsic limitations to adult CNS axon regeneration (for an extensive review see [137]). In a recent study, Park et al. used the optic nerve injury system to study the effects of ablating an number of evolutionarily conserved genes responsible for controlling cell growth and size, including Rb, p53, Smad4, Dicer, LKB1, and PTEN [138]. PTEN deletion results in both increased retinal ganglion cell (RGC) survival and sustained axon regeneration following an optic nerve crush. Deletion of TSC1, which, like PTEN, negatively regulates the mTOR pathway, was also beneficial to RGC survival and regeneration. While axotomy appeared to reduce phosphorylation levels of ribosomal s6—a protein downstream of mTOR kinase—deletion of PTEN maintains the level of p-s6, suggesting that retaining the mTOR protein translation system active is essential for sustained axon regeneration [138].
In the spinal cord injury model, the intrinsic growth capacity of regenerating axons of the dorsal column can be improved when a simultaneous or preceding sciatic nerve lesion is performed [139]. The molecular basis of this is a peripheral lesion-induced increase in cAMP levels, which also allows regenerating fibers to overcome myelin inhibition [131, 132]. Activating transcription factor-3 (ATF-3) also enhances the intrinsic growth capacity of regenerating dorsal column fibers, but it does not overcome myelin inhibition [140]. A recent study by Dill et al. also points to glycogen synthase kinase-3β (GSK-3β) inactivation as increasing the intrinsic growth potential of adult DRG neurons [141]. These findings add to the constant expanding list of genes found that can elicit axonal extension of the DRG neurons such as GAP-34 and CAP-23, two major growth cone proteins [142]. Additionally, IL-1β has been also shown overcome MAG-induced neurite outgrowth inhibition via p38 MAPK pathway that deactivates RhoA [143].
Concluding Remarks
The molecular mechanisms mediating the inhibition of CNS regeneration appear to be highly redundant. There are upwards of 11 known, or proposed, cell surface receptors on axons interacting with at least seven distinct ligands. Interestingly, many are integral to axonal guidance so that, at least superficially, it appears that the same molecules that initially allow for the systemic generation of complex topographical circuits may be a hindrance to effective regeneration. Yet these molecules can only be part of the conundrum that is CNS regeneration as they exist in a variety of organisms where effective regeneration does occur. Perhaps then it is wise to consider the organism as a whole and the contributions of non-neuronal cells, in particular, on regenerative outcomes.
References
David S, Aguayo AJ (1981) Axonal elongation into peripheral nervous system "bridges" after central nervous system injury in adult rats. Science 214(4523):931–933
Eftekharpour E, Karimi-Abdolrezaee S, Fehlings MG (2008) Current status of experimental cell replacement approaches to spinal cord injury. Neurosurg Focus 24(3–4):E19
Yiu G, He Z (2006) Glial inhibition of CNS axon regeneration. Nat Rev Neurosci 7(8):617–627
Carulli D et al (2005) Chondroitin sulfate proteoglycans in neural development and regeneration. Curr Opin Neurobiol 15(1):116–120
Silver J, Miller JH (2004) Regeneration beyond the glial scar. Nat Rev Neurosci 5(2):146–156
Fawcett JW, Asher RA (1999) The glial scar and central nervous system repair. Brain Res Bull 49(6):377–391
Gris P et al (2007) Transcriptional regulation of scar gene expression in primary astrocytes. Glia 55(11):1145–1155
Jones LL, Margolis RU, Tuszynski MH (2003) The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol 182(2):399–411
Bradbury EJ et al (2002) Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416(6881):636–640
Schmalfeldt M et al (2000) Brain derived versican V2 is a potent inhibitor of axonal growth. J Cell Sci 113(Pt 5):807–816
Ughrin YM, Chen ZJ, Levine JM (2003) Multiple regions of the NG2 proteoglycan inhibit neurite growth and induce growth cone collapse. J Neurosci 23(1):175–186
Fidler PS et al (1999) Comparing astrocytic cell lines that are inhibitory or permissive for axon growth: the major axon-inhibitory proteoglycan is NG2. J Neurosci 19(20):8778–8788
Laabs TL et al (2007) Inhibiting glycosaminoglycan chain polymerization decreases the inhibitory activity of astrocyte-derived chondroitin sulfate proteoglycans. J Neurosci 27(52):14494–14501
Kwok JC et al (2008) Proteoglycans in the central nervous system: plasticity, regeneration and their stimulation with chondroitinase ABC. Restor Neurol Neurosci 26(2–3):131–145
Crocker PR, Varki A (2001) Siglecs in the immune system. Immunology 103(2):137–145
Arquint M et al (1987) Molecular cloning and primary structure of myelin-associated glycoprotein. Proc Natl Acad Sci USA 84(2):600–604
Lai C et al (1987) Neural protein 1B236/myelin-associated glycoprotein (MAG) defines a subgroup of the immunoglobulin superfamily. Immunol Rev 100:129–151
Schachner M, Bartsch U (2000) Multiple functions of the myelin-associated glycoprotein MAG (siglec-4a) in formation and maintenance of myelin. Glia 29(2):154–165
McKerracher L et al (1994) Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 13(4):805–811
Mukhopadhyay G et al (1994) A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13(3):757–767
Bartsch S et al (1997) Increased number of unmyelinated axons in optic nerves of adult mice deficient in the myelin-associated glycoprotein (MAG). Brain Res 762(1–2):231–234
Bartsch U et al (1995) Multiply myelinated axons in the optic nerve of mice deficient for the myelin-associated glycoprotein. Glia 14(2):115–122
Li C et al (1998) Myelin associated glycoprotein modulates glia–axon contact in vivo. J Neurosci Res 51(2):210–217
Lassmann H et al (1997) Dying-back oligodendrogliopathy: a late sequel of myelin-associated glycoprotein deficiency. Glia 19(2):104–110
Shen YJ et al (1998) Myelin-associated glycoprotein in myelin and expressed by Schwann cells inhibits axonal regeneration and branching. Mol Cell Neurosci 12(1–2):79–91
Bartsch U et al (1995) Lack of evidence that myelin-associated glycoprotein is a major inhibitor of axonal regeneration in the CNS. Neuron 15(6):1375–1381
Caroni P, Schwab ME (1988) Two membrane protein fractions from rat central myelin with inhibitory properties for neurite growth and fibroblast spreading. J Cell Biol 106(4):1281–1288
Caroni P, Schwab ME (1988) Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron 1(1):85–96
Bregman BS et al (1995) Recovery from spinal cord injury mediated by antibodies to neurite growth inhibitors. Nature 378(6556):498–501
Schnell L, Schwab ME (1990) Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature 343(6255):269–272
Schnell L, Schwab ME (1993) Sprouting and regeneration of lesioned corticospinal tract fibres in the adult rat spinal cord. Eur J Neurosci 5(9):1156–1171
Chen MS et al (2000) Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403(6768):434–439
GrandPre T et al (2000) Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403(6768):439–444
Prinjha R et al (2000) Inhibitor of neurite outgrowth in humans. Nature 403(6768):383–384
Dodd DA et al (2005) Nogo-A, -B, and -C are found on the cell surface and interact together in many different cell types. J Biol Chem 280(13):12494–12502
Josephson A et al (2001) NOGO mRNA expression in adult and fetal human and rat nervous tissue and in weight drop injury. Exp Neurol 169(2):319–328
Huber AB et al (2002) Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J Neurosci 22(9):3553–3567
Oertle T et al (2003) Nogo-A inhibits neurite outgrowth and cell spreading with three discrete regions. J Neurosci 23(13):5393–5406
Voeltz GK et al (2006) A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124(3):573–586
Zheng B et al (2003) Lack of enhanced spinal regeneration in Nogo-deficient mice. Neuron 38(2):213–224
Kim JE et al (2003) Axon regeneration in young adult mice lacking Nogo-A/B. Neuron 38(2):187–199
Simonen M et al (2003) Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron 38(2):201–211
Dimou L et al (2006) Nogo-A-deficient mice reveal strain-dependent differences in axonal regeneration. J Neurosci 26(21):5591–5603
Cafferty WB et al (2007) Response to correspondence: Kim et al., “Axon regeneration in young adult mice lacking Nogo-A/B.” Neuron 38, 187–199. Neuron 54(2):195–199
Cafferty WB, Strittmatter SM (2006) The Nogo–Nogo receptor pathway limits a spectrum of adult CNS axonal growth. J Neurosci 26(47):12242–12250
Steward O et al (2007) Response to: Kim et al., “Axon regeneration in young adult mice lacking Nogo-A/B.” Neuron 38, 187–199. Neuron 54(2):191–195
Lee JK et al (2009) Reassessment of corticospinal tract regeneration in Nogo-deficient mice. J Neurosci 29(27):8649–8654
Wang KC et al (2002) Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 417(6892):941–944
Kottis V et al (2002) Oligodendrocyte-myelin glycoprotein (OMgp) is an inhibitor of neurite outgrowth. J Neurochem 82(6):1566–1569
Mikol DD, Gulcher JR, Stefansson K (1990) The oligodendrocyte-myelin glycoprotein belongs to a distinct family of proteins and contains the HNK-1 carbohydrate. J Cell Biol 110(2):471–479
Kobe B, Deisenhofer J (1994) The leucine-rich repeat: a versatile binding motif. Trends Biochem Sci 19(10):415–421
Habib AA et al (1998) Expression of the oligodendrocyte-myelin glycoprotein by neurons in the mouse central nervous system. J Neurochem 70(4):1704–1711
Huang JK et al (2005) Glial membranes at the node of Ranvier prevent neurite outgrowth. Science 310(5755):1813–1817
Ji B et al (2008) Assessment of functional recovery and axonal sprouting in oligodendrocyte-myelin glycoprotein (OMgp) null mice after spinal cord injury. Mol Cell Neurosci 39(2):258–267
Moore SW, Tessier-Lavigne M, Kennedy TE (2007) Netrins and their receptors. Adv Exp Med Biol 621:17–31
Hedgecock EM, Culotti JG, Hall DH (1990) The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron 4(1):61–85
Serafini T et al (1994) The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell 78(3):409–424
Manitt C et al (2001) Widespread expression of netrin-1 by neurons and oligodendrocytes in the adult mammalian spinal cord. J Neurosci 21(11):3911–3922
Low K et al (2008) Netrin-1 is a novel myelin-associated inhibitor to axon growth. J Neurosci 28(5):1099–1108
Koncina E et al (2007) Role of semaphorins during axon growth and guidance. Adv Exp Med Biol 621:50–64
Pasterkamp RJ, Verhaagen J (2006) Semaphorins in axon regeneration: developmental guidance molecules gone wrong? Philos Trans R Soc Lond B Biol Sci 361(1473):1499–1511
Luo Y, Raible D, Raper JA (1993) Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell 75(2):217–227
Cohen RI et al (2003) A role for semaphorins and neuropilins in oligodendrocyte guidance. J Neurochem 85(5):1262–1278
Moreau-Fauvarque C et al (2003) The transmembrane semaphorin Sema4D/CD100, an inhibitor of axonal growth, is expressed on oligodendrocytes and upregulated after CNS lesion. J Neurosci 23(27):9229–9239
Pasterkamp RJ, Ruitenberg MJ, Verhaagen J (1999) Semaphorins and their receptors in olfactory axon guidance. Cell Mol Biol (Noisy-le-grand) 45(6):763–779
De Winter F, Holtmaat AJ, Verhaagen J (2002) Neuropilin and class 3 semaphorins in nervous system regeneration. Adv Exp Med Biol 515:115–139
Kaneko S et al (2006) A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat Med 12(12):1380–1389
Du J, Fu C, Sretavan DW (2007) Eph/ephrin signaling as a potential therapeutic target after central nervous system injury. Curr Pharm Des 13(24):2507–2518
Benson MD et al (2005) Ephrin-B3 is a myelin-based inhibitor of neurite outgrowth. Proc Natl Acad Sci USA 102(30):10694–10699
Wang KC et al (2002) P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420(6911):74–78
Liu BP et al (2002) Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science 297(5584):1190–1193
Fournier AE, GrandPre T, Strittmatter SM (2001) Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 409(6818):341–346
Pignot V et al (2003) Characterization of two novel proteins, NgRH1 and NgRH2, structurally and biochemically homologous to the Nogo-66 receptor. J Neurochem 85(3):717–728
Venkatesh K et al (2005) The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J Neurosci 25(4):808–822
Wong ST et al (2002) A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci 5(12):1302–1308
Mi S et al (2004) LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci 7(3):221–228
Park JB et al (2005) A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron 45(3):345–351
Shao Z et al (2005) TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron 45(3):353–359
Fournier AE et al (2002) Truncated soluble Nogo receptor binds Nogo-66 and blocks inhibition of axon growth by myelin. J Neurosci 22(20):8876–8883
GrandPre T, Li S, Strittmatter SM (2002) Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature 417(6888):547–551
Li S et al (2004) Blockade of Nogo-66, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein by soluble Nogo-66 receptor promotes axonal sprouting and recovery after spinal injury. J Neurosci 24(46):10511–10520
Harvey PA et al (2009) Blockade of Nogo receptor ligands promotes functional regeneration of sensory axons after dorsal root crush. J Neurosci 29(19):6285–6295
Fischer D, He Z, Benowitz LI (2004) Counteracting the Nogo receptor enhances optic nerve regeneration if retinal ganglion cells are in an active growth state. J Neurosci 24(7):1646–1651
Wang X et al (2006) Delayed Nogo receptor therapy improves recovery from spinal cord contusion. Ann Neurol 60(5):540–549
Kim JE et al (2004) Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron 44(3):439–451
Zheng B et al (2005) Genetic deletion of the Nogo receptor does not reduce neurite inhibition in vitro or promote corticospinal tract regeneration in vivo. Proc Natl Acad Sci USA 102(4):1205–1210
Chivatakarn O et al (2007) The Nogo-66 receptor NgR1 is required only for the acute growth cone-collapsing but not the chronic growth-inhibitory actions of myelin inhibitors. J Neurosci 27(27):7117–7124
McGee AW et al (2005) Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science 309(5744):2222–2226
Lee H et al (2008) Synaptic function for the Nogo-66 receptor NgR1: regulation of dendritic spine morphology and activity-dependent synaptic strength. J Neurosci 28(11):2753–2765
Atwal JK et al (2008) PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322(5903):967–970
Syken J et al (2006) PirB restricts ocular-dominance plasticity in visual cortex. Science 313(5794):1795–1800
Yang LJ et al (1996) Gangliosides are neuronal ligands for myelin-associated glycoprotein. Proc Natl Acad Sci USA 93(2):814–818
Vyas AA et al (2002) Gangliosides are functional nerve cell ligands for myelin-associated glycoprotein (MAG), an inhibitor of nerve regeneration. Proc Natl Acad Sci USA 99(12):8412–8417
DeBellard ME et al (1996) Myelin-associated glycoprotein inhibits axonal regeneration from a variety of neurons via interaction with a sialoglycoprotein. Mol Cell Neurosci 7(2):89–101
Mehta NR et al (2007) Gangliosides and Nogo receptors independently mediate myelin-associated glycoprotein inhibition of neurite outgrowth in different nerve cells. J Biol Chem 282(38):27875–27886
Venkatesh K et al (2007) Molecular dissection of the myelin-associated glycoprotein receptor complex reveals cell type-specific mechanisms for neurite outgrowth inhibition. J Cell Biol 177(3):393–399
Williams G et al (2008) Ganglioside inhibition of neurite outgrowth requires Nogo receptor function: identification of interaction sites and development of novel antagonists. J Biol Chem 283(24):16641–16652
Yamashita T, Higuchi H, Tohyama M (2002) The p75 receptor transduces the signal from myelin-associated glycoprotein to Rho. J Cell Biol 157(4):565–570
Hu F, Strittmatter SM (2008) The N-terminal domain of Nogo-A inhibits cell adhesion and axonal outgrowth by an integrin-specific mechanism. J Neurosci 28(5):1262–1269
Goh EL et al (2008) beta1-integrin mediates myelin-associated glycoprotein signaling in neuronal growth cones. Mol Brain 1(1):10
Loschinger J et al (1997) Retinal axon growth cone responses to different environmental cues are mediated by different second-messenger systems. J Neurobiol 33(6):825–834
Song H et al (1998) Conversion of neuronal growth cone responses from repulsion to attraction by cyclic nucleotides. Science 281(5382):1515–1518
Hasegawa Y et al (2004) Promotion of axon regeneration by myelin-associated glycoprotein and Nogo through divergent signals downstream of Gi/G. J Neurosci 24(30):6826–6832
Sivasankaran R et al (2004) PKC mediates inhibitory effects of myelin and chondroitin sulfate proteoglycans on axonal regeneration. Nat Neurosci 7(3):261–268
Snow DM et al (1994) Chondroitin sulfate proteoglycan elevates cytoplasmic calcium in DRG neurons. Dev Biol 166(1):87–100
Koprivica V et al (2005) EGFR activation mediates inhibition of axon regeneration by myelin and chondroitin sulfate proteoglycans. Science 310(5745):106–110
Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420(6916):629–635
Jalink K et al (1994) Inhibition of lysophosphatidate- and thrombin-induced neurite retraction and neuronal cell rounding by ADP ribosylation of the small GTP-binding protein Rho. J Cell Biol 126(3):801–810
Niederost B et al (2002) Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. J Neurosci 22(23):10368–10376
Lehmann M et al (1999) Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci 19(17):7537–7547
Fournier AE, Takizawa BT, Strittmatter SM (2003) Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci 23(4):1416–1423
Dergham P et al (2002) Rho signaling pathway targeted to promote spinal cord repair. J Neurosci 22(15):6570–6577
Domeniconi M et al (2005) MAG induces regulated intramembrane proteolysis of the p75 neurotrophin receptor to inhibit neurite outgrowth. Neuron 46(6):849–855
Yamashita T, Tohyama M (2003) The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci 6(5):461–467
Lingor P et al (2007) Inhibition of Rho kinase (ROCK) increases neurite outgrowth on chondroitin sulphate proteoglycan in vitro and axonal regeneration in the adult optic nerve in vivo. J Neurochem 103(1):181–189
Monnier PP et al (2003) The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol Cell Neurosci 22(3):319–330
Borisoff JF et al (2003) Suppression of Rho-kinase activity promotes axonal growth on inhibitory CNS substrates. Mol Cell Neurosci 22(3):405–416
Sahin M et al (2005) Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron 46(2):191–204
Moore SW et al (2008) Rho inhibition recruits DCC to the neuronal plasma membrane and enhances axon chemoattraction to netrin 1. Development 135(17):2855–2864
Alabed YZ et al (2006) Neuronal responses to myelin are mediated by rho kinase. J Neurochem 96(6):1616–1625
Kubo T et al (2008) Myosin IIA is required for neurite outgrowth inhibition produced by repulsive guidance molecule. J Neurochem 105(1):113–126
Hsieh SH, Ferraro GB, Fournier AE (2006) Myelin-associated inhibitors regulate cofilin phosphorylation and neuronal inhibition through LIM kinase and Slingshot phosphatase. J Neurosci 26(3):1006–1015
Bamburg JR, Bernstein BW (2008) ADF/cofilin. Curr Biol 18(7):R273–R275
Mimura F et al (2006) Myelin-associated glycoprotein inhibits microtubule assembly by a Rho-kinase-dependent mechanism. J Biol Chem 281(23):15970–15979
Alabed YZ et al (2007) Identification of CRMP4 as a convergent regulator of axon outgrowth inhibition. J Neurosci 27(7):1702–1711
Fukata Y et al (2002) CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol 4(8):583–591
Quinn CC et al (2003) TUC-4b, a novel TUC family variant, regulates neurite outgrowth and associates with vesicles in the growth cone. J Neurosci 23(7):2815–2823
Cai D et al (2001) Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci 21(13):4731–4739
Gao Y et al (2003) Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. J Neurosci 23(37):11770–11777
Cai D et al (1999) Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron 22(1):89–101
Qiu J et al (2002) Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34(6):895–903
Neumann S et al (2002) Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34(6):885–893
Gao Y et al (2004) Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 44(4):609–621
Cai D et al (2002) Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron 35(4):711–719
Cao Z et al (2006) The cytokine interleukin-6 is sufficient but not necessary to mimic the peripheral conditioning lesion effect on axonal growth. J Neurosci 26(20):5565–5573
Wen Z et al (2007) BMP gradients steer nerve growth cones by a balancing act of LIM kinase and Slingshot phosphatase on ADF/cofilin. J Cell Biol 178(1):107–119
Benowitz L, Yin Y (2008) Rewiring the injured CNS: lessons from the optic nerve. Exp Neurol 209(2):389–398
Park KK et al (2008) Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322(5903):963–966
Neumann S, Woolf CJ (1999) Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23(1):83–91
Seijffers R, Allchorne AJ, Woolf CJ (2006) The transcription factor ATF-3 promotes neurite outgrowth. Mol Cell Neurosci 32(1–2):143–154
Dill J et al (2008) Inactivation of glycogen synthase kinase 3 promotes axonal growth and recovery in the CNS. J Neurosci 28(36):8914–8928
Bomze HM et al (2001) Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat Neurosci 4(1):38–43
Temporin K et al (2008) IL-1beta promotes neurite outgrowth by deactivating RhoA via p38 MAPK pathway. Biochem Biophys Res Commun 365(2):375–380
Acknowledgments
We would like to thank Dr. Aaron McGee and Corrie Fox for their comments and suggestions on the manuscript and graphic work, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nash, M., Pribiag, H., Fournier, A.E. et al. Central Nervous System Regeneration Inhibitors and their Intracellular Substrates. Mol Neurobiol 40, 224–235 (2009). https://doi.org/10.1007/s12035-009-8083-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-009-8083-y