
Abstract
The insulin-like growth factor receptor type 1 (IGF1R) signalling pathway is activated in the mammalian nervous system from early developmental stages. Its major effect on developing neural cells is to promote their growth and survival. This pathway can integrate its action with signalling pathways of growth and morphogenetic factors that induce cell fate specification and selective expansion of specified neural cell subsets. This suggests that during developmental and adult neurogenesis cellular responses to many signalling factors, including ligands of Notch, sonic hedgehog, fibroblast growth factor family members, ligands of the epidermal growth factor receptor, bone morphogenetic proteins and Wingless and Int-1, may be modified by co-activation of the IGF1R. Modulation of cell migration is another possible role that IGF1R activation may play in neurogenesis. Here, I briefly overview neurogenesis and discuss a role for IGF1R-mediated signalling in the developing and mature nervous system with emphasis on crosstalk between the signalling pathways of the IGF1R and other factors regulating neural cell development and migration. Studies on neural as well as on non-neural cells are highlighted because it may be interesting to test in neurogenic paradigms some of the models based on the information obtained in studies on non-neural cell types.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurogenesis is regulated by a complex network of growth factors and morphogens. Effects of discreet signalling factors on early patterning of the nervous system and on neural cell growth and differentiation are well-documented, but their combinatorial effects are only now starting to be elucidated. Since factors regulating neurogenesis may activate overlapping signalling pathways, the outcome of their combined action may be different from a simple sum of cellular responses elicited by each particular factor. Various modes of interaction between signalling pathways are conceivable, such as competition for a limited pool of a secondary messenger, a cumulative effect of two pathways if they converge on a component that cannot be fully activated by either of them, etc.
A well-recognised function of insulin-like growth factor (IGF)-I is to regulate cellular mass in various tissues by controlling both cell size and number. Complete genetic ablation of IGF-I results in a ~50% decrease in body size [1, 2]. Studies on mice with genetically manipulated expression of IGF system proteins (including IGF-I, IGF-II, IGF receptor type 1(IGF1R) and IGF-binding proteins) selectively in the nervous system revealed that the development of all neural cell lineages is highly dependent on the IGF1R signalling because a consistent finding in those studies was brain size reduction in mice with IGF1R signalling deficiency (by up to 70% in IGF1R knockouts) and brain overgrowth in mice with exaggerated IGF1R activation [3, 4].
Correct morphogenesis of the nervous system requires a tightly regulated localised and transient expression of signalling factors. The peculiarity of the IGF1R pathway is that it is activated early in the primordial central nervous system (CNS) and its activity is maintained, with some regional variation, throughout development into adulthood. Therefore, there are many instances during developmental neurogenesis or during the regenerative and adaptive responses of the adult CNS when the IGF1R and receptors for growth and morphogenetic factors with more localised distribution in the CNS are co-activated. This raises interest to the molecular events where the IGF1R and these pathways converge.
IGF1R Signalling in Neural Cells
IGF1R Signalling Complex Assembly
The IGF1R, whose ligands include IGF-I, IGF-II and insulin at a concentration in excess of 100 nM, is a receptor tyrosine kinase. It consists of covalently linked two α- and two β-subunits. Ligand binding to the α-subunits of the IGF1R induces tyrosine kinase activity in the β-subunits by the phosphorylation of three tyrosine residues within the receptor autocatalytic domain. This leads to the phosphorylation of other tyrosine residues in the IGF1R β-subunit and subsequent formation of the signalling complex by docking and tyrosine phosphorylation of several intracellular proteins, including insulin receptor substrates (IRS) and Src homology domain-containing adaptor proteins [5–8]. At least three IRS isoforms, IRS-1, IRS-2 and IRS-4, are expressed in the nervous system [9] and may play different roles in neural cell activation and development [10]. The activation of the two major signalling cascades, the phosphatidylinositol-3 (PI3) kinase/Akt and the Ras/Raf/mitogen-activated protein kinase (MAPK) pathways, resulting from the IGF1R signalling complex formation is well documented in neural cells.
Neuroprotective Pathways of the IGF1R
The serine threonine kinase Akt mediates pro-survival effects of the IGF1R signalling pathway by targeting a number of proteins regulating tissue growth, such as the glycogen synthase kinase 3 (GSK3), the mammalian target of rapamycin (mTOR) and proteins belonging to a subclass of forkhead transcription factors (FoxO) [11, 12] (Fig. 1). Akt activation is coupled with IGF1R ligation by the activation of PI3 kinase. PI3 kinase is activated by recruitment to the IGF1R signalling complex and generates phosphatidylinositol 3,4,5-triphosphate at the plasma membrane. This forms a binding site for the pleckstrin homology domain of Akt, which is activated following translocation to the plasma membrane. The PI3 kinase/Akt pathway is coupled to the IGF1R in neural cells [13] and may be developmentally regulated in neurogenesis [14].

Neuroprotective signalling by IGF1R. Three signalling pathways mediating cytoprotective effect of IGF1R signalling with a confirmed role in developing and mature neural cells are shown. IGF1R activation leads to the activation of Akt, which phosphorylates mTOR, GSK3β and FoxO, master regulators of pathways controlling cell growth. Positive regulation is shown as an arrowhead, negative as a black circle. References to the reports investigating a role of these pathways in neuroprotection mediated by IGF1R activation are given in brackets. PIP 3,4,5 phosphatidylinositol 3,4,5-triphosphate
The serine/threonine kinase GSK3 induces inhibitory phosphorylation of several proteins in pathways promoting cell growth and proliferation, including β-catenin and N-myc [15]. Akt was phosphorylated in response to IGF1R activation and inhibited GSK3β in different types of neurons [16–18]. In the study of Johnson-Farley et al. [16], IGF-I and brain-derived neurotrophic factor exhibited an additive effect on the activation of the PI3 kinase/Akt pathway that was translated into the additive effect on the inhibition of GSK. The authors explained the additive effect of the two growth factors by spatial segregation of their signalling pathways, suggesting a possible mode of interaction between signalling pathways in general.
In dephosphorylated form, FoxO transcription factors are translocated to the nucleus where they induce the expression of proteins inhibiting cell cycle progression, such as the cyclin D kinase (cdk) inhibitor p27kip1 and the retinoblastoma-related protein p107. They also inhibit the expression of cyclin D and upregulate eLF4E binding protein 1, a global transcriptional repressor sequestering eIF4E [19]. Akt-mediated FoxO protein phosphorylation followed by their nuclear exclusion and transcriptional inactivation [20] has been implicated in a neuroprotective effect of IGF1R activation [18, 21, 22].
mTOR is activated by Akt-mediated phosphorylation. mTOR activates p70 ribosomal S6 protein, a positive regulator of protein translation [23]. mTOR mediates effects of IGF-I and other growth factors on cell growth, but little experimental evidence is available to appreciate the significance of IGF1R/mTOR axis in neural cells. In cortical neurons, the mTOR pathway, in response to activation by IGF-I or insulin, induced the expression of the monocarboxylate transporter 2 [24], suggesting a role for this pathway in the regulation of neuronal energy metabolism.
Another downstream target of the IGF1R signalling cascade is the upstream binding factor 1 (UBF1), an important regulator of cell size. Cell size largely depends on protein biosynthesis, which, in turn, depends on the biosynthesis of ribosomes. UBF1 transcriptionally activates RNA polymerase I, a limiting factor in ribosome biosynthesis. UBF1 gene expression is activated by the nuclear translocation of IRS-1 in response to IGF-I stimulation (reviewed in reference [1]).
MAPK in IGF1R Signalling
The activation of Ras by the son-of-sevenless recruited to the IGF1R signalling complex is the major mechanism of MAPK activation in response to IGF-I. Additional mechanisms, such as the transactivation of G-proteins [25] or Src kinase activation [26], have also been shown. Which particular pathways link the Ras/Raf/MAPK to IGF1R activation in neural cells is not fully understood, but the activation of MAPK in response to IGF1R ligation has been clearly demonstrated in several, but not all, types of neural cells. This pathway was activated by the IGF1R in adult rat hippocampal neural stem and progenitor cells (NSPC) [27] and, to a lesser extent, in postmitotic neurons from embryonic rat hippocampus [16, 28]. No MAPK activation, however, was found in astrocytes of transgenic mice overexpressing IGF-I [13]. The involvement of the MAPK pathway in downstream signal transduction from activated IGF1R moderately increased in cells of oligodendroglial lineage as they matured [14], but an anti-apoptotic response of neonatal rat oligodendrocyte precursors (OPC) to IGF-I was independent of this pathway [29]. Extracellular response kinase inhibition may be required for an anti-apoptotic effect of IGF-I on neural cells [30]. The available experimental evidence suggests that the efficacy of IGF1R coupling to the Ras/Raf/MAPK signalling pathway varies between different neural cell subsets.
Anatomical Basis of Neurogenesis and a Role of Signalling Factors
Two models have been proposed to account for the patterning activity of morphogenetic factors produced in organising centres of the developing nervous system (Fig. 2). In one of them, different cellular identities are induced along the concentration gradient of a signalling factor secreted at one end of undifferentiated tissue mass. This mechanism, for example, accounts for the dorsoventral patterning of the spinal cord by sonic hedgehog (Shh) secreted from the notochord [31]. An alternative possibility is that a morphogenetic factor produced by a restricted group of cells regulates cell fate specification within the surrounding area. Such a mechanism may account for the patterning of the telencephalon [32, 33]. The patterning molecules impart positional information to neural progenitors by inducing specific sets of transcription factors in them [34, 35]. Patterns of transcription factor expression demarcate boundaries of neuroepithelial domains, which are particularly distinct at the embryonic stage of development (before E12.5 in mice). Several signalling molecules have been implicated in early CNS patterning on the basis of their expression in organising centres and ability to form concentration gradients, as well as on the basis of developmental abnormalities associated with their deficiency or ectopic expression. They include ligands of Notch, Shh, fibroblast growth factor (FGF) family members, ligands of the epidermal growth factor (EGF) receptor (EGFR), bone morphogenetic proteins (BMP) and Wingless and Int-1 (Wnt; Fig. 3a).

Early CNS patterning. Two models of early tissue patterning are schematically represented. In model 1, a signalling factor (black circles) secreted at one end of the tissue mass forms a concentration gradient. Cells (grey circles) along this gradient are exposed to different concentrations of the signalling factor. The set of transcription factors which a given cell will express depends on what concentration of the signalling factor it is exposed to. This will define its cellular identity (id1, id2, etc.). In model 2, a signalling factor is produced in a restricted area and induces a particular cellular identity in surrounding cells [31–33]

Signalling factors regulating neurogenesis. a Typical modes of signalling factor expression in the developing CNS are schematically represented. These include a concentration gradient of BMP, which mediates patterning of dorsal midline, local production of Wnt, which mediates the induction of dorsal identities, secretion of Notch ligands, which maintains multipotential progenitors in undifferentiated state, a concentration gradient of the ventralising morphogen Shh and tonic expression of IGF1R ligands throughout the developing CNS. b Signalling factors expressed in the adult SVZ are shown. IGF1R activation in the adult SVZ is maintained by local secretion and systemic delivery of its ligands. References to selected reports are shown in brackets
The pool of NSPC is reduced as development progresses, but a subset of self-renewing multipotential progenitors persists into adulthood residing at restricted anatomical locations of the mature CNS adult neurogenic zones [36]. The best characterised area of adult neurogenesis is in the subventricular zone (SVZ) [37–39]. Newly born neuroblasts tangentially migrate from the SVZ to the olfactory bulb (OB) to become interneurons [40–42]. In addition, a platelet-derived growth factor (PDGF) receptor α (PDGFRα)-expressing subset of astrocyte-like glial fibrillary acidic protein+ type B cells in SVZ may give rise to NG2 chondroitin sulphate-positive OPC that migrate into the brain parenchyma [43, 44] to become myelinating oligodendrocytes [45]. The expression of Sox2 and Pax6 may define subsets specified for glial and neuronal fate, respectively, in the human SVZ [46]. Additional areas of adult neurogenesis include the subgranular layer of the dentate gyrus (SGZ) in the hippocampus [47] and cerebellum [48, 49] where neurogenesis may continue until puberty [50]. Self-renewing cell populations are also present in the normal CNS parenchyma and include pyramidal cells in the rat pyriform cortex [51], O4+ OPC and multipotential A2B5+ cells [44, 52].
Adult neurogenic zones have some features characteristic for the developing CNS, such as the basal lamina, proximity to the vascular bed and polarised niche cells, but there is no evidence of organising centre formation in them. Many proteins regulating developmental neurogenesis are expressed in the adult brain and have been shown to play a role in adult neurogenesis (Fig. 3b). They include leukaemia inhibitory factor (LIF), which stimulates growth of primitive NSPC in early embryogenesis (before E8.5 in mice) [53], and Numb/Numblike, a protein with a cell fate-determining function in the developing CNS [54]. The expression of such proteins in the adult CNS may be regulated by external stimuli, with LIF expression in the adult SVZ in response to hypoxic conditions being one of such examples [55].
IGF1R in Neurogenesis
The IGF1R and its ligands IGF-I and IGF-II are widely expressed in the developing CNS starting from early embryogenesis and continue to be expressed, with some variation in intensity, during CNS development and into adulthood; cells of all three neural lineages have been shown to express the IGF1R and secrete its ligands [56–67]. IGF-I, produced locally or delivered to the CNS from the circulation, induces a constitutive level of IGF1R-mediated signalling in the adult CNS, which is important for brain functions and adult neurogenesis [68]. This level can increase in response to a pathological process in the CNS [69] or as a result of adaptive response to exercise and correlates with increased hippocampal neurogenesis [70]. Secretion of IGF1R ligands in organising centres or its expression in a graded manner is not characteristic for the developing mammalian CNS whose patterning, by contrast to the neurogenesis in Xenopus laevis [71–73], is not known to depend on IGF1R activation.
Cell differentiation involves transcriptional activation of genes encoding proteins that mediate functions of mature cells of a given lineage. IGF1R signalling may play a role in terminal differentiation of oligodendrocytes because IGF-I induces the expression of the gene for myelin oligodendrocyte glycoprotein in OPC [74–76] and has been found to synergise with transferrin in the induction of myelin basic protein gene expression [77]. A pro-survival effect of IGF-I may complicate interpretation of experiments addressing its role in neural cell differentiation.
The recently emerged hypothesis that cellular proteins involved in the regulation of apoptosis, such as caspases, FoxO3a, mTOR and p53, may also be involved in the differentiation of neural cells [78–80] suggests a novel conceptual framework for understanding the role of cytoprotective factors, in particular IGF1R ligands, in neural cell differentiation. In this regard, recent reports on hypermyelination of the CNS in mice constitutively overexpressing active Akt in OPC [81] and the involvement of mTOR in the induction of genes for several myelin components in maturing OPC [82] are particularly interesting because, as said above, the Akt/mTOR axis is a target of the IGF1R signalling.
A role for the IGF1R signalling in the epigenetic control of gene expression in the brain [13] suggests a possible mode of co-operation between the IGF1R and signalling factors regulating neurogenesis. Epigenetic effects involve covalent modification of histone proteins by acetylation, phosphorylation and methylation. By promoting such modifications, IGF-I may loosen the structure of the nucleosome, making it more accessible to transcription factors induced by other signalling factors.
Some of the signalling factors with a patterning function in the developing CNS can also control the size of specific CNS structures because they may regulate survival and proliferation of the cell subsets whose fate they specify. It is incompletely understood how these pathways can integrate their action with the signalling pathway of the IGF1R, a global regulator of tissue mass, but some points of convergence between them have been elucidated and are discussed below.
Modulation of Signalling Pathways Regulating Neurogenesis by IGF1R
Shh
In the developing CNS, Shh is secreted in a spatially restricted manner from the ventral midline and displays a concentration gradient decreasing dorsal-ward (Fig. 3a). This morphogen defines ventral identities in the neural tube [83–85] and in the embryonic forebrain [35, 86–89]. Shh produced by Purkinje neurons in the neonatal cerebellum is a sole mitogen-expanding cerebellar granule cell precursors (reviewed in [90]), and in adulthood its activity is required for neurogenesis in the hippocampus [91, 92] and SVZ [93, 94] (Fig. 3b).
Shh has a mitogenic and pro-survival effect on subsets of those neuronal cells which it specifies. As well as its patterning activity, the growth-promoting effect of Shh is mediated by transcription factors of the Gli family [35, 95], which are unique for this signalling pathway. Unlike the anti-apoptotic response to IGF1R activation, the effect of Shh on cell survival does not involve a direct activation of the PI3 kinase/Akt pathway [96], a GSK3β inhibitor. Moreover, GSK3β itself can negatively regulate Shh-induced cell proliferation by inactivating N-myc, a proto-oncogene that is a direct transcriptional target of Shh and mediates its mitogenic effect [97, 98] (Fig. 4). Therefore, the effect of Shh may be dependent on co-operation with other factors that can activate the PI3 kinase/Akt pathway and thereby inhibit GSK3β, such as IGF1R ligands [90]. As discussed below, in addition to Shh, the IGF1R/PI3 kinase/Akt/GSK3β axis may regulate signalling pathways of several other morphogens. In addition, the interaction between Shh and IGF1R pathways may be bi-directional as suggested by the ability of Shh to stabilise IRS-1 by interfering with its mTOR-mediated degradation [99].

Convergence of the IGF1R pathway with signalling pathways implicated in neurogenesis. Schematic representation of interactions mediating integration of IGF1R signals into the pathways of signalling factors regulating neural cell growth and development. Solid arrows show pathways confirmed in neural cells; dash arrows show pathways demonstrated in non-neural cells only. Positive regulation is shown as an arrowhead, negative as a black circle. References to the reports confirming a given interaction between signalling pathways are given in brackets. Well-established interactions are shown unreferenced. The IGF1R/PI3K/Akt/GSK3β axis positively regulates signalling pathways activated by Wnt, BMP, Shh and Notch ligands. β-cat β-catenin; Ptc patched, Shh receptor; Smo Ptc inhibitor, which dissociates upon Shh binding; Fz frizzled, Wnt receptor; Lpr Fz co-receptor; Jag Jagged, Notch ligand; RTK a receptor tyrosine kinase
Wnt
Wnt is secreted by the dorsal midline and adjacent non-neural cells and is involved in dorsal telencephalon patterning (reviewed in [34, 100]; Fig. 3a); deficiency of this pathway leads to underdevelopment of dorsally derived structures of the brain, such as the hippocampus [101]. The major cellular receptor for Wnt is Frizzled, a seven-pass transmembrane protein, which functions in combination with a co-receptor, a single-pass transmembrane protein belonging to the family of low-density lipoproteins.
GSK3β negatively regulates the canonical Wnt signalling pathway in neural progenitors by the inactivation of its relatively specific component β-catenin [102–104] (Fig. 4). In the absence of Wnt stimulation, β-catenin associates with a complex of axin and adenomatous polyposis coli (APC) gene product, which pre-phosphorylates β-catenin, making it susceptible to the phosphorylation by GSK3β. GSK3β-mediated phosphorylation reduces the pool of β-catenin available for signalling by targeting it to degradation. When released from the axin/APC complex in response to Wnt stimulation, β-catenin becomes resistant to GSK3β-mediated phosphorylation and translocates to the nucleus where it forms a complex with members of the T cell factor (TCF)/lymphoid enhancer factor (LEF) family of DNA-binding proteins, which activates transcriptional targets of the Wnt signalling pathway. Wnt has also been shown to inhibit GSK3β [105], but significance of this inhibition in developing mammalian neural cells has not yet been confirmed. A role of the IGF1R/PI3 kinase/Akt/GSK3β axis in the activation of the canonical Wnt signalling in developing neural cells is incompletely understood, but insulin- or IGF-I-induced GSK3β inhibition, mediated by the PI3 kinase/Akt pathway, has been shown to stabilise β-catenin in cancer cell lines and in normal epithelial cells and fibroblasts and can induce the expression of Wnt target genes [106–108].
Activation of the canonical Wnt signalling pathway has diverse effects on developing neural cells ranging from the maintenance of self-renewing multipotential pool of progenitors to the induction of neuronal fate specification [109–113]. Whether progenitor cells proliferate or undergo neuronal specification in response to Wnt signal may be mediated by cell-autonomous mechanisms [114], as well as by modulation of cellular response to Wnt by growth factors that can activate the IGF1R/PI3 kinase/Akt/GSK3β axis. The latter possibility is suggested by the ability of FGF2 to divert NSPC overexpressing β-catenin from differentiation to self-renewal, which correlated with the nuclear translocation of β-catenin and GSK3β inhibition [115], but whether IGF-I can have a similar effect remains to be seen.
Studies on non-neural cells indicate that some of Wnt target genes are transcriptionally activated by the IGF1R independently of GSK3β inhibition via mechanisms that involve an Akt-independent action of PI3 kinase [116] or the Ras/Raf/MAPK pathway [106]. Another GSK3β-indepenent mode of co-operation between the IGF1R and Wnt pathways involves formation of a complex between IRS-1 phosphorylated by IGF1R activation and β-catenin. This complex was translocated to the nucleus and activated the cyclin D1 promoter in mouse embryonic fibroblasts [117] (Fig. 4).
It has been suggested that the competition for a limited pool of β-catenin between FoxO proteins and the transcription factor complex of the Wnt signalling pathway TCF/LEF constitutes a regulatory mechanism inhibiting the cell-growth-promoting Wnt signalling under conditions of cellular stress when FoxO proteins become activated [116, 118, 119]. Via this mechanism, factors causing cellular stress, for example at sites of CNS pathology [120], may compromise Wnt-dependent maintenance of NSPC. One could hypothesise that the IGF1R signalling pathway, via one of the above-said mechanisms, might promote Wnt target gene expression at the expense of FoxO-mediated transcriptional inhibition in NSPC, thereby rescuing their Wnt-dependent self-renewal at sites affected by a pathological process.
Notch
A well-documented function of the Notch receptor signalling pathway is to maintain the pool of NSPC by promoting their symmetrical division and preventing their neuronal differentiation [121–124]. This pathway also regulates neural cell death, and its activation has been shown to either promote [125] or inhibit [126, 127] apoptosis in different subsets of neural cells. Similar diversity of cellular responses to Notch activation has been reported in non-neural cells [128]. There are little published data on the role of GSK3 in Notch signalling pathway in neural cells. In non-neural cell lines, however, Notch has been shown to be a substrate of inhibitory phosphorylation by both isoforms of GSK3, α and β [129, 130], suggesting that Notch signalling, in similarity to the pathways of Shh and Wnt, may be influenced by the IGF1R/PI3 kinase/Akt/GSK3β axis (Fig. 4).
Notch and IGF1R pathways may also co-operate at levels other than GSK3β activation. The pro-apoptotic effect of Notch signalling on some of the NSPC subsets may be dependent on the activation of p53 and its transcriptional targets Bax and Noxa [125]. The p53-dependent pathway of cell death may be inhibited by IGF1R activation, as demonstrated in studies on transformed cells [131] and primary neurons [132], indicating that Notch and IGF1R signalling may have opposite effects on p53-mediated apoptosis. Furthermore, Akt-dependent activation of mTOR may also mediate the anti-apoptotic effect of Notch signalling [126]. This identifies p53 and mTOR as potential points of convergence between Notch and IGF1R signalling (Fig. 4).
The expression of Notch1 and its ligands in the postnatal mouse SVZ [133, 134] (Fig. 3b), the ability of adult NSPC to respond to Notch stimulation [126] and the reduced number of NSPC in neurogenic zones of adult Notch1-deficient mice [124] point to the importance of this pathway for adult neurogenesis. This suggests that the interplay between the Notch and IGF1R signalling pathways may play a role in the regenerative response in the postnatal CNS.
BMP
BMPs, members of the transforming growth factor beta superfamily, have diverse effects on NSPC including specification of neuronal or astroglial identities, cell proliferation, mitotic cycle arrest and apoptosis [135, 136]. They inhibit oligodendroglial specification at all developmental stages [135]. BMPs expressed in the signalling centre located along the dorsomedial edge of the cortical primordium [137] mediate patterning of the medial–lateral axis of the telencephalon [34, 138] (Fig. 3a). They also promote development of the choroid plexus [32] and specify dorsal identities in the hindbrain [139]. Adult neurogenesis in the SVZ [140] and hippocampus [141] may be in part regulated by a balance between BMPs, which are secreted by endothelial cells [142], and their inhibitor Noggin (Fig. 3b).
The type of cellular response to BMPs is determined by intensity of activation, different signalling capabilities of their receptors BMPR1 and BMPR2 [143] and convergence of their signalling pathways with signals delivered by other receptors, such as the receptors for LIF and, most importantly for this discussion, receptor tyrosine kinases [136]. Adding to this complexity is the appreciation that, dependent on the developmental stage of neural cells, the interaction of BMPs with such co-activating signals can be either co-operation or antagonism. For example, neural plate specification in the ectoderm in early gestation was dependent on the FGFR-mediated inhibition of BMP signalling [144], but a combination of BMP2 and FGF2 activities was required for specification of identities characteristic for choroid plexus and neural crest [145].
There are several events in the signalling pathways of the BMPRs and IGF1R where negative or positive interactions between these pathways are possible, and some of these interactions have been confirmed experimentally in neural cells. Intracellular signal from activated BMP receptors is transmitted in neural cells via the canonical pathway that involves activating phosphorylation of the signalling proteins Smad1, Smad5 and Smad8, which then associate with common-partner Smads and translocate to the nucleus where they function as transcription factors [146]. Non-canonical BMP signalling and its functional significance in developing neural cells have also been shown. The non-canonical pathways include the activation of STAT3 by S727 phosphorylation, which may be mTOR dependent [147] and a pathway leading to p38MAPK activation [148]. In addition, the activation of mTOR mediated by the PI3 kinase/Akt pathway in response to BMP-2 has been demonstrated in cancer cells [149], but its significance in developing mammalian neural cells has not been elucidated.
As first shown by Kretzschmar et al. [150], BMP-activated Smad1 can be inactivated by phosphorylation with MAPK at the linker region, a location different from the BMPR phosphorylation site. Functional significance of this inhibitory phosphorylation was confirmed in studies on the Xenopus embryo and suggested an explanation to the known ability of FGF2 and IGF-I to promote neurogenesis in this developmental paradigm by inhibiting the effect of BMP [71, 151]. To complicate the prediction of the effect that IGF1R signalling may have on cellular response to BMP in mammalian neural cells, a second phosphorylation event mediated by GSK3β, in addition to phosphorylation by MAPK, has been found to be required for targeting Smad1 to degradation [105]. Thus, IGF1R activation has the potential to inhibit Smad1 by activating MAPK cascade as well as the potential to protect this signalling molecule from inactivation via the IGF1R/PI3 kinase/Akt/GSK3β axis (Fig. 4).
MAPK-dependent Smad1 inhibition may play a role in mammalian neurogenesis because in the rat spinal cord the inhibition of BMP signalling via MAPK-dependent Smad1 phosphorylation mediated one of the ventralising effects of FGF2, the Shh-independent induction of oligodendroglial differentiation [152]. In the adult rat hippocampus, IGF-I promoted induction of oligodendroglial differentiation via a mechanism that also involved inhibition of BMP signalling [75]. A role of Smad1 phosphorylation was not reported in that study, but an increased expression of extracellular (Noggin) and cytoplasmic (Smad6) inhibitors of BMP signalling was demonstrated. Furthermore, the expression of the lipid phosphatase and tensin homolog deleted from chromosome 10 (PTEN), an inhibitor of PI3 kinase, in adult NSPC in response to BMP stimulation suggests a possible mechanism whereby a signal from BMPR may antagonise the IGF1R pathway [142].
Possible modes of interaction between BMP and IGF1R signalling are suggested by examples of crosstalk between these pathways during histogenesis of non-neural tissues. BMP-2 and BMP-9, which have no mitogenic effect on chondrocytes, may indirectly stimulate their proliferation by enhancing binding of IGF-I, which is a mitogen for these cells [153]. Furthermore, BMP-2 and IGF-I signalling pathways had a synergistic effect on the osteogenic differentiation of human mesenchymal [154, 155] and bone marrow [156] stem cells, with protein kinase D, MAPK and PI3 kinase identified as points of convergence between them.
Receptor Tyrosine Kinases
The IGF1R signalling complex contains components that are shared with other receptor tyrosine kinases regulating neural cell development, including FGFR family members, EGFR and PDGFRα. These shared components may mediate crosstalk between receptor tyrosine kinases. The Grb2-associated binder family adapter proteins may be interesting in this regard because they provide a major link between activated receptor tyrosine kinases and downstream signalling and play non-redundant functions in mammalian neurogenesis [157–159]. Furthermore, the downstream of tyrosine kinase (Dok) family adaptor proteins p62Dok-1 and p56Dok-2 have the potential to cross-inhibit activation of the IGF1R and other receptor tyrosine kinases (Fig. 4). When tyrosine phosphorylated by EGFR [160, 161], PDGFR [162] or IGF1R, Dok adaptors can inhibit the MAPK pathway, an effect that depends on their ability to bind several signalling proteins, most importantly RAS GTPase activating protein, and to modulate the activity of their binding partners [160, 162–167]. Neuronal cells in the adult hippocampus express p62Dok-1 with a pattern similar to their IGF1R expression [168].
FGFRs
At least 18 members of the FGF family play a role in neurogenesis [169], and at least three members of the FGFR family, FGFR1-3, mediate their effects ([170, 171]; reviewed in reference [144]). The analysis of FGFR signalling in neural cells is complicated because FGFRs activate different sets of downstream signalling molecules [172]; their expression is developmentally regulated [173–176]; they display different ligand binding affinities to a given FGF [169, 175, 177] and elicit different cellular responses in neural cells [173].
Examples of FGF-mediated early CNS patterning include specification of neuronal identities at the junction between the midbrain and hindbrain by FGF8, FGF17 and FGF18 (reviewed in [178]), anterior–posterior patterning of the telencephalon by FGF8 [34, 179, 180], medial–lateral patterning of the dorsal telencephalon by FGF7 [181], patterning of cerebellar structures by FGF8 and FGF17 [182], laminar patterning of the developing neocortex by FGF18 [183], ventral forebrain specification [184] and the Shh-independent specification of oligodendrocytes in the dorsal neural tube (reviewed in [185]). Those FGFs that induce cell fate specification of developing neural cells may also promote subsequent expansion of the cell subsets they have specified. FGF2 is, perhaps, more widely expressed in the embryonic nervous system than FGFs with patterning activity [186], and its expression continues in the adult CNS [187, 188]. Its wide distribution resembles that of the IGF1R and its ligands. In addition, reduced size of progenitor pool resulting from FGF2 deficiency [188–192] and its well-recognised role as a growth factor for NSPC in vitro suggest that FGF2 may function as an NSPC growth and maintenance factor in embryonic and mature CNS.
The ability of some of the FGFs to induce cell fate specification is in apparent disagreement with the ability of FGF2 to maintain a pool of self-renewing neural progenitors by promoting their symmetrical division. Amongst other possible explanations, which I do not discuss here, a plausible suggestion is that co-activation with other factors may bias the FGFR-induced cellular response either toward cell fate specification or toward self-renewing cell division. IGF1R ligands might play a role of such a factor for the following reasons. On the one hand, patterning activity of FGFs does not depend on IGF1R co-activation because there is no loss of particular structures in the IGF1R-signalling-deficient CNS. On the other hand, a mitogenic effect of FGFs on developing neural cells strongly depends on the co-activation of the IGF1R [193, 194]. Changing local availability of IGF1R ligands suggests a hypothetical mechanism to account for modulation of the cellular response to FGFs: cell fate specification occurs in the absence of IGF1R activation whereas a co-activation of IGF1R and FGFRs is mitogenic.
What is the molecular basis of interaction between the FGFRs and IGF1R signalling pathways? A direct promotion of mitotic cycle progression in FGF-stimulated neural cells by the co-activation of the IGF1R has been clearly demonstrated. The mitotic cycle restriction points regulating transition from G1 to S phase are possible convergence points of growth factor signalling pathways. Accelerated transition through G1 phase of the mitotic cycle in response to IGF1R activation has been observed in various cell types including embryonic [195] and adult neural progenitors [196]. The formation of the early G1 complex containing cyclin D1, cdk4 and the cdk inhibitor p27kip1 is required for the cell to pass the restriction points and proceed to S phase [197, 198]. IGF-I applied on its own was unable to induce early G1 complex formation in primary OPC, but the formation of this complex in response to FGF2 was enhanced by co-stimulation with IGF-I and so was the association of cyclin E with cdk4 in late G1, which was required for progression of the cell to peak S phase [199].
Furthermore, FGFR activation can prevent apoptotic death of non-dividing neural cells, with the PI3 kinase/Akt signalling pathway playing a role in this neuroprotection [200]. The interaction between neuroprotective signalling pathways has been addressed in a number of recent reports [16, 28, 201, 202]. Johnson-Farley et al. demonstrated that FGF2 and IGF-I co-treatment had a cumulative effect on the phosphorylation of Akt in cultured hippocampal neurons, which partially correlated with the increased survival of cells co-treated with both growth factors in comparison with cells treated with FGF2 alone [28].
EGFR and PDGFRα
Two other receptor tyrosine kinases, EGFR and PDGFRα, play an important role in neurogenesis. EGF promotes expansion of NSPC clones whose multipotentiality may be restricted in comparison with FGF-responsive clones because they emerge at a later developmental stage [203], and in the adult SVZ they have the morphology of transit amplifying progenitor cells and neuroblasts [204]. EGF-responsive precursors may be biassed to the astroglial lineage [192] or to the oligodendroglial lineage, as suggested by the ability of EGF to expand pre-progenitors positive for the polysialylated form of the neural cell adhesion molecule (PSA-NCAM), a subset in the rat neonatal neocortex that gives rise to OPC [205, 206]. EGFR ligands may also have a patterning activity because they are expressed in organising centres of the embryonic CNS [181].
A combination of IGF-I and EGF exerts a far more potent growth-promoting effect on the PSA-NCAM+ pre-progenitors than either factor alone [206] suggesting a possibility of co-operation between their signalling pathways. The molecular basis of this co-operation in neural cells is largely unknown. In mammary epithelial cells, in which a synergistic effect of IGF-I and EGF on cell proliferation was also observed, IGF1R and EGF promoted Akt phosphorylation via different signalling pathways and only stimulation through EGFR induced the activation of the Ras/Raf/MAPK cascade [207].
PDGFRα is expressed in distinct neuroepithelial domains specified by transcription factors Olig1 and Olig2 [208] in the embryonic spinal cord, hindbrain and ventral telencephalon. Early OPC transiently express high levels of PDGFRα, and its ligand, PDGFAA, is a growth factor for them [209]. PDGFRα expression and PDGFAA responsiveness rapidly decrease to zero as these cells mature to become OPC, through the stage of the PSA-NCAM+ pre-progenitors [206, 209].
A combination of PDGFAA and FGF2 was mitogenic for the multipotential A2B5+ progenitors from the adult rat corpus callosum and IGF-I enhanced the effect of these growth factors. The more mature O4+ OPC from the same source proliferated only when IGF-I was applied in addition to the other two growth factors [74] suggesting that the dependency of PDGFAA- and FGF2-responsive progenitors on IGF1R signalling may increase as they mature.
IGF1R and Developmental Changes in Neural Cell Motility
Molecular Basis of Neural Cell Motility
The motility of developing neural cells changes according to the requirements of a given developmental stage. In order to ensure correct positioning of a progenitor cell within a concentration gradient of a signalling factor, this cell has to remain immotile up to a strictly defined developmental stage when it leaves the boundaries of the neuroepithelial domain and migrates, radially or tangentially, to its final destination [210]. Neural cell migration is usually guided by phenotypically distinct tissues including radial glia of the embryonic CNS [211], Bergman glia, which mediates final layering of the cerebellum shortly after birth, and the rostral migratory stream [41] of the adult brain. Normal mature brain parenchyma, however, is permissive for neural cell migration too [212, 213]. The transition between motile and sessile phenotype in developing neural cells is regulated by changes in the ligand binding affinities of adherence junctions and focal adhesions, two types of multiprotein complexes on their cytoplasmic membrane [214, 215].
The function of adherence junctions is to mediate intercellular adhesion. Increased ligand binding affinity of adherence junctions is often associated with decreased cell motility. Cadherins are adhesion molecules in adherence junctions. Their multiple isoforms are expressed in neural cells [216–220]; their expression is developmentally regulated and correlates with regional cell specification [221–223]. Correlation between increased cadherin activity in neural cells and decreased cell motility has been shown [217, 224].
The function of cadherins as cell adhesion molecules is mediated by homophilic interaction between calcium-binding repeats of two cadherin molecules on the cytoplasmic membranes of apposing cells. Cadherin cytoplasmic domains are linked to the cytoskeleton and a number of intracellular proteins. For example, one of the most common cadherin isoforms expressed in neural cells, E-cadherin, is associated with α-, β- and γ-catenins [225]. Functional regulation of adherence junctions involves modulation of cadherin expression and their interaction with the cytoskeleton rather than conformation-dependent affinity modulation [226]. Redistribution of intracellular pool of β-catenin to adherence junctions stabilises E-cadherin, thereby increasing ligand binding affinity of adherence junctions. Since, as said above, β-catenin is also involved in the Wnt canonical signalling pathway, it may play an important role in the co-ordination of neural cell development and their motility [111].
Focal adhesions mediate binding of cells to the extracellular matrix (ECM). Their ligand-biding components are β integrins, heterodimers formed by several combinations of a limited number of α- and β-subunit. The ectodomains of β integrins bind to ECM proteins, such as laminins, fibronectin, collagen, etc., and their cytoplasmic domains are associated with a variety of cytoskeletal, adaptor and signalling molecules constituting focal adhesions. Focal adhesions link the actin cytoskeleton to the ECM and transmit signals generated by β integrin ligand occupancy. Cell migration fully depends on oscillating changes in β integrin ligand-binding affinity, which are translated, via a number of intermediate interactions, into dynamic remodelling of the cytoskeleton. NSPC express several sub-classes of β integrins that are functionally important for their maintenance and survival [227–229]. The expression of these molecules is developmentally regulated, as for example shown in cells of the oligodendroglial lineage, which express β1, β3 or β5 integrin subunits at different stages of maturation, with a positive correlation between the expression of β1 and motile phenotype [230].
Cell migration through solid tissues is a common biological processes studied in many paradigms including embryogenesis, haematopoiesis, inflammation, tumour metastasis, etc. Metastasis of some tumours strongly depends on the activation of receptor tyrosine kinases, which can induce separation of transformed cells from an epithelial monolayer and stimulate their motility and invasion of tissue parenchyma. IGF-I is amongst the most potent scattering factors for breast cancer cells [231] and can also modify migratory behaviour of other transformed and untransformed cells, such as glioblastoma [232] and cardiac fibroblasts [233]. The activation of the receptor tyrosine kinases of FGFR family [234], PDGFRα [235, 236] and IGF1R [237–239] has been shown to stimulate migration of neural cells at different developmental stages. Molecular events underlying the ability of receptor tyrosine kinases to promote cell migration have been elucidated in transformed cells, but their nature in neural cells is largely unknown.
Disruption of Intercellular Adhesion by IGF1R Activation
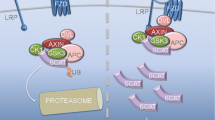
Invasive growth of transformed cells and epithelial-to-mesenchymal transition in response to IGF1R activation is thought to result from disintegration of adherence junctions caused by phosphorylation of β-catenin and its translocation from the E-cadherin-bound pool to the nucleus [240]. This mechanism, however, is not uniformly confirmed [241]. A possibility of cross-signalling between the IGF1R and adherence junctions is suggested by the formation of multimolecular complexes containing components of the IGF1R signalling pathway together with adherence junction components in a number of cell types, including mouse embryonic stem cells [117, 242]. Reorganisation of α-actinin mediated by the PI3 kinase/Akt pathway may also promote disintegration of adherence junction in response to IGF1R activation [231] (Fig. 5; Table 1).

Pathways mediating increased cell motility in response to IGF1R activation. Schematic representation of possible pathways transmitting the signal from the activated IGF1R to focal adhesions (FA) and adherence junctions (AJ), multiprotein cytoplasmic membrane complexes mediating cell adhesion and motility. Disruption of AJ followed by increased cell motility may result from Akt-mediated β-catenin phosphorylation. The scaffolding protein RACK1, which shuttles signalling proteins between different cellular compartments, may play a role in the increased adhesion turnover in FA in response to IGF1R activation. Disintegration of AJ and oscillating changes of β-integrin ligand binding affinity in FA are required for increased cell motility
Little is known about reorganisation of adherence junctions in response to IGF1R activation in neural cells, but a role of the PI3 kinase/Akt pathway in the regulation of neural cell motility has been indirectly suggested by the phenotype of mice with genetically ablated expression of PTEN, a natural inhibitor of the PI3 kinase/Akt pathway whose effect is mediated by degradation of phospholipids generated by PI3 kinase. Its hereditary deficiencies manifest by increased cancer incidence and aberrant tissue development [239, 243, 244].
Aberrant layering was a common finding in mice with genetically ablated PTEN. This includes the overgrown and displastic cerebellum in mice with conditional PTEN inactivation in precursors of cells populating the vermis of the cerebellum [244], the impaired ability of PTEN-deficient Bergmann glia to guide migration of granular cells inward from the Purkinje cell layer [243] and accumulation of Purkinje cells, in which PTEN was selectively inactivated, in clusters above the fourth ventricle instead of migrating to the cerebellar surface [244]. These results point to a possibility that the PI3 kinase pathway is involved in the regulation of neural cell migration without, however, excluding contribution of other PTEN-dependent pathways [245–247] to the observed effects.
IGF1R Activation and Neural Cell Migration
Modulation of β Integrin Activity by IGF1R
β integrins play a role in modulation of cell migration induced by IGF1R activation. For example, a migratory response of smooth muscle cells to IGF1R activation was dependent on the interaction of their αvβ3 integrin with vitronectin [248]. The migration-promoting effect of IGF1R activation requires that the activation of IGF1R pathway components and β integrin ligand occupancy are spatially and temporally co-ordinated and is mediated mainly by the PI3 kinase/Akt pathway [249].
Changes in the activation status of focal adhesion components have been shown to occur in response to IGF1R activation. This includes changes in the phosphorylation status of focal adhesion kinase (FAK) in transformed epithelial cells, which are indicative of adhesion turnover in focal adhesions [250, 251], and tyrosine phosphorylation of several other focal adhesion components, which correlated with lamellipodia motility, in fibroblast [252] and neural [253] cell lines. Furthermore, αV integrin was redistributed from adhesion junctions to focal adhesions in response to IGF-I stimulation in transformed epithelial cells, suggesting a mechanisms to account for the increased migration of these cells in response to this stimulation [251] (Fig. 5; Table 1). Modulation of β integrin activity and changes in focal adhesions induced by IGF1R activation in developing neural cells remain to be assessed.
Adaptor Proteins Mediate Association of IGF1R Signalling with Focal Adhesions
One of the proteins supporting structural integrity of focal adhesions and facilitating their functions is the receptor for activated C kinase (RACK)1, a scaffolding protein [254] whose major function is to regulate cellular responses by targeting signalling molecules to specific intracellular locations. Protein kinase C (PKC) family members and other signalling molecules, including β-integrin cytoplasmic domains [255], have been identified as RACK1 binding partners (reviewed in [256]). RACK1 is required for targeting PKC isoforms [257], Src [258] and ERK [259] to focal adhesions and for their participation in adhesion dynamics and cell migration.
Elucidating the mechanism to account for the crosstalk between IGF1R activation and focal adhesion dynamics, two research groups identified independently that IGF1R binds to RACK1 [260, 261]. This has led to the hypothesis that RACK1 mediates association of IGF1R signalling cascade with focal adhesions. In the MCF-7 breast carcinoma cell line, translocation of the complex formed by IGF1R and RACK1 to focal adhesions was dependent on β1 integrin binding to ECM, an event which induced release of RACK1 from an inhibitory action of the serine threonine phosphatase, the protein phosphatase 2A (PP2A). RACK1-mediated translocation of IGF1R to focal adhesions resulting from PP2A dissociation led to increased cell migration [262]. In a cardiomyocyte cell line, however, modulation of cell migration by IGF1R signalling was RACK1 independent [263]. The authors identified a tyrosine residue in the RACK1 molecule that was critical for its ability to associate IGF1R activation with the function of focal adhesions [264].
Little is known about a role of RACK1 as a link between IGF1R activation and reorganisation of focal adhesions in neural cells. The importance of this scaffolding protein for neuronal functions, however, has been shown. RACK1 binding partners in this cell lineage include proteins involved in localised protein synthesis [265] and receptors for neurotransmitters [266]. RACK1 is widely expressed in the developing and mature CNS, with regional variations in expression levels, and forms concentration gradients, suggesting its involvement in CNS patterning [267]. RACK1-mediated translocation of a Na+/H+ exchanger to focal adhesions in neurons has been shown [268], implicating this adaptor in functions of focal adhesions in cells of the neural lineage.
Modulation of Cellular Responses to IGF-I by β Integrin Activation
Co-operation between the IGF1R and β integrin signalling pathways is bi-directional because β integrins activated by ligation with ECM components can modify cellular response to IGF-I. A growth-promoting effect of IGF-I was enhanced by ligation of α5β1 integrin with fibronectin in chondrocytes [269] and by co-activation of a vitronectin receptor in transformed and non-transformed epithelial cells [270]. Co-activation of αVβ3 integrin in IGF-I-stimulated smooth muscle cells enhanced their proliferative response via a mechanism that involved an increase in the duration of IGF1R phosphorylation [271, 272]. Combinatorial effects of short-range signals delivered to NSPC by adhesive interactions with the components of the basal lamina and signals from activated growth factor receptors have been proposed to play an important role in NSPC maintenance within neurogenic zones [227–229, 273]. IGF1R might be an important component in such regulatory signalling networks.
IGF1R and Cellular Responses that Involve Cytoplasmic Membrane Remodelling
Accumulating evidence suggests that IGF1R signalling may promote outgrowth of cytoplasmic membrane extensions in neurons and glial cells. The importance of this effect for neurogenesis and functions of the CNS is suggested by recent studies. The IGF-I and IGF-II have been shown to be expressed in a graded manner in a developing mouse OB and mediate mapping of its neuronal connectivity by directing growth of neuronal processes [274]. The exercise-induced induction of dendritic spine outgrowth in specific subsets of hippocampal neurons depends on IGF-I [275].
Cytoplasmic membrane remodelling in neural cells is regulated by a variety of signals, including attractive and repulsive guidance cues, in many cases without the induction of new gene expression. The effects of the IGF1R activation on cytoplasmic membrane remodelling are likely to be in conjunction with these guidance molecules. Analysis of signalling pathways mediating stimulatory effects of IGF-I on the outgrowth of cytoplasmic membrane derivatives showed that the Ras/Raf/MAPK and PI3 kinase/Akt pathways mediated axon outgrowth in corticospinal motor neurons [276] and exocytosis of plasmalemmal precursor vesicle during growth cone extension [277]. Transcription-independent cytoskeleton remodelling mediated by the Rho family GTPases was involved in such effects of IGF-I as the induction of neurite outgrowth in SH-SY5Y neuroblastoma [278] and the establishment of polarity and initiation of axonal outgrowth in hippocampal neurons [279].
Conclusion
IGF1R signalling pathway plays a non-redundant role in neural cell development. Its major biological function in the nervous system is to promote increase in cellular mass via a variety of mechanisms which involve suppression of pathways inhibiting cell growth, such as FoxO transcription factors and GSK3β, stimulation of mitotic cycle progression by enhancing G1 to S phase transition in cells proliferating in response to other mitogens and promoting growth of cytoplasmic derivatives in neural cells. IGF1R signalling may be involved in the combinatorial regulation of neurogenesis at different developmental stages by cumulative or antagonistic interactions with pathways of several signalling factors, including receptor tyrosine kinases, Shh, BMP, Notch and Wnt. The Akt-mediated inactivation of GSK3β has emerged as a common mechanism integrating the action of the IGF1R with other signalling pathways. A role of the IGF1R pathway in the developmental regulation of neural cell motility is suggested by its well-documented effects on migration in a number of transformed and non-transformed cells, including mouse embryonic stem cells. It may be interesting to investigate changes in adherence junctions and focal adhesions induced by IGF1R activation in neural cells and implications that such changes may have for the regulation of neural cell migration during development and in CNS pathology. A better understanding of molecular basis to account for co-operation between IGF1R and other signalling pathways in neural cells may lead to identification of new targets for neuroprotective and neuroregenerative therapy, as well as to the development of new approaches to promote engraftment and dissemination of NSPC transplanted in the CNS, thereby increasing their therapeutic efficiency.
References
Baserga R (2007) Is cell size important? Cell Cycle 6:814–816
Sun H, Tu X, Baserga R (2006) A mechanism for cell size regulation by the insulin and insulin-like growth factor-I receptors. Cancer Res 66:11106–11109
Beck KD, Powell-Braxton L, Widmer HR, Valverde J, Hefti F (1995) Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron 14:717–730
D’Ercole AJ, Ye P, O’Kusky JR (2002) Mutant mouse models of insulin-like growth factor actions in the central nervous system. Neuropeptides 36:209–220
LeRoith D, Werner H, Beitner-Johnson D, Roberts C Jr (1995) Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev 16:143–163
Laviola L, Natalicchio A, Giorgino F (2007) The IGF-I signaling pathway. Curr Pharm Des 13:663–669
Li W, Miller WT (2006) Role of the activation loop tyrosines in regulation of the insulin-like growth factor I receptor tyrosine kinase. J Biol Chem 281:23785–23791
Adams TE, Epa VC, Garrett TP, Ward CW (2000) Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol Life Sci 57:1050–1093
Ye P, D’Ercole AJ (2006) Insulin-like growth factor actions during development of neural stem cells and progenitors in the central nervous system. J Neurosci Res 83:1–6
Freude S, Leeser U, Muller M, Hettich MM, Udelhoven M, Schilbach K, Tobe K, Kadowaki T, Kohler C, Schroder H, Krone W, Bruning JC, Schubert M (2008) IRS-2 branch of IGF-1 receptor signaling is essential for appropriate timing of myelination. J Neurochem 107:907–917
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789
Whittaker J, Whittaker L (2005) Characterization of the functional insulin binding epitopes of the full-length insulin receptor. J Biol Chem 280:20932–20936
Sun LY, D’Ercole AJ (2006) Insulin-like growth factor-I (IGF-I) stimulates histone H3 and H4 acetylation in the brain in vivo. Endocrinology 147:5480–5490
Broughton SK, Chen H, Riddle A, Kuhn SE, Nagalla S, Roberts CT, Back SA (2007) Large-scale generation of highly enriched neural stem-cell-derived oligodendroglial cultures: maturation-dependent differences in insulin-like growth factor-mediated signal transduction. J Neurochem 100:628–638
Wada A, Yokoo H, Yanagita T, Kobayashi H (2005) New twist on neuronal insulin receptor signaling in health, disease, and therapeutics. J Pharmacol Sci 99:128–143
Johnson-Farley NN, Travkina T, Cowen DS (2006) Cumulative activation of Akt and consequent inhibition of glycogen synthase kinase-3 by brain-derived neurotrophic factor and insulin-like growth factor-1 in cultured hippocampal neurons. J Pharmacol Exp Ther 316:1062–1069
Duarte AI, Santos P, Oliveira CR, Santos MS, Rego AC (2008) Insulin neuroprotection against oxidative stress is mediated by Akt and GSK-3β signaling pathways and changes in protein expression. Biochim Biophys Acta 1783:994–1002
Leinninger GM, Backus C, Uhler MD, Lentz SI, Feldman EL (2004) Phosphatidylinositol 3-kinase and Akt effectors mediate insulin-like growth factor-I neuroprotection in dorsal root ganglia neurons. FASEB J 18:1544–1546
Neufeld TP (2003) Shrinkage control: regulation of insulin-mediated growth by FOXO transcription factors. J Biol 2:18.11–18.15
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868
Zheng W-H, Kar S, Quirion R (2002) Insulin-like growth factor-1-induced phosphorylation of transcription factor FKHRL1 is mediated by phosphatidylinositol 3-kinase/Akt kinase and role of this pathway in insulin-like growth factor-1-induced survival of cultured hippocampal neurons. Mol Pharmacol 62:225–233
Lixia G, Wenhua Z, Jean-Guy C, Terry GU, Remi Q (2005) Nuclear/cytoplasmic shuttling of the transcription factor FoxO1 is regulated by neurotrophic factors. J Neurochem 93:1209–1219
Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR (1999) Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J 344(Pt 2):427–431
Chenal J, Pierre K, Pellerin L (2008) Insulin and IGF-1 enhance the expression of the neuronal monocarboxylate transporter MCT2 by translational activation via stimulation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin pathway. Eur J Neurosci 27:53–65
El-Shewy HM, Johnson KR, Lee MH, Jaffa AA, Obeid LM, Luttrell LM (2006) Insulin-like growth factors mediate heterotrimeric G protein-dependent ERK1/2 activation by transactivating sphingosine-1-phosphate receptors. J Biol Chem 281:31399–31407
Lieskovska J, Ling Y, Badley-Clarke J, Clemmons DR (2006) The role of Src kinase in insulin-like growth factor dependent mitogenic signaling in vascular smooth muscle cells. J Biol Chem 281:25041–25053
Aberg MA, Aberg ND, Palmer TD, Alborn AM, Carlsson-Skwirut C, Bang P, Rosengren LE, Olsson T, Gage FH, Eriksson PS (2003) IGF-I has a direct proliferative effect in adult hippocampal progenitor cells. Mol Cell Neurosci 24:23–40
Johnson-Farley NN, Patel K, Kim D, Cowen DS (2007) Interaction of FGF-2 with IGF-1 and BDNF in stimulating Akt, ERK, and neuronal survival in hippocampal cultures. Brain Res 1154:40–49
Cui QL, Zheng WH, Quirion R, Almazan G (2005) Inhibition of Src-like kinases reveals Akt-dependent and -independent pathways in insulin-like growth factor I-mediated oligodendrocyte progenitor survival. J Biol Chem 280:8918–8928
Subramaniam S, Shahani N, Strelau J, Laliberte C, Brandt R, Kaplan D, Unsicker K (2005) Insulin-like growth factor 1 inhibits extracellular signal-regulated kinase to promote neuronal survival via the phosphatidylinositol 3-kinase/protein kinase A/c-Raf pathway. J Neurosci 25:2838–2852
Roelink H, Porter JA, Chiang C, Tanabe Y, Chang DT, Beachy PA, Jessell TM (1995) Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell 81:445–455
Hebert JM, Mishina Y, McConnell SK (2002) BMP signaling is required locally to pattern the dorsal telencephalic midline. Neuron 35:1029–1041
Ragsdale CW, Grove EA (2001) Patterning the mammalian cerebral cortex. Curr Opin Neurobiol 11:50–58
Takahashi H, Liu FC (2006) Genetic patterning of the mammalian telencephalon by morphogenetic molecules and transcription factors. Birth Defects Res C Embryo Today 78:256–266
Bertrand N, Dahmane N (2006) Sonic hedgehog signaling in forebrain development and its interactions with pathways that modify its effects. Trends Cell Biol 16:597–605
Riquelme PA, Drapeau E, Doetsch F (2007) Brain micro-ecologies: neural stem cell niches in the adult mammalian brain. Philos Trans R Soc Lond B Biol Sci 363:123–137
Ponti G, Aimar P, Bonfanti L (2006) Cellular composition and cytoarchitecture of the rabbit subventricular zone and its extensions in the forebrain. J Comp Neurol 498:491–507
Ayuso-Sacido A, Roy NS, Schwartz TH, Greenfield JP, Boockvar JA (2008) Long-term expansion of adult human brain subventricular zone precursors. Neurosurgery 62:223–229 discussion 229–231
Moe MC, Westerlund U, Varghese M, Berg-Johnsen J, Svensson M, Langmoen IA (2005) Development of neuronal networks from single stem cells harvested from the adult human brain. Neurosurgery 56:1182–1188 discussion 1188–1190
Alvarez-Buylla A, Lim DA (2004) For the long run: maintaining germinal niches in the adult brain. Neuron 41:683–686
Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A (1997) Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci 17:5046–5061
Barkho BZ, Munoz AE, Li X, Li L, Cunningham LA, Zhao X (2008) Endogenous matrix metalloproteinase (MMP)-3 and MMP-9 promote the differentiation and migration of adult neural progenitor cells in response to chemokines. Stem Cells 26:3139–3149
Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, Vandenberg S, Alvarez-Buylla A (2006) PDGFRα-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron 51:187–199
Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, Alvarez-Buylla A (2006) Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci 26:7907–7918
Zhao C, Zawadzka M, Roulois AJ, Bruce CC, Franklin RJ (2008) Promoting remyelination in multiple sclerosis by endogenous adult neural stem/precursor cells: defining cellular targets. J Neurol Sci 265:12–16
Baer K, Eriksson PS, Faull RL, Rees MI, Curtis MA (2007) Sox-2 is expressed by glial and progenitor cells and Pax-6 is expressed by neuroblasts in the human subventricular zone. Exp Neurol 204:828–831
Navarro-Quiroga I, Hernandez-Valdes M, Lin SL, Naegele JR (2006) Postnatal cellular contributions of the hippocampus subventricular zone to the dentate gyrus, corpus callosum, fimbria, and cerebral cortex. J Comp Neurol 497:833–845
Sottile V, Li M, Scotting PJ (2006) Stem cell marker expression in the Bergmann glia population of the adult mouse brain. Brain Res 1099:8–17
Alcock J, Lowe J, England T, Bath P, Sottile V (2008) Expression of Sox1, Sox2 and Sox9 is maintained in adult human cerebellar cortex. Neurosci Lett 450:114–116
Ponti G, Peretto P, Bonfanti L (2006) A subpial, transitory germinal zone forms chains of neuronal precursors in the rabbit cerebellum. Dev Biol 294:168–180
Pekcec A, Loscher W, Potschka H (2006) Neurogenesis in the adult rat piriform cortex. Neuroreport 17:571–574
Franklin RJ, Kotter MR (2008) The biology of CNS remyelination: the key to therapeutic advances. J Neurol 255(Suppl 1):19–25
Bauer S, Patterson PH (2006) Leukemia inhibitory factor promotes neural stem cell self-renewal in the adult brain. J Neurosci 26:12089–12099
Kuo CT, Mirzadeh Z, Soriano-Navarro M, Rasin M, Wang D, Shen J, Sestan N, Garcia-Verdugo J, Alvarez-Buylla A, Jan LY, Jan YN (2006) Postnatal deletion of numb/numblike reveals repair and remodeling capacity in the subventricular neurogenic niche. Cell 127:1253–1264
Covey MV, Levison SW (2007) Leukemia inhibitory factor participates in the expansion of neural stem/progenitors after perinatal hypoxia/ischemia. Neuroscience 148:501–509
Bohannon NJ, Corp ES, Wilcox BJ, Figlewicz DP, Dorsa DM, Baskin DG (1988) Localization of binding sites for insulin-like growth factor-I (IGF-I) in the rat brain by quantitative autoradiography. Brain Res 444:205–213
Sherrard RM, Richardson NA, Sara VR (1997) Localisation of insulin-like growth factor-I (IGF-I) immunoreactivity in the olivocerebellar system of developing and adult rats. Brain Res Dev Brain Res 98:102–113
Garcia-Segura LM, Rodriguez JR, Torres-Aleman I (1997) Localization of the insulin-like growth factor I receptor in the cerebellum and hypothalamus of adult rats: an electron microscopic study. J Neurocytol 26:479–490
Dore S, Kar S, Rowe W, Quirion R (1997) Distribution and levels of [125I]IGF-I, [125I]IGF-II and [125I]insulin receptor binding sites in the hippocampus of aged memory-unimpaired and -impaired rats. Neuroscience 80:1033–1040
Schechter R, Whitmire J, Beju D, Jackson KW, Harlow R, Gavin JR 3rd (1995) An immunohistochemical and in situ hybridization study of insulin-like growth factor I within fetal neuron cell cultures. Brain Res 670:1–13
Torres-Aleman I, Pons S, Arevalo MA (1994) The insulin-like growth factor I system in the rat cerebellum: developmental regulation and role in neuronal survival and differentiation. J Neurosci Res 39:117–126
Folli F, Bonfanti L, Renard E, Kahn CR, Merighi A (1994) Insulin receptor substrate-1 (IRS-1) distribution in the rat central nervous system. J Neurosci 14:6412–6422
Kar S, Chabot JG, Quirion R (1993) Quantitative autoradiographic localization of [125I]insulin-like growth factor I, [125I]insulin-like growth factor II, and [125I]insulin receptor binding sites in developing and adult rat brain. J Comp Neurol 333:375–397
Shinar Y, McMorris FA (1995) Developing oligodendroglia express mRNA for insulin-like growth factor-I, a regulator of oligodendrocyte development. J Neurosci Res 42:516–527
Wilkins A, Chandran S, Compston A (2001) A role for oligodendrocyte-derived IGF-1 in trophic support of cortical neurons. Glia 36:48–57
Salehi Z, Mashayekhi F, Naji M, Pandamooz S (2009) Insulin-like growth factor-1 and insulin-like growth factor binding proteins in cerebrospinal fluid during the development of mouse embryos. J Clin Neurosci 16:950–953
Davila D, Piriz J, Trejo JL, Nunez A, Torres-Aleman I (2007) Insulin and insulin-like growth factor I signalling in neurons. Front Biosci 12:3194–3202
Llorens-Martin M, Torres-Aleman I, Trejo JL (2009) Mechanisms mediating brain plasticity: IGF1 and adult hippocampal neurogenesis. Neuroscientist 15:134–148
Fushimi S, Shirabe T (2004) Expression of insulin-like growth factors in remyelination following ethidium bromide-induced demyelination in the mouse spinal cord. Neuropathology 24:208–218
Trejo JL, Llorens-Martin MV, Torres-Aleman I (2008) The effects of exercise on spatial learning and anxiety-like behavior are mediated by an IGF-I-dependent mechanism related to hippocampal neurogenesis. Mol Cell Neurosci 37:402–411
Pera EM, Ikeda A, Eivers E, De Robertis EM (2003) Integration of IGF, FGF, and anti-BMP signals via Smad1 phosphorylation in neural induction. Genes Dev 17:3023–3028
Pera EM, Wessely O, Li SY, De Robertis EM (2001) Neural and head induction by insulin-like growth factor signals. Dev Cell 1:655–665
Richard-Parpaillon L, Heligon C, Chesnel F, Boujard D, Philpott A (2002) The IGF pathway regulates head formation by inhibiting Wnt signaling in Xenopus. Dev Biol 244:407–417
Mason JL, Goldman JE (2002) A2B5+ and O4+ cycling progenitors in the adult forebrain white matter respond differentially to PDGF-AA, FGF-2, and IGF-1. Mol Cell Neurosci 20:30–42
Hsieh J, Aimone JB, Kaspar BK, Kuwabara T, Nakashima K, Gage FH (2004) IGF-I instructs multipotent adult neural progenitor cells to become oligodendrocytes. J Cell Biol 164:111–122
Palacios N, Sanchez-Franco F, Fernandez M, Sanchez I, Cacicedo L (2005) Intracellular events mediating insulin-like growth factor I-induced oligodendrocyte development: modulation by cyclic AMP. J Neurochem 95:1091–1107
Espinosa-Jeffrey A, Kumar S, Zhao PM, Awosika O, Agbo C, Huang A, Chang R, De Vellis J (2002) Transferrin regulates transcription of the MBP gene and its action synergizes with IGF-1 to enhance myelinogenesis in the md rat. Dev Neurosci 24:227–241
Chang MY, Sun W, Ochiai W, Nakashima K, Kim SY, Park CH, Kang JS, Shim JW, Jo AY, Kang CS, Lee YS, Kim JS, Lee SH (2007) Bcl-XL/Bax proteins direct the fate of embryonic cortical precursor cells. Mol Cell Biol 27:4293–4305
Fernando P, Megeney LA (2007) Is caspase-dependent apoptosis only cell differentiation taken to the extreme? FASEB J 21:8–17
Aranha MM, Sola S, Low WC, Steer CJ, Rodrigues CM (2009) Caspases and p53 modulate FOXO3A/Id1 signaling during mouse neural stem cell differentiation. J Cell Biochem 107:748–758
Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, Avila RL, Kirschner DA, Macklin WB (2008) Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci 28:7174–7183
Tyler WA, Gangoli N, Gokina P, Kim HA, Covey M, Levison SW, Wood TL (2009) Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J Neurosci 29:6367–6378
Ulloa F, Briscoe J (2007) Morphogens and the control of cell proliferation and patterning in the spinal cord. Cell Cycle 6:2640–2649
Kessaris N, Pringle N, Richardson WD (2008) Specification of CNS glia from neural stem cells in the embryonic neuroepithelium. Philos Trans R Soc Lond B Biol Sci 363:71–85
Briscoe J, Novitch BG (2008) Regulatory pathways linking progenitor patterning, cell fates and neurogenesis in the ventral neural tube. Philos Trans R Soc Lond B Biol Sci 363:57–70
Rallu M, Corbin JG, Fishell G (2002) Parsing the prosencephalon. Nat Rev Neurosci 3:943–951
Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA (1996) Cyclopia and defective axial patterning in mice lacking sonic hedgehog gene function. Nature 383:407–413
Wilson SW, Rubenstein JL (2000) Induction and dorsoventral patterning of the telencephalon. Neuron 28:641–651
Britto J, Tannahill D, Keynes R (2002) A critical role for sonic hedgehog signaling in the early expansion of the developing brain. Nat Neurosci 5:103–110
Knoepfler PS, Kenney AM (2006) Neural precursor cycling at sonic speed: N-Myc pedals, GSK-3 brakes. Cell Cycle 5:47–52
Lai K, Kaspar BK, Gage FH, Schaffer DV (2003) Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci 6:21–27
Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A (2008) Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci 11:277–284
Angot E, Loulier K, Nguyen-Ba-Charvet KT, Gadeau AP, Ruat M, Traiffort E (2008) Chemoattractive activity of sonic hedgehog in the adult subventricular zone modulates the number of neural precursors reaching the olfactory bulb. Stem Cells 26:2311–2320
Balordi F, Fishell G (2007) Mosaic removal of hedgehog signaling in the adult SVZ reveals that the residual wild-type stem cells have a limited capacity for self-renewal. J Neurosci 27:14248–14259
Cayuso J, Ulloa F, Cox B, Briscoe J, Marti E (2006) The Sonic hedgehog pathway independently controls the patterning, proliferation and survival of neuroepithelial cells by regulating Gli activity. Development 133:517–528
Peltier J, O’Neill A, Schaffer DV (2007) PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol 67:1348–1361
Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, Wickramasinghe R, Scott MP, Wechsler-Reya RJ (2003) Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci U S A 100:7331–7336
Kenney AM, Widlund HR, Rowitch DH (2004) Hedgehog and PI-3 kinase signaling converge on N-myc1 to promote cell cycle progression in cerebellar neuronal precursors. Development 131:217–228
Parathath SR, Mainwaring LA, Fernandez LA, Campbell DO, Kenney AM (2008) Insulin receptor substrate 1 is an effector of sonic hedgehog mitogenic signaling in cerebellar neural precursors. Development 135:3291–3300
Campbell K (2003) Dorsal–ventral patterning in the mammalian telencephalon. Curr Opin Neurobiol 13:50–56
Lee SM, Tole S, Grove E, McMahon AP (2000) A local Wnt-3a signal is required for development of the mammalian hippocampus. Development 127:457–467
Maurer MH, Bromme JO, Feldmann RE Jr, Jarve A, Sabouri F, Burgers HF, Schelshorn DW, Kruger C, Schneider A, Kuschinsky W (2007) Glycogen synthase kinase 3β (GSK3β) regulates differentiation and proliferation in neural stem cells from the rat subventricular zone. J Proteome Res 6:1198–1208
Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ, Tsai LH (2009) Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136:1017–1031
Kunke D, Bryja V, Mygland L, Arenas E, Krauss S (2009) Inhibition of canonical Wnt signaling promotes gliogenesis in P0-NSCs. Biochem Biophys Res Commun 386:628–633
Fuentealba LC, Eivers E, Ikeda A, Hurtado C, Kuroda H, Pera EM, De Robertis EM (2007) Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 131:980–993
Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ, Bertrand F, Cherqui G, Perret C, Capeau J (2001) Insulin and IGF-1 stimulate the β-catenin pathway through two signalling cascades involving GSK-3β inhibition and Ras activation. Oncogene 20:252–259
Satyamoorthy K, Li G, Vaidya B, Patel D, Herlyn M (2001) Insulin-like growth factor-1 induces survival and growth of biologically early melanoma cells through both the mitogen-activated protein kinase and β-catenin pathways. Cancer Res 61:7318–7324
Verras M, Sun Z (2005) β-catenin is involved in insulin-like growth factor 1-mediated transactivation of the androgen receptor. Mol Endocrinol 19:391–398
Kalani MY, Cheshier SH, Cord BJ, Bababeygy SR, Vogel H, Weissman IL, Palmer TD, Nusse R (2008) Wnt-mediated self-renewal of neural stem/progenitor cells. Proc Natl Acad Sci U S A 105:16970–16975
Nusse R (2008) Wnt signaling and stem cell control. Cell Res 18:523–527
Woodhead GJ, Mutch CA, Olson EC, Chenn A (2006) Cell-autonomous β-catenin signaling regulates cortical precursor proliferation. J Neurosci 26:12620–12630
Adachi K, Mirzadeh Z, Sakaguchi M, Yamashita T, Nikolcheva T, Gotoh Y, Peltz G, Gong L, Kawase T, Alvarez-Buylla A, Okano H, Sawamoto K (2007) β-Catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells 25:2827–2836
Hirsch C, Campano LM, Wohrle S, Hecht A (2007) Canonical Wnt signaling transiently stimulates proliferation and enhances neurogenesis in neonatal neural progenitor cultures. Exp Cell Res 313:572–587
Hirabayashi Y, Itoh Y, Tabata H, Nakajima K, Akiyama T, Masuyama N, Gotoh Y (2004) The Wnt/β-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 131:2791–2801
Israsena N, Hu M, Fu W, Kan L, Kessler JA (2004) The presence of FGF2 signaling determines whether β-catenin exerts effects on proliferation or neuronal differentiation of neural stem cells. Dev Biol 268:220–231
Jin T, George Fantus I, Sun J (2008) Wnt and beyond Wnt: multiple mechanisms control the transcriptional property of β-catenin. Cell Signal 20:1697–1704
Chen J, Wu A, Sun H, Drakas R, Garofalo C, Cascio S, Surmacz E, Baserga R (2005) Functional significance of type 1 insulin-like growth factor-mediated nuclear translocation of the insulin receptor substrate-1 and β-catenin. J Biol Chem 280:29912–29920
Hoogeboom D, Essers MA, Polderman PE, Voets E, Smits LM, Burgering BM (2008) Interaction of FOXO with β-catenin inhibits β-catenin/T cell factor activity. J Biol Chem 283:9224–9230
Manolagas SC, Almeida M (2007) Gone with the Wnts: β-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol 21:2605–2614
Fukunaga K, Ishigami T, Kawano T (2005) Transcriptional regulation of neuronal genes and its effect on neural functions: expression and function of forkhead transcription factors in neurons. J Pharmacol Sci 98:205–211
Chambers C, Peng Y, Nguyen H, Gaiano N, Fishell G, Nye J (2001) Spatiotemporal selectivity of response to Notch1 signals in mammalian forebrain precursors. Development 128:689–702
Gaiano N, Nye JS, Fishell G (2000) Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron 26:395–404
Louvi A, Artavanis-Tsakonas S (2006) Notch signalling in vertebrate neural development. Nat Rev Neurosci 7:93–102
Alexson TO, Hitoshi S, Coles BL, Bernstein A, van der Kooy D (2006) Notch signaling is required to maintain all neural stem cell populations—irrespective of spatial or temporal niche. Dev Neurosci 28:34–48
Yang X, Klein R, Tian X, Cheng HT, Kopan R, Shen J (2004) Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev Biol 269:81–94
Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, McKay RD (2006) Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442:823–826
Mason HA, Rakowiecki SM, Gridley T, Fishell G (2006) Loss of notch activity in the developing central nervous system leads to increased cell death. Dev Neurosci 28:49–57
Mandinova A, Lefort K, Tommasi di Vignano A, Stonely W, Ostano P, Chiorino G, Iwaki H, Nakanishi J, Dotto GP (2008) The FoxO3a gene is a key negative target of canonical Notch signalling in the keratinocyte UVB response. EMBO J 27:1243–1254
Jin YH, Kim H, Oh M, Ki H, Kim K (2009) Regulation of Notch1/NICD and Hes1 expressions by GSK-3alpha/beta. Mol Cells 27:15–19
Espinosa L, Ingles-Esteve J, Aguilera C, Bigas A (2003) Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. J Biol Chem 278:32227–32235
Xiong L, Kou F, Yang Y, Wu J (2007) A novel role for IGF-1R in p53-mediated apoptosis through translational modulation of the p53-Mdm2 feedback loop. J Cell Biol 178:995–1007
Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M (2001) Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem 276:5256–5264
Givogri MI, de Planell M, Galbiati F, Superchi D, Gritti A, Vescovi A, de Vellis J, Bongarzone ER (2006) Notch signaling in astrocytes and neuroblasts of the adult subventricular zone in health and after cortical injury. Dev Neurosci 28:81–91
Nyfeler Y, Kirch RD, Mantei N, Leone DP, Radtke F, Suter U, Taylor V (2005) Jagged1 signals in the postnatal subventricular zone are required for neural stem cell self-renewal. EMBO J 24:3504–3515
Mehler MF, Mabie PC, Zhu G, Gokhan S, Kessler JA (2000) Developmental changes in progenitor cell responsiveness to bone morphogenetic proteins differentially modulate progressive CNS lineage fate. Dev Neurosci 22:74–85
Chen HL, Panchision DM (2007) BMP pleiotropism in neural stem cells and their derivatives—alternative pathways, convergent signals. Stem Cells 25:63–68
Furuta Y, Piston DW, Hogan BL (1997) Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development 124:2203–2212
Cheng X, Hsu CM, Currle DS, Hu JS, Barkovich AJ, Monuki ES (2006) Central roles of the roof plate in telencephalic development and holoprosencephaly. J Neurosci 26:7640–7649
Arkell R, Beddington RS (1997) BMP-7 influences pattern and growth of the developing hindbrain of mouse embryos. Development 124:1–12
Lim DA, Tramontin AD, Trevejo JM, Herrera DG, Garcia-Verdugo JM, Alvarez-Buylla A (2000) Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron 28:713–726
Bonaguidi MA, Peng CY, McGuire T, Falciglia G, Gobeske KT, Czeisler C, Kessler JA (2008) Noggin expands neural stem cells in the adult hippocampus. J Neurosci 28:9194–9204
Mathieu C, Sii-Felice K, Fouchet P, Etienne O, Haton C, Mabondzo A, Boussin FD, Mouthon MA (2008) Endothelial cell-derived bone morphogenetic proteins control proliferation of neural stem/progenitor cells. Mol Cell Neurosci 38:569–577
Panchision DM, Pickel JM, Studer L, Lee SH, Turner PA, Hazel TG, McKay RD (2001) Sequential actions of BMP receptors control neural precursor cell production and fate. Genes Dev 15:2094–2110
Weinstein DC, Hemmati-Brivanlou A (1999) Neural induction. Annu Rev Cell Dev Biol 15:411–433
Sailer MH, Hazel TG, Panchision DM, Hoeppner DJ, Schwab ME, McKay RD (2005) BMP2 and FGF2 cooperate to induce neural-crest-like fates from fetal and adult CNS stem cells. J Cell Sci 118:5849–5860
Itoh S, Itoh F, Goumans MJ, Ten Dijke P (2000) Signaling of transforming growth factor-β family members through Smad proteins. Eur J Biochem 267:6954–6967
Rajan P, Panchision DM, Newell LF, McKay RD (2003) BMPs signal alternately through a SMAD or FRAP-STAT pathway to regulate fate choice in CNS stem cells. J Cell Biol 161:911–921
Kendall SE, Battelli C, Irwin S, Mitchell JG, Glackin CA, Verdi JM (2005) NRAGE mediates p38 activation and neural progenitor apoptosis via the bone morphogenetic protein signaling cascade. Mol Cell Biol 25:7711–7724
Langenfeld EM, Kong Y, Langenfeld J (2005) Bone morphogenetic protein-2-induced transformation involves the activation of mammalian target of rapamycin. Mol Cancer Res 3:679–684
Kretzschmar M, Doody J, Massague J (1997) Opposing BMP and EGF signalling pathways converge on the TGF-β family mediator Smad1. Nature 389:618–622
Eivers E, Fuentealba LC, De Robertis EM (2008) Integrating positional information at the level of Smad1/5/8. Curr Opin Genet Dev 18:304–310
Bilican B, Fiore-Heriche C, Compston A, Allen ND, Chandran S (2008) Induction of Olig2 precursors by FGF involves BMP signalling blockade at the Smad level. PLoS ONE 3:e2863
Takahashi T, Morris EA, Trippel SB (2007) Bone morphogenetic protein-2 and -9 regulate the interaction of insulin-like growth factor-I with growth plate chondrocytes. Int J Mol Med 20:53–57
Celil AB, Campbell PG (2005) BMP-2 and insulin-like growth factor-I mediate Osterix (Osx) expression in human mesenchymal stem cells via the MAPK and protein kinase D signaling pathways. J Biol Chem 280:31353–31359
Celil AB, Hollinger JO, Campbell PG (2005) Osx transcriptional regulation is mediated by additional pathways to BMP2/Smad signaling. J Cell Biochem 95:518–528
Osyczka AM, Leboy PS (2005) Bone morphogenetic protein regulation of early osteoblast genes in human marrow stromal cells is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase signaling. Endocrinology 146:3428–3437
Nishida K, Hirano T (2003) The role of Gab family scaffolding adapter proteins in the signal transduction of cytokine and growth factor receptors. Cancer Sci 94:1029–1033
Mao Y, Lee AW (2005) A novel role for Gab2 in bFGF-mediated cell survival during retinoic acid-induced neuronal differentiation. J Cell Biol 170:305–316
Hayakawa-Yano Y, Nishida K, Fukami S, Gotoh Y, Hirano T, Nakagawa T, Shimazaki T, Okano H (2007) Epidermal growth factor signaling mediated by grb2 associated binder1 is required for the spatiotemporally regulated proliferation of olig2-expressing progenitors in the embryonic spinal cord. Stem Cells 25:1410–1422
Jones N, Dumont DJ (1999) Recruitment of Dok-R to the EGF receptor through its PTB domain is required for attenuation of Erk MAP kinase activation. Curr Biol 9:1057–1060
Belsches AP, Haskell MD, Parsons SJ (1997) Role of c-Src tyrosine kinase in EGF-induced mitogenesis. Front Biosci 2:d501–518
Zhao M, Schmitz AA, Qin Y, Di Cristofano A, Pandolfi PP, Van Aelst L (2001) Phosphoinositide 3-kinase-dependent membrane recruitment of p62(dok) is essential for its negative effect on mitogen-activated protein (MAP) kinase activation. J Exp Med 194:265–274
Yoshida K, Yamashita Y, Miyazato A, Ohya K, Kitanaka A, Ikeda U, Shimada K, Yamanaka T, Ozawa K, Mano H (2000) Mediation by the protein-tyrosine kinase Tec of signaling between the B cell antigen receptor and Dok-1. J Biol Chem 275:24945–24952
Songyang Z, Yamanashi Y, Liu D, Baltimore D (2001) Domain-dependent function of the rasGAP-binding protein p62Dok in cell signaling. J Biol Chem 276:2459–2465
Di Cristofano A, Carpino N, Dunant N, Friedland G, Kobayashi R, Strife A, Wisniewski D, Clarkson B, Pandolfi PP, Resh MD (1998) Molecular cloning and characterization of p56dok-2 defines a new family of RasGAP-binding proteins. J Biol Chem 273:4827–4830
Van Slyke P, Coll ML, Master Z, Kim H, Filmus J, Dumont DJ (2005) Dok-R mediates attenuation of epidermal growth factor-dependent mitogen-activated protein kinase and Akt activation through processive recruitment of c-Src and Csk. Mol Cell Biol 25:3831–3841
Pamonsinlapatham P, Hadj-Slimane R, Lepelletier Y, Allain B, Toccafondi M, Garbay C, Raynaud F (2009) P120-Ras GTPase activating protein (RasGAP): a multi-interacting protein in downstream signaling. Biochimie 91:320–328
Smith A, Wang J, Cheng CM, Zhou J, Weickert CS, Bondy CA (2004) High-level expression of Dok-1 in neurons of the primate prefrontal cortex and hippocampus. J Neurosci Res 75:218–224
Ford-Perriss M, Abud H, Murphy M (2001) Fibroblast growth factors in the developing central nervous system. Clin Exp Pharmacol Physiol 28:493–503
Thisse B, Thisse C (2005) Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev Biol 287:390–402
Itoh N, Ornitz DM (2004) Evolution of the Fgf and Fgfr gene families. Trends Genet 20:563–569
Zhang Y, McKeehan K, Lin Y, Zhang J, Wang F (2008) Fibroblast growth factor receptor 1 (FGFR1) tyrosine phosphorylation regulates binding of FGFR substrate 2alpha (FRS2alpha) but not FRS2 to the receptor. Mol Endocrinol 22:167–175
Maric D, Fiorio Pla A, Chang YH, Barker JL (2007) Self-renewing and differentiating properties of cortical neural stem cells are selectively regulated by basic fibroblast growth factor (FGF) signaling via specific FGF receptors. J Neurosci 27:1836–1852
Bansal R, Lakhina V, Remedios R, Tole S (2003) Expression of FGF receptors 1, 2, 3 in the embryonic and postnatal mouse brain compared with PDGFRα, Olig2 and Plp/dm20: implications for oligodendrocyte development. Dev Neurosci 25:83–95
Fortin D, Rom E, Sun H, Yayon A, Bansal R (2005) Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. J Neurosci 25:7470–7479
Oh LYS, Denninger A, Colvin JS, Vyas A, Tole S, Ornitz DM, Bansal R (2003) Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J Neurosci 23:883–894
Hecht D, Zimmerman N, Bedford M, Avivi A, Yayon A (1995) Identification of fibroblast growth factor 9 (FGF9) as a high affinity, heparin dependent ligand for FGF receptors 3 and 2 but not for FGF receptors 1 and 4. Growth Factors 12:223–233
Nakamura H, Katahira T, Matsunaga E, Sato T (2005) Isthmus organizer for midbrain and hindbrain development. Brain Res Brain Res Rev 49:120–126
Fukuchi-Shimogori T, Grove EA (2001) Neocortex patterning by the secreted signaling molecule FGF8. Science 294:1071–1074
Garel S, Huffman KJ, Rubenstein JLR (2003) Molecular regionalization of the neocortex is disrupted in Fgf8 hypomorphic mutants. Development 130:1903–1914
Assimacopoulos S, Grove EA, Ragsdale CW (2003) Identification of a Pax6-dependent epidermal growth factor family signaling source at the lateral edge of the embryonic cerebral cortex. J Neurosci 23:6399–6403
Xu J, Liu Z, Ornitz D (2000) Temporal and spatial gradients of Fgf8 and Fgf17 regulate proliferation and differentiation of midline cerebellar structures. Development 127:1833–1843
Hasegawa H, Ashigaki S, Takamatsu M, Suzuki-Migishima R, Ohbayashi N, Itoh N, Takada S, Tanabe Y (2004) Laminar patterning in the developing neocortex by temporally coordinated fibroblast growth factor signaling. J Neurosci 24:8711–8719
Gutin G, Fernandes M, Palazzolo L, Paek H, Yu K, Ornitz DM, McConnell SK, Hebert JM (2006) FGF signalling generates ventral telencephalic cells independently of SHH. Development 133:2937–2946
Ligon KL, Fancy SP, Franklin RJ, Rowitch DH (2006) Olig gene function in CNS development and disease. Glia 54:1–10
Gómez-Pinilla F, Lee JW-K, Cotman CW (1994) Distribution of basic fibroblast growth factor in the developing rat brain. Neuroscience 61:911–923
Gomez-Pinilla F, Lee J, Cotman C (1992) Basic FGF in adult rat brain: cellular distribution and response to entorhinal lesion and fimbria-fornix transection. J Neurosci 12:345–355
Zheng W, Nowakowski RS, Vaccarino FM (2004) Fibroblast growth factor 2 is required for maintaining the neural stem cell pool in the mouse brain subventricular zone. Dev Neurosci 26:181–196
Korada S, Zheng W, Basilico C, Schwartz ML, Vaccarino FM (2002) Fibroblast growth factor 2 is necessary for the growth of glutamate projection neurons in the anterior neocortex. J Neurosci 22:863–875
Chen K, Ohkubo Y, Shin D, Doetschman T, Sanford LP, Li H, Vaccarino FM (2008) Decrease in excitatory neurons, astrocytes and proliferating progenitors in the cerebral cortex of mice lacking exon 3 from the Fgf2 gene. BMC Neurosci 9:94–102
Yoshimura S, Takagi Y, Harada J, Teramoto T, Thomas SS, Waeber C, Bakowska JC, Breakefield XO, Moskowitz MA (2001) FGF-2 regulation of neurogenesis in adult hippocampus after brain injury. Proc Natl Acad Sci U S A 98:5874–5879
Kuhn HG, Winkler J, Kempermann G, Thal LJ, Gage FH (1997) Epidermal growth factor and fibroblast growth factor-2 have different effects on neural progenitors in the adult rat brain. J Neurosci 17:5820–5829
Erickson RI, Paucar AA, Jackson RL, Visnyei K, Kornblum H (2008) Roles of insulin and transferrin in neural progenitor survival and proliferation. J Neurosci Res 86:1884–1894
Arsenijevic Y, Weiss S, Schneider B, Aebischer P (2001) Insulin-like growth factor-I is necessary for neural stem cell proliferation and demonstrates distinct actions of epidermal growth factor and fibroblast growth factor-2. J Neurosci 21:7194–7202
Hodge RD, D’Ercole AJ, O’Kusky JR (2004) Insulin-like growth factor-I accelerates the cell cycle by decreasing G1 phase length and increases cell cycle reentry in the embryonic cerebral cortex. J Neurosci 24:10201–10210
Kalluri HS, Vemuganti R, Dempsey RJ (2007) Mechanism of insulin-like growth factor I-mediated proliferation of adult neural progenitor cells: role of Akt. Eur J Neurosci 25:1041–1048
Sherr CJ, Roberts JM (1995) Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 9:1149–1163
Elledge SJ (1996) Cell cycle checkpoints: preventing an identity crisis. Science 274:1664–1672
Frederick TJ, Wood TL (2004) IGF-I and FGF-2 coordinately enhance cyclin D1 and cyclin E-cdk2 association and activity to promote G1 progression in oligodendrocyte progenitor cells. Mol Cell Neurosci 25:480–492
Bonthius DJ, Karacay B, Dai D, Pantazis NJ (2003) FGF-2, NGF and IGF-1, but not BDNF, utilize a nitric oxide pathway to signal neurotrophic and neuroprotective effects against alcohol toxicity in cerebellar granule cell cultures. Brain Res Dev Brain Res 140:15–28
Zheng WH, Quirion R (2004) Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J Neurochem 89:844–852
Zheng WH, Kar S, Dore S, Quirion R (2000) Insulin-like growth factor-1 (IGF-1): a neuroprotective trophic factor acting via the Akt kinase pathway. J Neural Transm Suppl 261–272
Tropepe V, Sibilia M, Ciruna BG, Rossant J, Wagner EF, van der Kooy D (1999) Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol 208:166–188
Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A (2002) EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron 36:1021–1034
Grinspan JB, Franceschini B (1995) Platelet-derived growth factor is a survival factor for PSA-NCAM+ oligodendrocyte pre-progenitor cells. J Neurosci Res 41:540–551
Gago N, Avellana-Adalid V, Evercooren AB, Schumacher M (2003) Control of cell survival and proliferation of postnatal PSA-NCAM+ progenitors. Mol Cell Neurosci 22:162–178
Fleming JM, Desury G, Polanco TA, Cohick WS (2006) Insulin growth factor-I and epidermal growth factor receptors recruit distinct upstream signaling molecules to enhance AKT activation in mammary epithelial cells. Endocrinology 147:6027–6035
Liu R, Cai J, Hu X, Tan M, Qi Y, German M, Rubenstein J, Sander M, Qiu M (2003) Region-specific and stage-dependent regulation of Olig gene expression and oligodendrogenesis by Nkx6.1 homeodomain transcription factor. Development 130:6221–6231
Marmur R, Mabie PC, Gokhan S, Song Q, Kessler JA, Mehler MF (1998) Isolation and developmental characterization of cerebral cortical multipotent progenitors. Dev Biol 204:577–591
Marin O, Rubenstein JLR (2001) A long remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci 2:780–790
Schmechel DE, Rakic P (1979) Arrested proliferation of radial glial cells during midgestation in rhesus monkey. Nature 277:303–305
Cayre M, Bancila M, Virard I, Borges A, Durbec P (2006) Migrating and myelinating potential of subventricular zone neural progenitor cells in white matter tracts of the adult rodent brain. Mol Cell Neurosci 31:748–758
Jain M, Armstrong RJ, Elneil S, Barker RA (2006) Transplanted human neural precursor cells migrate widely but show no lesion-specific tropism in the 6-hydroxydopamine rat model of Parkinson’s disease. Cell Transplant 15:579–593
Pokutta S, Drees F, Yamada S, Nelson WJ, Weis WI (2008) Biochemical and structural analysis of α-catenin in cell–cell contacts. Biochem Soc Trans 36:141–147
Niessen CM, Gottardi CJ (2008) Molecular components of the adherens junction. Biochim Biophys Acta 1778:562–571
Lobo MV, Alonso FJ, Redondo C, Lopez-Toledano MA, Caso E, Herranz AS, Paino CL, Reimers D, Bazan E (2003) Cellular characterization of epidermal growth factor-expanded free-floating neurospheres. J Histochem Cytochem 51:89–103
Seki T, Namba T, Mochizuki H, Onodera M (2007) Clustering, migration, and neurite formation of neural precursor cells in the adult rat hippocampus. J Comp Neurol 502:275–290
Hughson E, Dowler S, Geall K, Johnson G, Rumsby M (1998) Rat oligodendrocyte O-2A precursor cells and the CG-4 oligodendrocyte precursor cell line express cadherins, β-catenin and the neural cell adhesion molecule, NCAM. Neurosci Lett 251:157–160
Payne HR, Hemperly JJ, Lemmon V (1996) N-cadherin expression and function in cultured oligodendrocytes. Brain Res Dev Brain Res 97:9–15
Angst B, Marcozzi C, Magee A (2001) The cadherin superfamily: diversity in form and function. J Cell Sci 114:629–641
Rubenstein JL, Anderson S, Shi L, Miyashita-Lin E, Bulfone A, Hevner R (1999) Genetic control of cortical regionalization and connectivity. Cereb Cortex 9:524–532
Nakagawa S, Takeichi M (1998) Neural crest emigration from the neural tube depends on regulated cadherin expression. Development 125:2963–2971
Takahashi M, Osumi N (2008) Expression study of cadherin7 and cadherin20 in the embryonic and adult rat central nervous system. BMC Dev Biol 8:87–105
Paez PM, Marta CB, Moreno MB, Soto EF, Pasquini JM (2002) Apotransferrin decreases migration and enhances differentiation of oligodendroglial progenitor cells in an in vitro system. Dev Neurosci 24:47–58
Huber AH, Stewart DB, Laurents DV, Nelson WJ, Weis WI (2001) The cadherin cytoplasmic domain is unstructured in the absence of β-catenin. A possible mechanism for regulating cadherin turnover. J Biol Chem 276:12301–12309
Gumbiner BM (2000) Regulation of cadherin adhesive activity. J Cell Biol 148:399–404
Nagato M, Heike T, Kato T, Yamanaka Y, Yoshimoto M, Shimazaki T, Okano H, Nakahata T (2005) Prospective characterization of neural stem cells by flow cytometry analysis using a combination of surface markers. J Neurosci Res 80:456–466
Campos LS, Leone DP, Relvas JB, Brakebusch C, Fassler R, Suter U, Ffrench-Constant C (2004) β1 Integrins activate a MAPK signalling pathway in neural stem cells that contributes to their maintenance. Development 131:3433–3444
Flanagan LA, Rebaza LM, Derzic S, Schwartz PH, Monuki ES (2006) Regulation of human neural precursor cells by laminin and integrins. J Neurosci Res 83:845–856
Milner R, Edwards G, Streuli C, Ffrench-Constant C (1996) A role in migration for the αvβ1 integrin expressed on oligodendrocyte precursors. J Neurosci 16:7240–7252
Guvakova MA, Adams JC, Boettiger D (2002) Functional role of α-actinin, PI 3-kinase and MEK1/2 in insulin-like growth factor I receptor kinase regulated motility of human breast carcinoma cells. J Cell Sci 115:4149–4165
Schlenska-Lange A, Knupfer H, Lange TJ, Kiess W, Knupfer M (2008) Cell proliferation and migration in glioblastoma multiforme cell lines are influenced by insulin-like growth factor I in vitro. Anticancer Res 28:1055–1060
Kanekar S, Borg TK, Terracio L, Carver W (2000) Modulation of heart fibroblast migration and collagen gel contraction by IGF-I. Cell Adhes Commun 7:513–523
Osterhout DJ, Ebner S, Xu J, Ornitz DM, Zazanis GA, McKinnon RD (1997) Transplanted oligodendrocyte progenitor cells expressing a dominant-negative FGF receptor transgene fail to migrate in vivo. J Neurosci 17:9122–9132
Frost EE, Zhou Z, Krasnesky K, Armstrong RC (2008) Initiation of oligodendrocyte progenitor cell migration by a PDGF-A activated extracellular regulated kinase (ERK) signaling pathway. Neurochem Res 34:69–81
Milner R, Anderson HJ, Rippon RF, McKay JS, Franklin RJM, Marchionni MA, Reynolds R, Ffrench-Constant C (1997) Contrasting effects of mitogenic growth factors on oligodendrocyte precursor cell migration. Glia 19:85–90
Espinosa-Jeffrey A, Zhao P, Awosika W, Wu N, Macias F, Cepeda C, Levine M, de Vellis J (2006) Activation, proliferation and commitment of endogenous stem/progenitor cells to the oligodendrocyte lineage by TS1 in a rat model of dysmyelination. Dev Neurosci 28:488–498
Kumar S, Biancotti JC, Yamaguchi M, de Vellis J (2007) Combination of growth factors enhances remyelination in a cuprizone-induced demyelination mouse model. Neurochem Res 32:783–797
Guvakova MA (2007) Insulin-like growth factors control cell migration in health and disease. Int J Biochem Cell Biol 39:890–909
Playford MP, Bicknell D, Bodmer WF, Macaulay VM (2000) Insulin-like growth factor 1 regulates the location, stability, and transcriptional activity of β-catenin. Proc Natl Acad Sci U S A 97:12103–12108
Mauro L, Salerno M, Morelli C, Boterberg T, Bracke ME, Surmacz E (2003) Role of the IGF-I receptor in the regulation of cell–cell adhesion: implications in cancer development and progression. J Cell Physiol 194:108–116
Morali OG, Delmas V, Moore R, Jeanney C, Thiery JP, Larue L (2001) IGF-II induces rapid beta-catenin relocation to the nucleus during epithelium to mesenchyme transition. Oncogene 20:4942–4950
Yue Q, Groszer M, Gil JS, Berk AJ, Messing A, Wu H, Liu X (2005) PTEN deletion in Bergmann glia leads to premature differentiation and affects laminar organization. Development 132:3281–3291
Marino S, Krimpenfort P, Leung C, van der Korput HA, Trapman J, Camenisch I, Berns A, Brandner S (2002) PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development 129:3513–3522
Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM (1998) Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 280:1614–1617
Tamura M, Gu J, Danen EH, Takino T, Miyamoto S, Yamada KM (1999) PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem 274:20693–20703
Tamura M, Gu J, Takino T, Yamada KM (1999) Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res 59:442–449
Jones JI, Prevette T, Gockerman A, Clemmons DR (1996) Ligand occupancy of the αVβ3 integrin is necessary for smooth muscle cells to migrate in response to insulin-like growth factor. Proc Natl Acad Sci U S A 93:2482–2487
Hollier BG, Kricker JA, Van Lonkhuyzen DR, Leavesley DI, Upton Z (2008) Substrate-bound insulin-like growth factor (IGF)-I-IGF binding protein-vitronectin-stimulated breast cell migration is enhanced by coactivation of the phosphatidylinositide 3-Kinase/AKT pathway by αv-integrins and the IGF-I receptor. Endocrinology 149:1075–1090
Manes S, Mira E, Gomez-Mouton C, Zhao ZJ, Lacalle RA, Martinez AC (1999) Concerted activity of tyrosine phosphatase SHP-2 and focal adhesion kinase in regulation of cell motility. Mol Cell Biol 19:3125–3135
Canonici A, Steelant W, Rigot V, Khomitch-Baud A, Boutaghou-Cherid H, Bruyneel E, Van Roy F, Garrouste F, Pommier G, Andre F (2008) Insulin-like growth factor-I receptor, E-cadherin and αv integrin form a dynamic complex under the control of α-catenin. Int J Cancer 122:572–582
Casamassima A, Rozengurt E (1998) Insulin-like growth factor I stimulates tyrosine phosphorylation of p130(Cas), focal adhesion kinase, and paxillin. Role of phosphatidylinositol 3′-kinase and formation of a p130(Cas). Crk complex. J Biol Chem 273:26149–26156
Leventhal PS, Shelden EA, Kim B, Feldman EL (1997) Tyrosine phosphorylation of paxillin and focal adhesion kinase during insulin-like growth factor-I-stimulated lamellipodial advance. J Biol Chem 272:5214–5218
Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D (1994) Cloning of an intracellular receptor for protein kinase C: a homolog of the β subunit of G proteins. Proc Natl Acad Sci U S A 91:839–843
Liliental J, Chang DD (1998) Rack1, a receptor for activated protein kinase C, interacts with integrin β subunit. J Biol Chem 273:2379–2383
McCahill A, Warwicker J, Bolger GB, Houslay MD, Yarwood SJ (2002) The RACK1 scaffold protein: a dynamic cog in cell response mechanisms. Mol Pharmacol 62:1261–1273
Buensuceso CS, Woodside D, Huff JL, Plopper GE, O’Toole TE (2001) The WD protein Rack1 mediates protein kinase C and integrin-dependent cell migration. J Cell Sci 114:1691–1698
Doan AT, Huttenlocher A (2007) RACK1 regulates Src activity and modulates paxillin dynamics during cell migration. Exp Cell Res 313:2667–2679
Vomastek T, Iwanicki MP, Schaeffer HJ, Tarcsafalvi A, Parsons JT, Weber MJ (2007) RACK1 targets the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway to link integrin engagement with focal adhesion disassembly and cell motility. Mol Cell Biol 27:8296–8305
Hermanto U, Zong CS, Li W, Wang LH (2002) RACK1, an insulin-like growth factor I (IGF-I) receptor-interacting protein, modulates IGF-I-dependent integrin signaling and promotes cell spreading and contact with extracellular matrix. Mol Cell Biol 22:2345–2365
Kiely PA, Sant A, O’Connor R (2002) RACK1 is an insulin-like growth factor 1 (IGF-1) receptor-interacting protein that can regulate IGF-1-mediated Akt activation and protection from cell death. J Biol Chem 277:22581–22589
Kiely PA, O’Gorman D, Luong K, Ron D, O’Connor R (2006) Insulin-like growth factor I controls a mutually exclusive association of RACK1 with protein phosphatase 2A and β1 integrin to promote cell migration. Mol Cell Biol 26:4041–4051
O’Donovan HC, Kiely PA, O’Connor R (2007) Effects of RACK1 on cell migration and IGF-I signalling in cardiomyocytes are not dependent on an association with the IGF-IR. Cell Signal 19:2588–2595
Kiely PA, Baillie GS, Lynch MJ, Houslay MD, O’Connor R (2008) Tyrosine 302 in RACK1 is essential for insulin-like growth factor-I-mediated competitive binding of PP2A and β1 integrin and for tumor cell proliferation and migration. J Biol Chem 283:22952–22961
Angenstein F, Evans AM, Settlage RE, Moran ST, Ling SC, Klintsova AY, Shabanowitz J, Hunt DF, Greenough WT (2002) A receptor for activated C kinase is part of messenger ribonucleoprotein complexes associated with polyA-mRNAs in neurons. J Neurosci 22:8827–8837
Brandon NJ, Jovanovic JN, Smart TG, Moss SJ (2002) Receptor for activated C kinase-1 facilitates protein kinase C-dependent phosphorylation and functional modulation of GABA(A) receptors with the activation of G-protein-coupled receptors. J Neurosci 22:6353–6361
Ashique AM, Kharazia V, Yaka R, Phamluong K, Peterson AS, Ron D (2006) Localization of the scaffolding protein RACK1 in the developing and adult mouse brain. Brain Res 1069:31–38
Onishi I, Lin PJ, Diering GH, Williams WP, Numata M (2007) RACK1 associates with NHE5 in focal adhesions and positively regulates the transporter activity. Cell Signal 19:194–203
Pulai JI, Del Carlo M Jr, Loeser RF (2002) The α5β1 integrin provides matrix survival signals for normal and osteoarthritic human articular chondrocytes in vitro. Arthritis Rheum 46:1528–1535
Van Lonkhuyzen DR, Hollier BG, Shooter GK, Leavesley DI, Upton Z (2007) Chimeric vitronectin: insulin-like growth factor proteins enhance cell growth and migration through co-activation of receptors. Growth Factors 25:295–308
Maile LA, Aday AW, Busby WH, Sanghani R, Veluvolu U, Clemmons DR (2008) Modulation of integrin antagonist signaling by ligand binding of the heparin-binding domain of vitronectin to the αVβ3 integrin. J Cell Biochem 105:437–446
Maile LA, Clemmons DR (2002) The αVβ3 integrin regulates insulin-like growth factor I (IGF-I) receptor phosphorylation by altering the rate of recruitment of the Src-homology 2-containing phosphotyrosine phosphatase-2 to the activated IGF-I receptor. Endocrinology 143:4259–4264
Garcion E, Faissner A, Ffrench-Constant C (2001) Knockout mice reveal a contribution of the extracellular matrix molecule tenascin-C to neural precursor proliferation and migration. Development 128:2485–2496
Scolnick JA, Cui K, Duggan CD, Xuan S, Yuan XB, Efstratiadis A, Ngai J (2008) Role of IGF signaling in olfactory sensory map formation and axon guidance. Neuron 57:847–857
Glasper ER, Llorens-Martin MV, Leuner B, Gould E, Trejo JL (2009) Blockade of insulin-like growth factor-I has complex effects on structural plasticity in the hippocampus. Hippocampus (in press)
Ozdinler PH, Macklis JD (2006) IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat Neurosci 9:1371–1381
Laurino L, Wang XX, de la Houssaye BA, Sosa L, Dupraz S, Caceres A, Pfenninger KH, Quiroga S (2005) PI3K activation by IGF-1 is essential for the regulation of membrane expansion at the nerve growth cone. J Cell Sci 118:3653–3662
Shiraishi M, Tanabe A, Saito N, Sasaki Y (2006) Unphosphorylated MARCKS is involved in neurite initiation induced by insulin-like growth factor-I in SH-SY5Y cells. J Cell Physiol 209:1029–1038
Sosa L, Dupraz S, Laurino L, Bollati F, Bisbal M, Caceres A, Pfenninger KH, Quiroga S (2006) IGF-1 receptor is essential for the establishment of hippocampal neuronal polarity. Nat Neurosci 9:993–995
Acknowledgement
The author thanks the National Multiple Sclerosis Society for financial support and Yuti Chernajovsky for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Annenkov, A. The Insulin-Like Growth Factor (IGF) Receptor Type 1 (IGF1R) as an Essential Component of the Signalling Network Regulating Neurogenesis. Mol Neurobiol 40, 195–215 (2009). https://doi.org/10.1007/s12035-009-8081-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-009-8081-0