
Abstract
The solid solution row A1−3x Eu\(^{\mathrm {3+}}_ {x}\)Zr2(PO4)3, where A = Na, K, Rb, Cs and x ranging from 0 to 0.25 was obtained by methods based on sol–gel processes in the presence of the salting-out agent, C2H5OH. The formation of phases of the structural type NaZr2(PO4)3 (NZP) and their crystallization in the crystal forms belonging to the space group \({\boldsymbol {R\bar {{3}}c}}\) (0 ≤x≤ 0.1) and \(\boldsymbol {{P\bar {{3}}c}}\) (x= 0.25) were determined. Morphotropic transition occurred. Trivalent europium was included as a local probe. Its electric and magnetic dipole was used to characterize the substitution rule and the localization of this cation in the structure, which is in agreement with the structural characterization. It appears that europium is located in the M1 and M2 sites as a function of the Eu3+ concentration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Atom isomorphism in complex crystalline compounds with tetrahedral oxo-anions is one of the fundamental factors for developing expected multifunctional materials. In this regard, orthophosphates with a rhombohedral structure (type NaZr 2(PO 4) 3, NZP) and forming a large family of isostructural phases are of interest [1,2].
The NZP-type structure is characterized by a framework topology, [T 2(XO 4) 3], where T is the structure-forming octahedral cation with an oxidation degree from + 1 to + 5, and X is the anion-forming cation with tetrahedral rosettes of chemical bonds. The frame contains three-dimensional cavities which neutralize its charge. There are two types of cavities available for stocking cations, one position of M1 type and three positions of M2 type, which are different in size, shape and environment. In the general form, NZP-phosphates are described by a crystal-chemical formula of (M1) [6](M2)\(^{\mathrm {[8]}}_{\mathrm {3}}\){L\(^{\mathrm {[6]}}_{\mathrm {2}}\)(PO 4) 3} ∞ (figures in square brackets are the coordination numbers of cations in the corresponding positions) [1].
An important aspect in the chemistry of NZP-phosphates is the introduction of f-elements into the structure in order to develop crystalline luminescent materials having controlled emission characteristics for a wide range of applications [3–5].
It is known that lanthanides Ln can be arranged in the framework with the formation of Na 1 + x Zr 2−x Ln x (PO 4) 3 compounds (in this case, their concentration x is limited, the number of occupied positions x ≤ 1, depending on the cation radius Ln 3+) [3]. Lanthanides can also be included in the non-frame positions, like the series of phosphates Ln 0.33Zr 2(PO 4) 3, where Ln = Ce–Lu (space group \({P\bar {{3}}c}\)) [4]. Systems of solid solutions such as B\(^{\text {II}}_{0.5-x}\)Eu x Zr 2(PO 4) 3, (B II–Ca, Sr, Ba) [6–9], Ca 0.5−1.5x Sm x Zr 2(PO 4) 3[6] and Ca 0.5−1.5(x + y)Er x Yb y Zr 2(PO 4) 3[7] with the NZP structure containing calcium, strontium, barium and lanthanides Eu 2+, Sm 3+, Er 3+, Yb 3+ in the cavity positions M1 have been investigated as a basis for luminescent materials having emissions in different light spectrum ranges and for obtaining white light [6–9]. However, the available data on the role of the environment of Ln 3+, its arrangement in the positions of the NZP structure and on the influence of the environment on luminescence properties are limited.
In this work, trivalent europium Eu 3+ was used as a local probe. Its visible optical characteristics are due to the 4f–4f transitions [10]. In the emission range, the 4f–4f transitions of Eu 3+ in the visible range consist of the following:
-
The 5D0 → 7 F 0 transition, at about 570–580 nm, which is related to the amount of crystallographic sites where Eu 3+ is located.
-
The 5D0 → 7 F 1 transitions at about 585–600 nm. The splitting is proportional to the intensity of the crystal field. This transition is related to the location of europium in an environment with inversion centres.
-
The 5D0 → 7 F 2 transitions at about 600–625 nm. This transition is related to the location of europium in an environment without an inversion centre. The ratio of insensitivity of 5 D 0 → 7 F 2 and 5 D 0 → 7 F 1 transitions \({I_{^{5}\mathrm {D}_{0} -^{7}\mathrm {F}_{2}}}/{I_{^{5}\mathrm {D}_{0} -^{7}\mathrm {F}_{1}}}\) is used to characterize the centro-symmetry of the europium site [11].
Upon excitation, the 4f–4f transitions are composed of 7 F 0 → 5 D j and 7 F 0 → 5 L j between 280 and 600 nm. However, in our case, the most interesting point is the charge band transfer, which is related to the transfer of an electron between europium and surrounding oxygen atoms. This transition is directly proportional to the ease with which one electron can be extracted from europium ions, which is proportional to the covalence of the Eu–O bond [12].
Comparison of the intensities of the forbidden 4f–4f absorption lines with respect to the charge transfer bands is relevant to the configuration quenching that can give information about the efficiency of the radiative transfer [13].
The aim of the present study was to obtain substances of the orthophosphate class having a frame-forming unit [Zr 2(PO 4) 3] − and alkaline (Na, K, Rb, Cs) and trivalent europium cations in the cavities. We sought to investigate the influence of cavity position occupancies and the effect of the alkaline ion rate of synthesized individual phases on luminescence properties. To date, the luminescence properties of NZP phosphates have received little attention. Their structure is very promising since many new compositions with adjustable optical properties can be created.
2 Experimental
We used phosphates A 1−3x Eu x Zr 2(PO 4) 3, where A = Na, K, Rb and Cs; x = 0, 0.001, 0.01, 0.05, 0.1 and 0.25. The reagents used were as follows: NaCl, KCl, RbCl, CsCl, Eu(NO 3) 3, ZrOCl 2, H 3 PO 4 and ethyl alcohol.
The sol–gel process was used to prepare phosphate samples. To this end, an H 3 PO 4 solution (1 M) was immediately added to the mixture of metal salt solutions (1 M) taken in stoichiometrical quantities while stirring at room temperature. The obtained gel was heated up to 90 ∘C for 5–8 h until the formation of a dry residue. The dry powders were sequentially heated at 600, 800 and 900 ∘C for 24 h at each stage with dispersion in an agate mortar before heating.
The samples obtained were studied by X-ray phase analysis and IR spectroscopy using a Shimadzu LabX XRD-6000 X-ray diffractometer (Cu K α filtered radiation, λ= 1.54178 Å) and an IR Furje-spectrometer FSM-1201.
A full-profile structure analysis was carried out on the samples Na 0.7Eu 0.1Zr 2(PO 4) 3 and Na 0.25Eu 0.25Zr 2(PO 4) 3. Diffractograms were recorded in the angle range 2 𝜃 10 ∘ (13 ∘)–100 ∘ with 0.02 ∘ increments. The lag time in the point was 6 s. The diffractograms and refining of phosphate structures were performed with the Rietveld method using the ‘Rietan-97’ program [14,15]. Peak profiles were approximated with the modified Voit pseudo-function [16]. Crystalline structures were refined by gradual addition of refined parameters under constant graphic modelling of background until R-factor values stabilized.
Excitation and emission were measured using a spectrofluorimeter SPEX FL212. The light source was a 450 W xenon lamp. The emitted light was selected with an emission double monochromator and then measured with a thermoelectricity cooled photomultiplier tube. Measured signals (in emission and excitation mode) were corrected for a variation of the incident flux, transmission of the monochromator and the response of the photomultiplier as a function of the wavelength.
3 Results and discussion
3.1 Characterization of phosphates
Synthesized phosphate samples appeared as colourless fine powders. Data for X-ray analysis were acquired for all the compounds and are shown in figure 1. The powder XRD data of the single-phase samples showed a similar distribution of Bragg reflections in the patterns of Na- and K-phosphates with smooth changing in their relative intensities and 2 𝜃 values over the entire composition range studied. However, the Rb- and Cs-phosphate-related patterns were not identical to those of the Na and K compounds (figure 1c). Moreover, X-ray data for Cs-containing phosphates showed some peaks belonging to additive phases (Zr-pyrophosphate and others; figure 1d).

X-ray data: A 1−3x Eu x Zr 2(PO 4) 3, where A = Na (a), K (b), Rb (c), Cs (d); x= 0 (1), 0.001 (2), 0.01 (3), 0.05 (4), 0.1 (5) and 0.25 (6).
With increasing x, a transition from space group \({R\bar {{3}}c}\) to \({P\bar {{3}}c}\) was evidenced: 0 ≤x≤ 0.1 \({R\bar {{3}}c}\), x= 0.25 \({P\bar {{3}}c}\). The \({P\bar {{3}}c}\) space group differed from the \({R\bar {{3}}c}\) one only by the loss of R translation. On the X-ray diffraction patterns, the peak that was the most relevant to the \({P\bar {{3}}c}\) space group was the (0 0 1) peak between 2 𝜃: 11 ∘ and 12 ∘. This peak was well resolved on all the patterns acquired of samples having an amount of europium of x= 0.25. The (2 1 2) peak, between 2 𝜃: 21 ∘ and 22 ∘, and the (2 0 2), (2 1 4) and (1 0 6) peaks, between 2 𝜃: 24 ∘ and 27 ∘, were not sufficiently resolved to confirm the space group.
Regarding only X-ray data, it was not possible to confirm that A 0.25Eu 0.25Zr 2(PO 4) 3 was in a single phase. The compounds might be a mixture of \({R\bar {{3}}c}\) and \({P\bar {{3}}c}\) phases. The cell parameters were calculated with the ‘UnitCell’ program [17]. Parameter c tended to increase and a tended to decrease with x growth in the 0 ≤x≤ 0.1 interval (figure 2). The cell parameters showed that parameter c was proportional to the size of the alkaline cation (Na + 1.0 Å, K + 1.38 Å, Rb + 1.52 Å, Cs + 1.67 Å, coordination number c.n. 6). Parameter a was in the opposite proportion with a NaZr2(PO4)3>a KZr2(PO4)3>a RbZr2(PO4)3>a CsZr2(PO4)3. This was due to the fact that the M1 polyhedra were oriented along the c axes, with a ZrO 6 polyhedron surrounding the M1 site on the top and bottom. Directions a and b offered more freedom for the M1 polyhedron to adjust their size.

Composition dependences of unit cell parameters for A 1−3x Eu x Zr 2(PO 4) 3, A = Na (1), K (2), Rb (3), Cs (4); Eu 0.33Zr 2(PO 4) 3 (5) (without Cs-Eu-phosphate with x=0.25).
The IR spectra of phosphates A 1−3x Eu x Zr 2(PO 4), where A = Na and K, are shown in figure 3. The IR spectra of A 1−3x Eu x Zr 2(PO 4), where A = Rb and Cs were similar to the Na and K ones. Figure 3 shows that two different types of IR spectra were observed depending on the Eu content in the sample. Factor group analysis predicted six IR active stretching vibrations of the PO\(_{\mathrm {4}}^{\mathrm {3-}}\) unit: ν 1 – E u ; ν 3 – 2 A 2u + 3 E u and seven IR active bending vibrations: ν 2 – 2 E u ; ν 3 – 2 A 2u + 3 E u for the phosphates A +Zr 2(PO 4) 3 with the space group \({R\bar {{3}}c}\)[18].

IR spectra of A 1−3x Eu x Zr 2(PO 4), where A = Na (a), K (b); x=0 (1), 0.05 (2), 0.1 (3) and 0.25 (4).
The ordered distribution of lanthanide atoms in the M1 site of the structure NaZr 2(PO 4) 3 (sp. gr. \({R\bar {{3}}c})\) led to the reduction of the symmetry with the \({P\bar {{3}}c}\) space group [4]. Results of factor-group analysis for compounds Ln 0.33Zr 2(PO 4) 3, where Ln = Ce–Lu, showed a space group \({P\bar {{3}}c}\) with fourteen IR-active modes related to the ν 3 vibrations of the PO\(_{\mathrm {4}}^{\mathrm {3-}}\) unit: 5 A 2u + 9 E u and four IR-active modes related to the ν 1 vibrations: A 2u + 3 E u [5].
It should be noted that values of wavenumbers of ν 3 vibration as high as ca. 1205 and 1230 cm −1 are quite uncommon for orthophosphates. This could be due to the polarizing nature of the metal ion (Zr 4+). These bands can be considered as P–O/Zr–O interaction vibrations.
The IR spectra of the phosphates with the space group \({R\bar {{3}}c}\) remained essentially the same, irrespective of the nature of the alkaline cation (figure 3). In these spectra, the asymmetrical stretching vibrations ν 3 of the PO\(_{\mathrm {4}}^{\mathrm {3-}}\) unit produced one broad band at ca. 1030–1040 cm −1 with one or two shoulders on its high-frequency side at ca. 1100 and ca. 1065 cm −1. The shoulder on its low-frequency side at ca. 1015–1020 cm −1 was assigned to symmetrical stretching vibrations ν 1 of the PO\(_{\mathrm {4}}^{\mathrm {3-}}\) unit as predicted by factor group analysis for this space group. The bending vibrations of the PO\(_{\mathrm {4}}^{\mathrm {3-}}\) unit are to be found in the region of 640–550 cm −1 with two or three bands.
The IR spectra of A 0.25Eu 0.25Zr 2(PO 4) 3 (x= 0.25), where A = Na, K, Rb and Cs, differed from the spectra of phosphates with 0 ≤x≤ 0.1 (figure 3). The bands of asymmetrical stretching vibrations of the PO\(_{\mathrm {4}}^{\mathrm {3-}}\) unit shifted to higher wavenumbers in the 1130–1060 cm −1 range. A larger number of modes in the region of the stretching vibrations ca. 1130–930 cm −1 were observed. The region 990–940 cm −1 contained two (in the case of Na) or three (in the case of K) signals which were assigned to symmetrical stretching vibrations of the PO\(_{\mathrm {4}}^{\mathrm {3-}}\) unit compared with four modes predicted by factor-group analysis for space group \({P\bar {{3}}c}\). The IR spectra of the phosphates A 0.25Eu 0.25Zr 2(PO 4) 3 were similar to those of R 0.33Zr 2(PO 4) 3, so they could be attributed to the space group \({P\bar {{3}}c}\)[4].
The alkaline metal atoms could occupy the M2 sites of the A 0.25Eu 0.25Zr 2(PO 4) 3 structure with space group \({P\bar {{3}}c}\) or they could be a mixture of the compounds belonging to the \({R\bar {{3}}c}\) and \({P\bar {{3}}c}\) space groups. In this case, the bands of the rhombohedral phase could overlap the bands of the compound with the \({P\bar {{3}}c}\) space group. However, the band of P–O/Zr–O interaction at 1205 cm −1, which is characteristic of IR spectra of the phosphates with the \({R\bar {{3}}c}\) space group, was absent in the IR spectra of phosphates A 0.25Eu 0.25Zr 2(PO 4) 3. Therefore, these compounds probably belong to a phase with the \({P\hspace *{-.3pt}\bar {{3}}\hspace *{-.3pt}c}\) space group and to M2 occupancy by the alkaline metal atoms.
Therefore, both X-ray and IR analyses are in agreement. The results suggest the formation of phosphate rows A 1−3x Eu x Zr 2(PO 4) 3 having an expected NZP structure (analogues: NaZr 2(PO 4) 3[19], KZr 2(PO 4) 3[20], RbZr 2(PO 4) 3[21], CsZr 2(PO 4) 3[22], space group \({R\bar {{3}}c}\) and Eu 0.33Zr 2(PO 4) 3 \({P\bar {{3}}c}\)) [4]. In these rows, the phosphates with 0 ≤x≤ 0.1 possess the space group \({R\bar {{3}}c}\) and those with x≥ 0.25 have the space group \({P\bar {{3}}c}\).
3.2 Rietveld analysis
We then refined the structures of this compound with the Rietveld method. Atomic coordinates in NaZr 2(PO 4) 3 (space group \({R\bar {{3}}c})\)[19] and Eu 0.33Zr 2(PO 4) 3 (space group \({P\bar {{3}}c})\)[4] were used as starting models to refine the structures of the Na 0.7Eu 0.1Zr 2(PO 4) 3 and Na 0.25Eu 0.25Zr 2(PO 4) 3 phosphates. The main refinement parameters and crystallographic data are given in table 1. The experimental and theoretical X-ray diffraction spectra of the phosphates are presented in figure 4. Excellent agreements between the calculated and fitted X-ray data were obtained.

Experimental (∘) and theoretical (—) XRD patterns of the compounds: (a) Na 0.7Eu 0.1Zr 2(PO 4) 3, sp. gr. \(R\bar {3}c\) and (b) Na 0.25Eu 0.25Zr 2(PO 4) 3, sp. gr. \(P\bar {3}c\).
Several additional diffraction maxima were observed on the X-ray diffraction pattern of the Na 0.25Eu 0.25Zr 2(PO 4) 3 phase. The atomic coordinates and isotropic thermal parameters are given in table 2. The data obtained confirm that the crystalline structures of the phosphates belong to the NZP type and are formed by the common topologic invariant [Zr 2(PO 4) 3] − (figure 5). As the \({P\bar {{3}}c}\) phase is the superstructure of the \({R\bar {{3}}c}\) phase, their unit cells are very similar. The sodium and europium atoms occupy non-framework sites. At the same time, this atoms are situated in the M1 sites (6b symmetry) in the Na 0.7Eu 0.1Zr 2(PO 4) 3 (space group \({R\bar {{3}}c}\)) structure and are distributed in the M1 (2b symmetry) and the M2 (6f symmetry) sites, so only atoms in the unit cell vertices become the cell-forming atoms. The calculated interatomic distances and bond angles are listed in table 3.

Fragment of structures of phosphates: (a) Na 0.7Eu 0.1Zr 2(PO 4) 3, sp. gr. R \(\bar {3}\) c and (b) Na 0.25Eu 0.25Zr 2(PO 4) 3, sp. gr. \(P\bar {3}c\).
3.3 Photoluminescence
All the compounds A 1−3x Eu x Zr 2(PO 4) 3 (A = Na, K, Rb, Cs, x= 0.001, 0.01, 0.05, 0.1, 0.25) were analysed in order to study the influence of the alkaline cation and the amount of europium on their optical properties. Eu 3+ photoluminescence in the phosphate series A 1−3x Eu x Zr 2(PO 4) 3 (A = Na, K, Rb, 0.001 ≤x≤ 0.25) was observed at various excitation and emission wavelengths (figure 6). In all cases, a broad band emission between 420 and 550 nm induced by an excitation between 300 and 400 nm was revealed. This band is indicative of the presence of divalent europium as already reported [23], meaning that a slight amount of europium is reduced in the divalent state. This amount does not seem to vary as a function of the europium concentration or the alkaline cation.

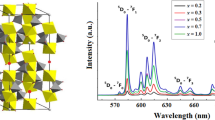
Photoluminescence spectra (excitation and emission) of Na 1−3x Eu x Zr 2(PO 4) 3, x=0.001, 0.01, 0.1, 0.25.
When analysing the trivalent europium signal (figure 6) from the point of view of emission, only one kind of emission related to a non-centrosymmetric site was evidenced. The peaks related to the transition 5 D 0– 7 F 2 (∼610 nm) were higher than those related to the transition 5 D 0– 7F 1 (∼590 nm).
The peak related to the transition 5 D 0– 7 F 0 was not observable (∼575 nm). With regard to excitation, only one charge band transfer was observed at high energy (low wavelength, <250 nm).
It can be deduced that the trivalent europium ions are incorporated in the kosnarite in a non-centrosymmetric site. This could correspond to the M2 site in the interframework. The high energy of the charge transfer band could be related to the low covalence of the Eu–O bond, which is in agreement with the localization of europium in the M2 site.
Regarding the compounds Na 0.97Eu 0.01Zr 2(PO 4) 3 and Na 0.85Eu 0.05Zr 2(PO 4) 3, the same observation as with Na 0.997Eu 0.001Zr 2(PO 4) 3 can be made, i.e., emission is clearly observed, relevant of europium in a non-centrosymmetric site and a charge band transfer at high energy.
The 5 D 0– 7 F 0 transition was now visible. By exciting at 210 nm, only one line was revealed. However, by exciting at other wavelengths (see the inset graph where the signal was normalized at 591 nm), a second contribution seemed to be apparent. It was not clear whether there was one or morepeaks relevant to the 5 D 0– 7 F 0 transition, especially with excitation at 250 nm. Furthermore, the 5 D 0– 7 F 1 transition was more widely split.
It could be deduced that most of the Eu 3+ ions are located in a non-centrosymmetric site corresponding to the M2 site. However, Eu 3+ seemed to be located elsewhere in a position that was not clear at this stage.
Regarding the compounds Na 0.7Eu 0.1Zr 2(PO 4) 3 and Na 0.25Eu 0.251Zr 2(PO 4) 3, two phenomena related to trivalent europium were evidenced. One concerned an excitation at 210 nm pertaining to europium located in a non-centrosymmetric site (intensity of 5 D 0– 7 F 2 was higher than that of 5 D 0– 7 F 1) with a charge transfer band at high energy, and another that was clearly evidenced by excitation at 260 nm, inducing an emission of 5 D 0– 7 F 1 that was higher than that of 5 D 0– 7 F 2. In addition, the 5 D 0– 7 F 1 peaks were more split, which was indicative of a higher crystal field and a charge transfer band at lower energy. The two sites were also evidenced by the 5 D 0– 7 F 0 transition as a function of excitation: 579.7 and 578.8 nm for excitation at 210 and 260 nm, respectively.
It can be deduced that trivalent europium is located in the M2 site like the previous compounds, but that it is also in a more symmetric site with a more covalent character in the Eu–O bond. This site corresponds to the M1 site in the interframework. Regarding the optical studies of the Na 1−3x Eu x Zr 2(PO 4) 3 compounds, it can be concluded that trivalent europium starts to incorporate kosnarite in the M2 site. Then, by increasing the amount of europium, Eu 3+ progressively incorporates a centro-symmetric site with a higher crystal field (M1). By adding europium, all the positions of the charge transfer bands shift to the lowest energy. This phenomenon is due to the antagonist bound of Eu–O. In the case of x= 0.001, polyhedra containing europium are surrounded by polyhedra containing sodium. By increasing the amount of europium, the surrounding environment consists gradually of polyhedra containing europium and vacancies. The shift to the lowest energy seems to indicate a global increase in the covalence of the europium–oxygen bond.
Photoluminescence of potassium, rubidium and caesium compounds was studied with the same approach. While many experiments were performed, only selected results are shown in figure 7. For potassium and rubidium, the findings were similar to those for the sodium compounds, i.e., the incorporation of europium starting in the M2 sites then gradually occurring in the M1 sites, with also a small amount of divalent europium.

Excitation and emission photoluminescence spectra of (K, Rb, Cs) 1−3x Eu x Zr 2(PO 4) 3.
Caesium compounds were slightly different in their optical properties. There was a high-energy charge band transfer (225 nm) inducing an emission due to europium in a non-centrosymmetric position and a lower energy charge band transfer (300 nm) inducing an emission related to a europium in a centrosymmetric site. However, another phenomenon was also evidenced.
With excitation at 208 nm, the 5 D 0– 7 F 1 transition was different than at 225 nm, but the intensity of 5 D 0– 7 F 1 was still lower than that of 5 D 0– 7 F 2. This indicated the presence of an additional non-centrosymmetric europium. There were three different 5 D 0– 7 F 0 lines corresponding to three different types of europium (see the inset graph in figure 7c).
3.4 X-ray and photoluminescence
As observed with X-ray and cell parameter measurements, it seems that the caesium-doped compounds were not like the other compounds, especially as there was a large amount of europium in the compound. For Na, K and Rb compounds, with the incorporation of Eu into the M2 site the M1 site could account for the change in cell parameters. The alkaline cations in the M1 site were substituted by europium cations in the M2 site. While the M1 site is an inverted bipyramid oriented along the c-axis, the M2 site is not so symmetric and could be described as a distorted pentahedron also oriented along the c-axis. Therefore, the incorporation of Eu induced a slight increase in the c parameter.
According to Bykov et al[4], the europium in the Eu 0.33Zr 2(PO 4)3 compound is only in the M1 site, which seems consistent with our work. However, with all the M2 sites empty, the cell parameters are almost the same as those of NaZr 2(PO 4) 3 compound, since Na + and Eu 3+ have a similar ionic radius.
The luminescence spectra of the phosphates confirm the results of infrared spectroscopy for Eu compositions of 0.05, where almost all the trivalent europium is in the M2 position. This result is quite new since the M2 site has until now been reported to be empty in most NZP phases.
When the amount of europium increases to x= 0.25, the amount of cations in the M2 cavities sites decreases. This leads to a lowering of the structure symmetry to space group \(P\bar {{3}}c\) and the splitting of the M1 (6b) and M2 (18f) sites into several types of lower multiplicity (2b and 6f) that are occupied by sodium and europium atoms.
4 Conclusion
The compounds A 1−3x Eu x Zr 2(PO 4) 3 (A = Na +, K +, Rb + and Cs +: x= 0–0.25) were investigated for the development of new phosphors. The transition from \({R\bar {{3}}c}\) to \({P\bar {{3}}c}\) as a function of x was evidenced by X-ray diffraction and infrared techniques. By understanding the incorporation of trivalent europium, it is possible to grasp the non-logical evolution of the cell parameters. Photoluminescence data showed that, during substitution, alkaline cations are removed from the M1 site and initially replaced by europium in the M2 site. Owing to the different symmetries of each site, the cell undergoes a decrease in parameter a and an increase in parameter c on adding more europium, while the volume remains almost constant. These results will prove fruitful for incorporating other cations in order to modulate their luminescence properties.
References
Alamo J 1993 J. Solid State Ion. 63–65 547
Orlova A I 2002 Radiochemistry 44 423
Miyajima Y, Muyoshi T, Tamaki J, Matsuoka M, Yamamoto Y, Masquelier C, Tabuchi M, Saito Y and Kageyama H 1999 J. Solid State Ion. 124 201
Bykov D M, Gobechiya E R, Kabalov Yu K, Orlova A I and Tomilin S V 2006 J. Solid State Chem. 179 3101
Kurazhkovskaya V S, Bykov D M, Borovikova E Y., Boldyrev N Y., Mikhalitsyn L and Orlova A I 2010 Vibr. Spectrosc. 52 137
Glorieux B, Jubera V, Orlova A I, Kanunov A E, Garcia A, Pallier C and Oleneva T A 2013 Inorg. Mater. 49 82
Kanunov A E, Gorshkova E N, Orlova A I, Shushunov A N, Mikheeva E R and Pleskova S N 2013 Phys. Procedia 44 224
Masui T, Koyabu K, Tamura S and Imanaka N 2006 J. Alloys Compd. 418 73
Hirayama M, Sonoyama N, Yamada A and Kanno R 2009 , J. Solid State Chem. 182 730
Laporte O and Meggers W F 1925 J. Opt. Soc. Am. 11 459
Tuan D C, Olazcuaga R, Guillen F, Garcia A, Moine B and Fouassier C 2005 J. Phys. IV 123 259
Fonger W H and Struck C W 1971 J. Electrochem. Soc. 118 273
Blasse G 1979 J. Solid State Chem. 27 3
Rietveld H M 1967 Acta Crystallogr. 22 151
Kim Y I and Izumi F 1994 J. Ceram. Soc. Jpn. 102 401
Izumi F 1993 In: R A Young (ed) The Rietveld method (New York: Oxford University Press Inc.)
Holland T J and Redfern S A T 1997 Mineral. Mag. 61 65
Tarte P, Rulmont A and Merckaert-Ansay C 1986 Spectrochim Acta A 42 1009
Hagman L and Kierkegaard P 1968 Acta Chem. Scand. 22 1822
Brownfield M E, Foord E E, Sutley S J and Botinelly T 1993 Am. Mineral. 78 653
Matkovic B, Prodic B and Sljukic M 1968 Bull. Soc. Chim. Fr. 4 1777
Gobichiya E R, Kabalov Yu K, Petkov V I and Sukhanov M V 2004 Crystallogr. Rep. 49 741
Boukhris A, Hidouri M, Glorieux B and Ben Amara M 2012 Mater. Chem. Phys. 137 26
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
KANUNOV, A., GLORIEUX, B., ORLOVA, A. et al. Synthesis, structure and luminescence properties of phosphates A1−3x Eu x Zr2(PO4)3 (A—alkali metal). Bull Mater Sci 40, 7–16 (2017). https://doi.org/10.1007/s12034-016-1337-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12034-016-1337-1