
Abstract
The ERF transcription factors belong to the AP2/ERF superfamily, one of the largest transcription factor families in plants. They play important roles in plant development processes, as well as in the response to biotic, abiotic, and hormone signaling. In the present study, 155 putative ERF transcription factor genes were identified from the potato (Solanum tuberosum) genome database, and compared with those from Arabidopsis thaliana. The StERF proteins are divided into ten phylogenetic groups. Expression analyses of five StERFs were carried out by semi-quantitative RT-PCR and compared with published RNA-seq data. These latter analyses were used to distinguish tissue-specific, biotic, and abiotic stress genes as well as hormone-responsive StERF genes. The results are of interest to better understand the role of the AP2/ERF genes in response to diverse types of stress in potatoes. A comprehensive analysis of the physiological functions and biological roles of the ERF family genes in S. tuberosum is required to understand crop stress tolerance mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plants are exposed to different types of stresses during their life cycle such as salinity, cold, drought, low temperatures, and pathogens that have adverse effects on growth and productivity [1]. It is therefore important to investigate the plant response to such stresses in order to improve crop productivity under stress conditions. Plant response to biotic and abiotic stresses is regulated by several signal transduction pathways that involve phytohormones and calcium for the activation of stress response genes. Transcription factors play pivotal functions in signal transduction by the activation or the repression of target genes [2]. The APETALA2/Ethylene Responsive Factor (AP2/ERF) superfamily is one of the largest groups of transcription factors in plants, which includes at least one APETALA2 (AP2) domain [3]. It is characterized by a conserved DNA-binding sequence of 60–70 amino acids [4]. The subdivision of these TFs into families is based on the number of AP2 domains present in their protein sequence. The AP2 family is characterized by a double-tandem-repeat AP2 domain [5], while the ERF family contains a single AP2 domain [6]. Recently, the RAV (related to ABI3/VP1) which contains a single AP2 domain and an additional B3 domain family was also identified [7]. Sakuma et al. [8] were the first to divide the ERF family into two major subfamilies in Arabidopsis thaliana, the ERF subfamily (group B) and the C-repeat binding factors/drought-responsive element binding factors (CBF/DREB) subfamily (group A). The highly conserved 14th and 19th amino acids distinguish the DREB from the ERF group. DREBs are characterized by a valine residue in position 14 and a glutamic acid in position 19. However, ERFs display an alanine and aspartic acid residue in position 14 and 19, respectively [8]. Nakano et al. [5] reported a new phylogenetic classification of ERF genes in Arabidopsis in which they were split into 10 clades, namely I–X.
The AP2/ERF transcription factors regulate diverse processes of plant development and stress responses (Table 1; [9–15]), such as vegetative and reproductive development, cell proliferation, abiotic and biotic stress responses, and plant hormone responses [16, 17].
The ERF transcription factors are involved in the ethylene signaling pathway and lead to the expression of various defense-related genes, such as pathogenesis-related (PR) genes [18] and abiotic stress-responsive genes [19]. These genes are characterized by the presence of a GCC-box (GCCGCC) or a dehydration responsive element/C-repeat element (DRE/CRT, CCGAC) located in their promoter sequences. It was reported that some DREB factors such as AtDREB2A [8], TINY2 [20], tobacco stress-induced gene 1 (Tsi1) [19], OsDREB2A [17], and BnDREBIII [21] can bind to both GCC and DRE boxes.
Recently, several plant genome sequences as well as expression data such as RNA-seq have become available publicly. These data have prompted in-depth bioinformatic investigations of the ERF transcription factor family in several plant species, such as A. thaliana [5], rice [17], Poplar [22], soybean [14], grapevine [6], Brassica rapa [23], barley and wheat [24], apple [25], and tomato [26]. The recent analysis of stress response in S. tuberosum Group phureja [27] measured through whole transcriptome sequencing allowed the identification of 210 AP2/ERF genes that are upregulated under stress conditions.
In this report, the identification and the characterization of the ERF transcription factors in potato (S. tuberosum) were attempted. The ERF transcription factor, phylogenetic relationships and orthologs genes between the S. tuberosum and A. thaliana genome were analyzed. The expression profile of some of these genes in vegetative tissues and under various stress conditions was also investigated using published RNA-seq data and semi-quantitative RT-PCR.
Materials and Methods
Plant material, In Vitro Culture, and Stress Treatments
Potato plants (S. tuberosum L. cv. Claustar) were cultivated in vitro and propagated in solid MS basal medium [28] supplemented with vitamins [29] in a growth chamber at 21 °C for 16/8 h photoperiods, under 250 µE/m2/s light intensity. This Claustar potato cultivar was chosen because of its important economic value in Tunisia [30].
For stress application, after 3 weeks of culture, the plants were transferred to an aqueous MS medium supplemented with 100 mM NaCl or 10 % (w/v) polyethylene glycol (PEG 8000) for salt and dehydration stress, respectively. All stress treatments were carried out for 3, 6, 9, and 24 h. Leaves, roots, and stems were harvested and immediately frozen in liquid nitrogen and stored at −80 °C until used.
RNA Isolation and cDNA Synthesis
Total RNAs were extracted from 0.5 to 1 g of frozen material as previously described [31]. The absence of genomic DNA was confirmed by PCR using the EF1α [PGSC0003DMG400005728] primers (EF1α-F:GATGCTACCACCCCGAAGTA, EF1α-R: ACGAAGGGGTTTGTCTGTTG) after DNase I treatment.
Primer Design and Semi-quantitative Analyses of Gene Expression by RT-PCR
The primers for semi-quantitative RT-PCR analyses (Table 2) were designed with melting temperatures of 60 °C using the Primer3 v.0.4.0 software.
Semi-quantitative RT-PCR was carried out as described by Degenhardt et al. [32]. Single-stranded cDNAs were synthesized from 2 µg of total RNA using the M-MuLV reverse transcriptase (200 U; BIO BASIC INC) in a final volume of 20 µl. The efficiency of reverse transcription was assessed using the EF1α mRNA pair of primers. PCR was performed on an Applied Biosystems 2700 machine as follows: initial step of 5 min at 95 °C, followed by 25 cycles of 20 s at 95 °C, 30 s at 60 °C, and 1 min at 72 °C. All PCR reactions were carried out in a final volume of 25 µl using Taq DNA polymerase (Promega). Three independent biological replications were performed. Each replicate consisted of pooled tissues from six plants cultivated under the same stress conditions.
PCR products were visualized on ethidium bromide-stained gels using the Gel DocXR Gel Documentation System (BioRad). The average intensity of the bands was quantified using Quantity One 1-D Analysis Software (BioRad) and graphed using Microsoft Excel. The error bar was determined from three separate biological replicates.
BLAST Searches and Sequence Analyses
AP2 domain-containing proteins in the S. tuberosum genome (genome annotation v3.4) were retrieved from Phytozome v9.0 (http://www.phytozome.net/potato.php) using BLASTp (Basic Local Alignment Search Tool) with a cutoff e value of 1e−5. The sequences of the A. thaliana AP2/ERF proteins were used as queries. All splice variants were removed, and only the primary transcript was retained for each gene. The resulting protein sequences were subjected to Pfam analysis (http://Pfam.sanger.ac.uk/) to confirm the presence of the corresponding conserved domains. Some genes encoding proteins lacking initiation and/or termination codons were corrected with FGENESH (http://linux1.softberry.com/berry.phtml) by using dicotyledonous plants as model. Gene models that could not be corrected were discarded.
Phylogenetic Analyses
Phylogenetic and molecular evolutionary analyses were conducted using MEGA software v.5 [33]. Full-length StERF proteins were aligned using the ClustalW software. The phylogenetic tree was generated by the neighbor-joining method using the pairwise deletion option. The reliability of the trees obtained was tested using bootstrapping with 1,000 replicates and amino acid p-distance parameters. Sequences of Arabidopsis ERF and annotation were used as described [5].
Chromosomal Location of the Potato AP2/ERF Genes
Informations on the chromosomal location were retrieved from the potato genome database. MapChart 2.2 [34] was used for chromosomal linkage visualization. A codon-based Z test was performed for each block in MEGA Software using the Pamilo–Bianchi–Li substitution model, the bootstrap variance estimation method (1,000 replicates), and pairwise deletion [33].
Prediction of Molecular Parameters
The isoelectric point (IP) and the molecular weight of potato ERF deduced proteins were calculated using the ProtParam tool (http://us.expasy.org/tools/protparam.html). The Fold Index program (http://bioportal.weizmann.ac.il/fldbin/findex) was used to investigate the state of protein folding. The location of the sub-cellular compartment was performed using the TargetP software (http://www.psort.nibb.ac.jp/form.html).
In Silico Gene Expression Analyses
Fragments Per Kilobase of transcript per Million fragments mapped values (FPKM values) were retrieved from the Illumina RNA-seq data that were previously generated and analyzed by Massa et al. [27]. The expression values were clustered using the QT clustering tool in the Multiple Array Viewer MeV v4.4.1 [35].
Determination of Conserved Protein Motifs and Gene Structural Analysis
Conserved motifs were investigated by multiple alignment analyses using Multiple Expectation Maximization for Motif Elicitation (MEME) v.4.7.0 [36] with the following parameters: distribution of motifs: zero or one per sequence; maximum number of motifs: 25; minimum number of sites: 2; the maximum number of sites: 167; minimum motif width: 6; maximum motif width: 50.
The diagrams of the exon–intron structure were obtained using the online Gene Structure Display Server (GSDS: http://gsds.cbi.pku.edu.cn) [37]. Both Coding Sequence (CDS) and genomic sequences were downloaded from Phytozome v9.0 (http://www.phytozome.net/potato.php).
Results and Discussion
Identification of the ERF Gene in S. tuberosum
Gene models containing an AP2 domain in the S. tuberosum genome were identified using BLASTp (see “Materials and Methods” section) using known A. thaliana genes encoding AP2/ERF as queries. Alternative splice variants were removed. Short sequences or those that did not present start and/or stop codons were re-predicted using the Fgenesh software. Sequences with no reliable prediction were removed. The remaining proteins were inspected for a significant match to the AP2 domain (PF00847; E value <0.001) using the Pfam database (http://Pfam.sanger.ac.uk/). Sequences with insignificant match were further removed. This process yielded 181 non-redundant candidate gene models containing an AP2 domain (Table 3). Indeed, 13 gene models harbor double complete or incomplete AP2 domains, and one gene model was assigned as a RAV gene. Finally, 155 gene models were assigned as ERFs, of which 65, 90, and 11 genes encoded CBF/DREB, ERF, and incomplete AP2 domains, respectively (Table 3). Among these genes, six ERF (StERF185, StERF84, StERF111, StERF94, StERF93, and StERF51) and four DREB (StERF44, StERF19, StERF37, and StERF186) genes were previously identified and characterized in the laboratory [38, 39]. These data revealed that potato possesses more ERF genes than other fully sequenced eudicotyledon plants. Indeed, 155 ERF genes were identified here, while the analysis of S. tuberosum Group phureja transcriptome in response to biotic and abiotic stresses allowed the detection of 210 upregulated AP2/ERF genes [27]. A total of 146 genes were recorded in tomato [26], 120 in Glycine max [14], and 122 in Arabidopsis [5]. Among monocot species, bread wheat (Triticum aestivum) contains 117 putative AP2/ERF genes that were identified via in silico analyses based on the presence of conserved AP2/ERF domain [24] and rice contains 139 ERF genes [5].
Phylogenetic Relationships Between the ERF Family Genes in Potato
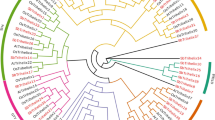
The evolutionary relationship between potato and Arabidopsis based on AP2/ERF transcription factors was assessed by phylogenetic reconstruction using the AP2 domain. The resulting phylogenetic tree (Fig. 1) showed that the StERFs were mainly subdivided into two major groups, the CBF/DREB and the ERF group. They are themselves divided into 10 clades designed clade I–X, as classified by Nakano et al. [5].

An unrooted phylogenetic tree of ERF proteins of potato, Arabidopsis, and rice. Multiple sequence alignments of the AP2 domains were performed using ClustalW2, and the phylogenetic tree was constructed using MEGA5.05 by the Neighbor-joining method with 1,000 bootstrap replicates. The tree was divided into the CBF/DREB subfamily (group I–V) and the ERF subfamily (group VI–X) (Color figure online)
Phylogenetic analyses showed that potato possesses 88 and 68 gene models encoding CBF/DREB and ERF, respectively (Supplementary material 1), while Arabidopsis possesses 70 CBF/DREB and 52 ERF genes.
Concerning the CBF/DREB groups, they split into 5 clades (Fig. 1; Supplementary material 1) containing 8, 7, 28, 9, and 14 gene models, respectively. The ERF group contained four clades (clade VII–X) with 17, 5, 16, 41, and 11 gene models, respectively. The expansion of the CBF/DREB and ERF genes in potato, compared to Arabidopsis, could be due to genome duplication by tetraploidization in potato. Indeed, gene duplication often involves stress response genes, suggesting that important genetic events occurred for adaptive evolution in dynamically changing environments [40]. The explanation of the AP2/ERF gene number in potato suggests a sub-functionalization of this superfamily in response to stress.
Molecular Characteristics of the Potato AP2/ERF Proteins
We have employed bioinformatic tools to investigate the physical properties of the ERF proteins from potato, such as the molecular weight (MW), the isoelectric point (pI), and the folding index. The majority of the StERF proteins were predicted to have a low MW, ranging from 1.2 to 11.8 kDa and an average of 247 amino acids, going from 118 to 1,054 amino acids (Supplementary material 2). Furthermore, 65.8 % StERFs were predicted to be acid, whereas 34.2 % were predicted to be basic or neutral. The fold index determination showed that 53 % of StERFs were folded proteins. All StERFs seemed to be nuclear (Supplementary material 2).
Chromosomal Location of the StERF Genes
In silico mapping of the gene loci showed that the 155 potato ERF genes were distributed across all twelve potato chromosomes (Fig. 2). Chromosome XII encompassed 25 ERF genes, the largest number of ERF genes followed by 24 and 23 on chromosome III and I, respectively. In contrast, 11 ERF genes were found on chromosome VI and 9 on chromosome VII. Only four ERF genes were located on chromosome VII (Fig. 2). Moreover, the following genes seemed to be in sequence clusters: StERF133 and StERF139 on chromosome VI; StERF156, StERF162, and StERF51 on chromosome XII; StERF26 and StERF27 on chromosome VIII; StERF61 and StERF65 on chromosome VIII; StERF62 and StERF64 on chromosome VIII; StERF64 and StERF63 on chromosome VIII; and StERF81 and StERF82 on chromosome III (Fig. 2).

Chromosomal distribution of the potato ERF genes identified in this study. The chromosome number is indicated above each chromosome. The chromosomal positions of the ERF genes are indicated by their generic name. Identical colored circles or squares indicate duplicated gene pairs, and the thick lines correspond to joined tandem-repeated genes (Color figure online)
The Relationship Between Gene Structure and Phylogenetic Classification
Gene structure analysis was carried to investigate the conservation of intron and exon arrangements in the StERF genes (Supplementary material 4b). The total number of exons ranged from 1 to 3. Moreover, 130 ERF genes (83 %) did not contain introns in their sequences. This finding corroborates a previous report that showed that most ERF genes in Arabidopsis are intronless and only four of them have one intron [8]. For the remaining genes, (StERF21, 3 and 1), they harbor one, 2, or 3 exons, respectively. The number and position of exons were similar between closely related genes. These data further validated the reliability of the potato ERF gene family classification presented here.
Conserved Motifs Outside of the AP2/ERF Domain
The regions outside the DNA-binding domain in transcription factors generally contain functionally important domains involved in transcriptional activity, protein–protein interactions, and nuclear localization [41]. Such conserved motifs found in the StERF family are summarized in Supplementary material 4a.
Eight groups possess nine conserved motifs except groups II and III that possess seven and eight conserved motifs, respectively. Moreover, the ERF proteins that cluster in the same clade share a similar motif composition, suggesting functional similarities among members of the same subgroup.
Sequence alignment of the AP2/ERF domains showed that all potato ERF family members contain the WLG (tryptophan, lysine, glycine) motif, and most of them contain the Gly-4, Arg-6, Arg-8, Glu-16, Arg-26, Ala-38, Asp-43, and Asn-57 residues (Supplementary Fig. 1). A previous report showed that the Ala-38 residue is essential for DNA binding to the AP2/ERF domain [21].
These analyses also showed the presence of the conserved AYD (alanine, tyrosine, aspartic acid) element in the ERF family, whereas the DREB family has conserved AHD (alanine, histidine, aspartic acid) element. These two elements within the AP2/ERF domain have been reported to be critical in activating DNA binding to modulate the expression of target genes. They are highly conserved in most of AP2/ERF in plants, implying their structural and functional necessity [8].
Motif analyses revealed the presence in group VIII of an ERF-associated amphiphilic repression (EAR) motif that has been shown to function as a repression domain [5]. The EAR motif (L/F)DLN(L/F)XP was identified as a conserved sequence in the C-terminal region of repressor-type ERF proteins.
Expression Profiling of StERF Genes
Several ERF genes play important roles in biotic and abiotic stress responses in plants [42]. The publicly available RNA-seq data previously generated from reproductive and vegetative libraries [27] were used to investigate the expression profiles of the StERF genes in these tissues. The FPKM value for each gene was retrieved, and the data were presented as Heatmap for each StERF clade (Fig. 3). Some genes showed a tissue-specific expression pattern; among them StERF6, StERF89, StERF63, StERF104, StERF92, and StERF7 from the CBF/DREB group, and StERF21 and StERF25 from the ERF group were expressed only in control roots, while StERF42 was expressed only in control leaves. These genes seemed to be involved in developmental processes, since they were only expressed in vegetative tissues and not in stressed tissues. On the other hand, StERF147, StERF169, StERF120, and StERF110 were only expressed under heat stress (35 °C, Fig. 3). Only one StERF gene (StERF114) reveals specific and low expression under salt stress. The StERF71, StERF47, StERF67, and StERF70 genes were specifically expressed in Phytophtora infestans (P.I) infected leaves. These factors belong to the same phylogenetic clade IX suggesting that most members of this clade could be involved in biotic stress response in potato.

Heat map representation and hierarchical clustering of the StERF genes. The FPKM values retrieved from published RNA-seq data were log2 transformed and the heat map generated using TIGR MeV v4.1.1. The data correspond to leaves (L), roots (R), shoots (S) in control conditions and after treatment for 24 h with 150 mM NaCl, 35 °C, ABA and Phytophtora infestans (P.I) infection in leaves (L). Color scale at the top of each dendrogram represents relative expression levels: white represents low level, and blue indicates high level. Asterisk represents the five selected genes for semi q-RT-PCR (Color figure online)
The RNA-seq expression pattern of the ERF genes showed that most of them are environmentally controlled in potato. Compared to control leaves, 5 and 20 StERFs were modulated by abiotic and biotic stress in the DREB and ERF group, respectively. The DREB group of genes belongs mainly to clade I (StERF41), clade II (StERF37, 19 and 149), and clade III (StERF8, 14, and 82), while the StERF genes are from clade VI (StERF145 and 86), clade VII (StERF43 and 148), clade VIII (StERF66, 45, 75, 84, and 115), clade IX (StERF46, 49, 135, 136, 11, 101, 117, 94, 50, 95, 100, 87, 51, 93, 156, and 134), and clade X (StERF32). Furthermore, a number of genes belonging to clade I (StERF41), clade II (StERF37, 19, 149, and 16), clade III (StERF14, 62, 2, 36, 82, and 129), clade V (StERF133, 139, 3, and 144), clade VI (StERF60, 145, 1, 12, and 154), clade VII (StERF141, 43, and 148), clade VIII (StERF75, 84, 115, and 143), clade IX (StERF49, 135, 117, 94, 50, 95, 100, 87, 51, 93, 156, 162, 127, and 134), and StERF 10 of clade X were induced in NaCl- and ABA-treated leaves compared to control leaves (Fig. 3). This suggests that these genes could be involved in the response to salt stress via an ABA-mediated signaling transduction pathway. StERF186 was previously isolated and labeled as StDREB1 [39]. This study showed that it is expressed in leaves, stems, and roots under stress conditions and is greatly induced by NaCl, drought, low temperature, and ABA treatments. Moreover, the overexpression of this gene in potato plants improved salt and drought stress tolerance [39]. These results are in agreement with the RNA-seq expression pattern of this gene. Another gene named StDREB2 was also isolated previously by Bouaziz et al. [38]. It corresponds to StERF37. This gene is expressed in leaves, stems, and roots and is induced by NaCl, ABA, and heat treatments. The overexpression of this gene in potato improves tolerance to abiotic stress in agreement with the data obtained in RNA-seq.
Eight additional StERF cDNAs were previously identified by Bouaziz et al. [38]; they were divided into 6 StERF and 2 DREB genes. They correspond to StERF1,StERF2, StERF3, StERF4, StERF5, and StERF6, which correspond to StERF185, StERF111, StERF93, StERF84, StERF94, and StERF51, respectively, and to StDREB3 and StDREB4, which correspond to StERF19 and StERF44, repectively [43]. All these genes were shown to be upregulated by ethylene treatment. All these factors except StERF2 were upregulated by NaCl and ABA treatment. These data are in agreement with those of RNA-seq analysis.
It was reported that the ERF family members are responsive to drought, ABA, and saline conditions and can serve as activators or repressors of ABA signaling under salt stress [44]. Data from RNA-seq suggested that StERFs may be associated with different functions and phytohormone-mediated stress signaling pathways as suggested by Gutha and Reddy [45].
Expression Analysis of Certain StERF Genes Using Semi-quantitative RT-PCR
Five StERF genes were selected for semi q-RT-PCR analyses, based on phylogenetic and RNA-seq data. These genes are representatives of the StERF groups. They belong to five different clades, and their RNA-seq profiles show that they are differently expressed under biotic and abiotic stress conditions. Semi-quantitative RT-PCR analyses under salt (100 mM NaCl) and dehydration (PEG 8000) stress conditions at different times during treatment ranging from 3 to 24 h were performed using primers specific for these genes. However, the results showed that none of the selected ERF genes was expressed in control leaves (Fig. 4). However, these genes seemed to be induced by salt stress in leaves after 3 h. Indeed, StERF94, 80, and 12 reached their maximum expression at 6 h of salt stress in leaves, while StERF85 and 141 were highly induced at 3 and 9 h, respectively. The expression profile of all the selected StERF genes except StERF185 is in agreement with the RNA-seq data. The selected ERF genes, except StERF94, were also upregulated by salt stress in stems.

Normalized semi-quantitative RT-PCR of selected StERF genes. a Relative expression under NaCl treatments (100 mM NaCl) for 3, 6, 9, and 24 h in leaves, stems, and roots; b Relative expression under stress treatments (PEG 8000) for 3, 6, 9, and 24 h in leaves, stems, and roots. RT-PCR data were normalized using the potato elongation factor EF1α gene. Standard error (SE) was determined from three independent biological replicates, and band densities were calculated by the Quantity One 1D analysis software (see “Materials and Methods” section)
These genes displayed different expression patterns in leaves of plants submitted to salt stress (Fig. 4a). Expression of StERF185 was observed only after 3 h of 100 mM NaCl application, while the remaining StERF genes were induced in leaves after 3 h of salt stress, and their expression remained higher than in the controls until 24 h of stress.
Concerning dehydration stress, these genes seemed to be induced by PEG treatment in leaves. Indeed, StERF12 and StERF141 were highly induced after 3 h of PEG application (Fig. 4b), and StERF141 induction continued from 3 to 24 h of PEG application in leaves. StERF94 showed stable expression in plants submitted to PEG treatment from 3 to 24 h. As opposed to other genes (StERF185, StERF80, and StERF12), the expression decreased significantly at 24 h in comparison with 3 and 6 h of stress applications.
Unlike in leaves, StERF12, StERF141, and StERF185 were expressed in both control stems and roots. The expression of StERF94 was not detectable in roots of PEG-treated plants, and it was not expressed in control and salt-stressed stems after 24 h of 100 mM NaCl application. StERF12, StERF80, and StERF185 were upregulated by both dehydration and salt stress in leaves. These data corroborate those reported on RAP2.6 of Arabidopsis that belongs to the same group as StERF80 and that plays a central role in the response to salinity [46]. On the other hand, AtERF3, the ortholog of StERF185, was reported to be regulated by high salinity and drought in Arabidopsis [47].
In stems, StERF12 and StERF141 seemed to be expressed in both control and stress conditions (Fig. 4). The StERF12 gene was highly upregulated by salt stress in stems, while StERF185 was induced only by dehydration in stems.
The RT-PCR results of the two StERF genes selected here (StERF80, StERF12) after 100 mM NaCl treatment corroborate the expression profile revealed by RNA-seq. For the three remaining ERF genes (StERF94, 141, and 185), the data obtained by RT-PCR were slightly different from those revealed by RNA-seq. Indeed, no expression of this gene was detected in control leaves, while the RNA-seq suggested a basal expression level of this gene in leaves of control plants with an increase in stress conditions. These differences could be due to the plant genotype. In addition, all the samples for the RNA-seq analyses were collected from greenhouse-grown plants, except for root and shoot tissues, which were collected from in vitro-grown plants [27], whereas in the present study, all the samples were collected from in vitro-cultivated plants. This difference in plant cultivar technique used for RNA-seq and those applied in this study may explain the differences observed.
These data confirmed those described (in silico) that also showed that these StERF genes are involved in different stress response pathways in potato. Moreover, their expression showed distinct expression profiles, suggesting that they are related to different signaling pathways in the different organs and during different stresses.
In conclusion, in the present study, we have identified 155 genes encoding ERF transcription factors from the S. tuberosum genome. Phylogenetic analyses showed that they were dispersed in two major groups (ERF and DREB) as described [5, 8]. Based on RNA-seq data, the expression analysis of five representative genes carried out in control and in stress conditions revealed divergent expression patterns in NaCl and PEG treatments. The expression patterns of some of these genes corroborated the RNA-seq data and previous reports [38, 39], suggesting their involvement in salt and dehydration response and signaling. Thus, overexpression of these ERF genes in the salt-sensitive Claustar potato cultivar is very helpful to carry out functional analyses in planta and the creation of new, salt-tolerant varieties.
References
Wang, W., Vinocur, B., & Altman, A. (2003). Plant responses to drought, salinity and extreme temperatures: towards genetic engineering for stress tolerance. Planta, 218, 1–14.
Xu, Z. S., Chen, M., Li, L. C., & Ma, Y. Z. (2011). Functions and application of the AP2/ERF transcription factor family in crop improvement. Journal of Integrative Plant Biology, 53, 570–585.
Wessler, S. R. (2005). Homing into the origin of the AP2 DNA binding domain. Trends in Plant Science, 10, 54–56.
Zhou, M. L., Ma, J. T., Pang, J. F., Zhang, Z. L., Tang, Y. X., & Wu, Y. M. (2010). Regulation of plant stress response by dehydration responsive element binding (DREB) transcription factors. African Journal of Biotechnology, 9, 9255–9269.
Nakano, T., Suzuki, K., Fujimura, T., & Shinshi, H. (2006). Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiology, 140, 411–432.
Licausi, F., Giorgi, F. M., Zenoni, S., Osti, F., Pezzotti, M., & Perata, P. (2010). Genomic and transcriptomic analysis of the AP2/ERF superfamily in Vitis vinifera. BMC Genomics, 11, 719.
Hu, L. F., & Liu, S. Q. (2011). Genome-wide identification and phylogenetic analysis of the ERF gene family in cucumbers. Genetics and Molecular Biology, 34, 624–633.
Sakuma, Y., Liu, Q., Dubouzet, J. G., Abe, H., Shinozaki, K., & Yamaguchi-Shinozaki, K. (2002). DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochemical and Biophysical Research Communications, 290, 998–1009.
Lin, R. C., Park, H. J., & Wang, H. Y. (2008). Role of Arabidopsis RAP2.4 in regulating light- and ethylene-mediated developmental processes and drought stress tolerance. Molecular Plant, 1, 42–57.
Liu, N., Zhong, N. Q., Wang, G. L., Li, L. J., Liu, X. L., He, Y. K., & Xia, G. X. (2007). Cloning and functional characterization of PpDBF1 gene encoding a DRE-binding transcription factor from Physcomitrella patens. Planta, 226, 827–838.
Qin, F., Kakimoto, M., Sakuma, Y., Maruyama, K., Osakabe, Y., Tran, L. S. P., et al. (2007). Regulation and functional analysis of ZmDREB2A in response to drought and heat stresses in Zea mays L. Plant J, 50, 54–69.
Wang, X. M., Chen, X. F., Liu, Y., Gao, H. W., Wang, Z., & Sun, G. Z. (2011). CkDREB gene in Caragana korshinskii is involved in the regulation of stress response to multiple abiotic stresses as an AP2/EREBP transcription factor. Molecular Biology Reports, 38, 2801–2811.
Yamaguchi-Shinozaki, K., & Shinozaki, K. (1993). Characterization of the expression of a desiccation-responsive rd29 gene of Arabidopsis thaliana and analysis of its promoter in transgenic plants. Molecular & General Genetics: MGG, 236, 331–340.
Zhang, G. Y., Chen, M., Chen, X. P., Xu, Z. S., Guan, S., Li, L. C., et al. (2008). Phylogeny, gene structures, and expression patterns of the ERF gene family in soybean (Glycine max L.). Journal of Experimental Botany, 59, 4095–4107.
Kim, Y. H., Jeong, J. C., Park, S., Lee, H. S., & Kwak, S. S. (2012). Molecular characterization of two ethylene response factor genes in sweetpotato that respond to stress and activate the expression of defense genes in tobacco leaves. Journal of Plant Physiology, 169, 1112–1120.
Rae, L., Lao, N. T., & Kavanagh, T. A. (2011). Regulation of multiple aquaporin genes in Arabidopsis by a pair of recently duplicated DREB transcription factors. Planta, 234, 429–444.
Sharoni, A. M., Nuruzzaman, M., Satoh, K., Shimizu, T., Kondoh, H., Sasaya, T., et al. (2011). Gene structures, classification and expression models of the AP2/EREBP transcription factor family in rice. Plant and Cell Physiology, 52, 344–360.
Zarei, A., Korbes, A. P., Younessi, P., Montiel, G., Champion, A., & Memelink, J. (2011). Two GCC boxes and AP2/ERF-domain transcription factor ORA59 in jasmonate/ethylene-mediated activation of the PDF1.2 promoter in Arabidopsis. Plant Molecular Biology, 75, 321–331.
Park, J. M., Park, C. J., Lee, S. B., Ham, B. K., Shin, R., & Paek, K. H. (2001). Overexpression of the tobacco Tsi1 gene encoding an EREBP/AP2-type transcription factor enhances resistance against pathogen attack and osmotic stress in tobacco. Plant Cell, 13, 1035–1046.
Wei, G., Pan, Y., Lei, J., & Zhu, Y. X. (2005). Molecular cloning, phylogenetic analysis, expressional profiling and in vitro studies of TINY2 from Arabidopsis thaliana. Journal of Biochemistry and Molecular Biology, 38, 440–446.
Liu, Y., Zhao, T. J., Liu, J. M., Liu, W. Q., Liu, Q., Yan, Y. B., & Zhou, H. M. (2006). The conserved Ala37 in the ERF/AP2 domain is essential for, binding with the DRE element and the GCC box. FEBS Letters, 580, 1303–1308.
Zhuang, J., Cai, B., Peng, R. H., Zhu, B., Jin, X. F., Xue, Y., et al. (2008). Genome-wide analysis of the AP2/ERF gene family in Populus trichocarpa. Biochemical and Biophysical Research Communications, 371, 468–474.
Song, X. M., Li, Y., & Hou, X. L. (2013). Genome-wide analysis of the AP2/ERF transcription factor superfamily in Chinese cabbage (Brassica rapa ssp. pekinensis). BMC Genomics, 14, 573.
Gil-Humanes, J., Piston, F., Martin, A., & Barro, F. (2009). Comparative genomic analysis and expression of the APETALA2-like genes from barley, wheat, and barley-wheat amphiploids. BMC Plant Biology, 9, 66.
Girardi, C. L., Rombaldi, C. V., Dal Cero, J., Nobile, P. M., Laurens, F., Bouzayen, M., & Quecini, V. (2013). Genome-wide analysis of the AP2/ERF superfamily in apple and transcriptional evidence of ERF involvement in scab pathogenesis. Scientia Horticulturae-Amsterdam, 151, 112–121.
Pirrello, J., Prasad, B. C. N., Zhang, W. S., Chen, K. S., Mila, I., Zouine, M., et al. (2012). Functional analysis and binding affinity of tomato ethylene response factors provide insight on the molecular bases of plant differential responses to ethylene. BMC Plant Biology, 12, 190.
Massa, A. N., Childs, K. L., Lin, H., Bryan, G. J., Giuliano, G., & Buell, C. R. (2011). The transcriptome of the reference potato genome Solanum tuberosum Group Phureja clone DM1-3 516R44. PLoS One, 6, e26801.
Murashige, T., & Skoog, F. (1962). A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiologia Plantarum, 15, 473–497.
Morel, G., & Wetmore, R. H. (1951). Fern callus tissue culture. American Journal of Botany, 38, 141–143.
Hassairi, A., Masmoudi, K., Albouy, J., Robaglia, C., Jullien, M., & Ellouz, R. (1998). Transformation of two potato cultivars ‘Spunta’ and ‘Claustar’ (Solanum tuberosum) with lettuce mosaic virus coat protein gene and heterologous immunity to potato virus Y. Plant Science, 136, 31–42.
Verwoerd, T. C., Dekker, B. M., & Hoekema, A. (1989). A small-scale procedure for the rapid isolation of plant RNAs. Nucleic Acids Research, 17, 2362.
Degenhardt, J., Al-Masri, A. N., Kurkcuoglu, S., Szankowski, I., & Gau, A. E. (2005). Characterization by suppression subtractive hybridization of transcripts that are differentially expressed in leaves of apple scab-resistant and susceptible cultivars of Malus domestica. Molecular Genetics and Genomics, 273, 326–335.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., & Kumar, S. (2011). MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739.
Voorrips, R. E. (2002). MapChart: Software for the graphical presentation of linkage maps and QTLs. The Journal of Heredity, 93, 77–78.
Saeed, A. I., Hagabati, N. K., Braisted, J. C., Liang, W., Sharov, V., Howe, E. A., et al. (2006). TM4 microarray software suite. Methods in Enzymology, 411, 134–193.
Bailey, T. L., & Elkan, C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers (Vol. 2). In Proceedings/… International Conference on Intelligent Systems for Molecular Biology, ISMB. International Conference on Intelligent Systems for Molecular Biology (pp. 28–36).
Guo, A. Y., Zhu, Q. H., Chen, X., & Luo, J. C. (2007). GSDS: A gene structure display server. Yi chuan = Hereditas/Zhongguo yi chuan xue hui bian ji, 29, 1023–1026.
Bouaziz, D., Pirrello, J., BenAmor, H., Hammami, A., Charfeddine, M., Dhieb, A., et al. (2012). Ectopic expression of dehydration responsive element binding proteins (StDREB2) confers higher tolerance to salt stress in potato. Plant Physiology and Biochemistry, 60, 98–108.
Bouaziz, D., Pirrello, J., Charfeddine, M., Hammami, A., Jbir, R., Dhieb, A., et al. (2013). Overexpression of StDREB1 transcription factor increases tolerance to salt in transgenic potato plants. Molecular Biotechnology, 54, 803–817.
Hanada, K., Zou, C., Lehti-Shiu, M. D., Shinozaki, K., & Shiu, S. H. (2008). Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiology, 148, 993–1003.
Liu, L. S., White, M. J., & MacRae, T. H. (1999). Transcription factors and their genes in higher plants—Functional domains, evolution and regulation. European Journal of Biochemistry, 262, 247–257.
Sharma, M. K., Kumar, R., Solanke, A. U., Sharma, R., Tyagi, A. K., & Sharma, A. K. (2010). Identification, phylogeny, and transcript profiling of ERF family genes during development and abiotic stress treatments in tomato. Molecular Genetics and Genomics, 284, 455–475.
Bouaziz, D. (2013). Isolement, caractérisation, étude fonctionelle de facteurs de transcription de la famille des ERF et surexpression de deux membres chez la pomme de terre. Thesis at University of Sfax.
Pandey, G. K., Grant, J. J., Cheong, Y. H., Kim, B. G., Li, L. G., & Luan, S. (2005). ABR1, an APETALA2-domain transcription factor that functions as a repressor of ABA response in Arabidopsis. Plant Physiology, 139, 1185–1193.
Gutha, L. R., & Reddy, A. R. (2008). Rice DREB1B promoter shows distinct stress-specific responses, and the overexpression of cDNA in tobacco confers improved abiotic and biotic stress tolerance. Plant Molecular Biology, 68, 533–555.
Zhu, Q., Zhang, J. T., Gao, X. S., Tong, J. H., Xiao, L. T., Li, W. B., & Zhang, H. X. (2010). The Arabidopsis AP2/ERF transcription factor RAP2.6 participates in ABA, salt and osmotic stress responses. Gene, 457, 1–12.
Fujimoto, S. Y., Ohta, M., Usui, A., Shinshi, H., & Ohme-Takagi, M. (2000). Arabidopsis ethylene-responsive element binding factors act as transcriptional activators or repressors of GCC box-mediated gene expression. Plant Cell, 12, 393–404.
Acknowledgments
This study was financed by the Tunisian Ministry of High Education and Scientific Research. The authors are grateful to Anne-Lise Haenni from Institute Jacques Monod (CNRS-France) for her kind help with the English language.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Charfeddine, M., Saïdi, M.N., Charfeddine, S. et al. Genome-Wide Analysis and Expression Profiling of the ERF Transcription Factor Family in Potato (Solanum tuberosum L.). Mol Biotechnol 57, 348–358 (2015). https://doi.org/10.1007/s12033-014-9828-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-014-9828-z