
Abstract
β-Glucosidases are of pivotal importance in bioconversion of carbonic biomass into fermentable and other useful metabolites, food industry, biotransformation, glyco-trimming of metabolome, etc. Trichoderma citrinoviride when grown on delignified Lantana camara produced a β-glucosidase and secreted it out in the medium. The extracellularly secreted β-glucosidase of T. citrinoviride was homogeneity purified and then characterized for its kinetic properties and proteomic characteristics. The 90 kDa enzyme was monomeric in nature, optimally active at pH 5.5 and the catalytic reaction rate was highest at 55°C. Uniquely, the enzyme was insensitive to inhibition by glucose (up to 5 mM). It also possessed catalytic ability of transglycosylation, as it could catalyze conversion of geraniol into its glucoside. MALDI-TOF assisted proteomic analysis revealed its high degree of sequence similarity with family 3 glycoside hydrolases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
β-Glucosidases are of pivotal importance in bioconversion of carbonic biomass into fermentable and other useful metabolites, food industry, biotransformation, glyco-trimmimg of metabolome, etc. They also have a key role/significance in (i) the release of fragrant and volatile lower terpenoids and phenylpropanoids from their odorless and non-volatile glycosides, (ii) fruit processing industries and winery, (iii) release of phenolic compounds with antioxidant and/or nutraceutical, (iv) altering the taste characteristics of fruits and vegetables, (v) cassava detoxification, etc. In the bio-energy production, they are helpful in processing ligno-cellulosic biomass by releasing phenolic compounds, to facilitate improved action of cellulose degrading enzymes. Particularly, β-glucosidases of bacterial and fungal origin are functional components of cellulase enzyme system and catalyze hydrolysis of short chain oligosaccharides and cellobiose (resulting from synergistic action of endoglucanases and cellobiohydrolases) into glucose. Therefore, these enzymes are being exploited in several biotechnological applications [1] like improvement of enzymatic saccharification of cellulosics, amelioration of cellobiose mediated inhibition of endoglucanase and exoglucanase process.
Large amounts of agro-industrial residues that are generated annually invoke exploration and recruitment of biotechnological means of their utilization as sources of energy and other useful materials [1]. β-Glucosidase is usually integral part of these processes. Production of β-glucosidase is still uneconomic as pure cellulose is used as substrate for their production in submerged fermentation conditions. Therefore, we have recently identified Trichoderma citrinoviride as a very useful fungus for processing agro-wastes into fermentable substrates [2, 3]. In this investigation, we noted that T. citrinoviride grown on delignified Lantana camara under submerged fermentation conditions produced a β-glucosidase that was secreted into the medium and we have studied its catalytic and proteomic properties some of which are novel.
Results and Discussion
We have studied a β-glucosidase that was produced and secreted extracellularly by T. citrinoviride, when grown under submerged fermentation on delignified L. camara as carbon source. The enzyme was harvested, purified and biochemically characterized in detail including its proteomics. β-Glucosidase activity in the crude culture filtrate (referred to as crude enzyme preparation) was about 1.1 IU/ml.
Homogeneity Purification
Differential (NH4)2SO4 precipitation (40–75% saturation) of the crude enzyme preparation resulted in 82% recovery of the enzyme and 2.7-fold enrichment of its specific activity (Table 1). The subsequent step of gel filtration (Sephadex G-100) gave a single peak of β-glucosidase activity (Fig. 1) and the enzyme preparation (61% recovery) had 4.3-fold higher specific activity. The subsequent ion exchange chromatography step (Q-Sepharose) resulted into 28.7% recovery of the overall activity but with only little higher (4.7-fold) specific activity. However, subsequent purification step with hydrophobic interaction chromatography (elution of the enzyme activity at 0.5 M (NH4)2SO4) increased the degree of purity by 12-fold with 16% recovery. Chromatography of the enzyme preparation through a Sephacryl HR-200 gel filtration column also gave a limited gain in purification level (13.5-fold) with only 3.5% of the crude enzyme activity in hand. Finally, when protein preparation was applied to an affinity chromatography (Fig. 1) resin (concanavalin A–agarose eluted with substrate analog, methyl α-d-glucopyranoside), homogeneity purification (>54-fold, 167.1 IU mg/protein with pNPG as substrate at 55°C and pH 4.8) was achieved (Table 1; Fig. 2).

Purification of β-glucosidase of Trichoderma citrinoviride through different chromatography columns. PB phosphate buffer; enzyme activity, IU/ml

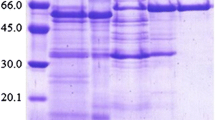
SDS-PAGE of purified β-glucosidase from Trichoderma citrinoviride. The enzyme was electrophoresed through 12% acrylamide gel and stained with Commassie Brilliant Blue R-250 (a) as well as silver stain (b). Lane M molecular weight markers; lane BG purified β-glucosidase (BG); red arrow BG polypeptide (Color figure online)
Molecular Mass
The native molecular mass of the T. citrinoviride β glucosidase as measured by gel filtration (Sephacryl HR 200) was observed to be almost the same (~94 kDa) as the one of the single polypeptide (90 kDa) observed on SDS-PAGE (Fig. 2) in the final enzyme preparation. Therefore, the enzyme was monomeric in nature. In terms of high molecular mass, the novel enzyme is similar to many extracellularly secreted β-glucosidases from other fungi [4]. Molecular mass and composition of β-glucosidases from aerobic fungi vary widely: native molecular mass up to 400 kDa with monomeric to homo-multimeric composition [5, 6], e.g., an octamer like 380 kDa β-glucosidase from Botryodiplodia theobromae [7]. Probably, it is reasonable to believe that ~90 kDa is a standard catalytically active form that may be multi-merized for in vivo functional efficiency and/or substrate specificity. Extracellular secreted enzyme may be preferably remain monomeric for maintaining catalytic activity in the variable extracellular medium that may not be as conducive to ordered multimerization as the intracellular environment.
Kinetic Properties
Optimum pH and Temperature
The pH optimum of the enzyme activity was 4.8 (100 mM sodium acetate buffer) with half-maximal velocities at pH 3.5 and 6.0 (Fig. 3). This pH of optimal activity is slightly acidic in comparison to the pH optimum values known (5.0–6.5) for the most of fungal β-glucosidases [8] with sodium phosphate as buffer. The pH stability of the enzyme, discerned from measurements of residual activity after 24 h of incubation at different pH, was better in the pH range from 4.0 to 5.5. Narrow and acidic pH range for optimal activity appears to be a common characteristic of several microbial β-glucosidases.

Effect of pH on catalytic activity (IU) of purified β-glucosidase of Trichoderma citrinoviride. Values written near the points represent respective pH values at which catalytic activity was measured
Thermostability of the enzyme (measured in the range from 20 to 80°C) revealed the enzyme to be substantially stable (for 8 h) up to 50°C (0.1 M borate buffer, pH 6.5). This feature is better than thermostability of many other microbial β-glucosidases like H. grisea, S. thermophilum [9] where stability at 50°C has been reported to be much shorter (60 min) with Mc Ilavaine (sodium phosphate-citrate) as assay buffer and Achatina fulica β-glucosidase with sodium phosphate–citrate as the buffer. Higher temperature (≥60°C) incubations, even for short durations, were detrimental for the catalytic stability of the enzyme: 4, 45, and 60% loss of activity on incubation at 60°C, respectively, for 10, 20, and 30 min; 60 and 100% loss of activity on incubation at 70°C for 5 and 10 min, respectively. These data categorize the enzyme as mesostable. The enzyme in solution (0.1 M sodium borate buffer pH 6.5) remained active for more than a year at 4°C. The mechanism of thermostability and pH stability are closely related to the structure and amino acid composition of an enzyme. Replacement of amino acid residues of the C-terminal domain is known to significantly affect the heat stability and such residues are distributed over the entire region of the C-terminal domain [10].
The optimum temperature for catalytic activity of T. citrinoviride β-glucosidase was 55°C (Fig. 4, inset), with 100 mM acetate buffer, pH 4.8 as the assay buffer, similar to many β-glucosidases from other fungi. The temperature versus activity data when used to generate Arrhenius plot, gave a single straight line Arrhenius plot for the enzyme catalyzed reaction (Fig. 4) that is typical of the non-membranous enzymes. The plot gave 5.10 kcal/mol as an estimate of activation energy for the enzyme. This was almost equal to that reported (5.08 kcal/mol) for cassava cortex β-glucosidase and half of that (9.5 kcal/mol) known for the enzyme from Alcaligenes faecalis [11].

Arrhenius plot (inverse of absolute (kelvin, K) temperature (T) versus natural log of rate of catalytic reaction (ln k) catalyzed by purified β-glucosidase of Trichoderma citrinoviride. Inset Temperature versus catalytic activity of the enzyme
Substrate Saturation Kinetics
The substrate curve of the enzyme for β-pNPG was normal hyperbolic (Fig. 5), typical of Michaelis–Menten kinetics of reaction catalysis. Lineweaver–Burk plot (supplementary Fig. 1) of the enzyme revealed K m and V max of the enzyme for β-pNPG as 0.27 mM and 200 μmol/min/mg, respectively. These data show similarity with β-glucosidase from sources like Neocallimastix frontalis (K m 0.26 mM) and Aspergillus oryzae (K m 0.29 mM) [12] but much lower than the enzyme from Piromyces sp. (K m 1.5 mM) [13]. K m values of β-glucosidases from aerobic fungi [14] are generally much higher (0.75–3.6 mM) than those from anaerobic fungi. However, enzyme catalytic constant or turn over number (K cat) and catalytic efficiency (K m/K cat) are the true kinetic parameters for comparison and ranking of catalytic performance of enzymes. Unfortunately, these parameters have been assessed for very few of the β-glucosidases reported from diverse organisms (Table 2). T. citrinovirdie β-glycosidase enzyme had a K cat of 8,953/s, far higher than that observed for its counterparts from other sources like 370/s for A. oryzae [15]. The enzyme showed also better catalytic efficiency (3.19 × 107/M/s) than other β-glucosidases [16–26] reported so far (Table 2).

Substrate–saturation curve of the purified Trichoderma citrinoviride β-glucosidase with β-paranitrophenyl glucose (β-pNPG) as substrate
Substrate Specificity
The enzyme substrate specificity was tested using substrates with different types (α or β) of sugar linkage as well as those with alternate position of the substituent group (–NO2) in the aglycone moiety of the molecule, e.g., p-nitrophenyl-β-d-glucopyranoside, (β-pNPGlu), p-nitrophenyl-β-d-galactopyranoside (β-pNPGal), p-nitrophenyl-β-d-mannopyranoside (β-pNPMan), p-nitrophenyl-β-d-fucopyranoside (β-pNPFuc), p-nitrophenyl-β-d-lactopyranoside (pNPLac), p-nitrophenyl-α-d-glucopyranoside (α-pNPGlu), p-nitrophenyl-α-d-galactopyranoside (α-pNPGlu) o-nitrophenyl-β-d-glucopyranoside (β-oNPGlu), and o-nitrophenyl-β-d-galactopyranoside (β-oNPGal). Interestingly, the enzyme showed significant deglycosylating activity only with β-pNPGlu implying that it had absolute substrate specificity for β-linkage and with glucose as the conjugated sugar. In general, fungal β-glucosidases are particularly specific for β-pNPGlu [22, 27]. Mechanistically, β-glucosidases catalyze hydrolysis of their substrates, retaining anomeric configuration, by double-displacement mechanism with intermediacy of oxocarbenium ion-like transition state involving an acid/base (proton donor) system comprised of two (a catalytic and a nucleophilic) glutamate residues housed in the active site located at C-terminal end of β-strands 4 and 7 [28].
Effect of Metal Ions
Trichoderma citrinoviride β-glucosidase activity was significantly inhibited by divalent cations like Ag2+, Hg2+, Cu2+, Li2+ and Mn2+ at 5 mM concentration. Cd2+, however, exhibited substantial increase in activity (Supplementary Fig. 2). Surprisingly, a multifold (>5) increase in the catalytic rate was noticed in presence of 1–2 mM of Fe2+, while Fe3+ was relatively less (threefold) effective as an activator (Supplementary Table 1).
Inhibition by Sugars
Glucose, mannose and sucrose caused, respectively, 23, 26 and 19% inhibition of the enzyme activity at 5 mM concentration. The inhibition by glucose was considerably less at lower concentrations (Fig. 6). Similarly, cellobiose had no inhibitory effect up to 4 mM and caused low inhibition even at higher concentrations (20 and 40% at 5 and 10 mM, respectively) (Fig. 6). However, gluconolactone completely inhibited the enzyme activity at 5 mM concentration (Supplementary Fig. 3). Inhibition of β-glucosidases by glucose and cellobiose is a commonly observed feature of β-glucosidases, and it is considered a major obstacle in glucose production from cellulose by the cellulase–(β-glucosidase) system [1, 13]. Therefore, it forms an important parameter for selection of the appropriate β-glucosidase for this application. Thus, T. citrinoviride enzyme joins a few of the promising β-glucosidase enzymes insensitive to inhibition by sugars [29]. This suggests that the enzyme is potentially useful for biotechnological use.

Effect of different concentrations (mM) of glucose and cellobiose on catalytic activity of purified β-glucosidase of Trichoderma citrinoviride, relative to control (100%, in absence of sugar—glucose or cellobiose)
Effect of Alcohols
The effect of alcohols on β-glucosidase activity was studied using pNPG as substrate (Supplementary Fig. 4). Increase of activity was observed with several alcohols viz. ethanol, methanol propanol, butanol, pentanol, hexanol, octanol, geraniol, and linalool increased the activity of β-glucosidase from 1.2- to 1.4-fold similar to the effect of ethanol on other glucosidases including the ones from fungi [30]. This can possibly be considered as a manifestation of glucosyltransferase (transglycosylation) activity of enzyme whereby alcohol may be used, in addition to water, as an acceptor for glucosyl moiety from pNPG contributing to higher rate of pNP formation being monitored in the typical glucosidase assay. However, glycosyltransferase activity of T. citrinoviride β-glucosidases was noted with only geraniol in the tranglycosylation detection and not with other alcohols. Should it occur, a phenomenon of direct activation of glucohydrolase activity by ethanol could be advantageous by facilitating single pot saccharification and fermentation of cellulose into ethanol. However, it is a moot point if alcohol effect represents activation or it is a manifestation consequent to contribution of the glycosyltransferase activity.
Transglycosylation
Transglucosylation of geraniol by T. citriniviride β-glucosidase was observed with pNPG as donor (Fig. 7), but not of other alcohols viz., methanol, ethanol, propanol, butanol, pentanol, hexanol, heptanol, octanol and linalool (acyclic primary alcohols of chain length from C1 to C10 except linalool which is a C10 acyclic tertiary alcohol). Probably shorter chain length or non-primary nature of acceptor hydroxyl group could be restrictive of their transglycosylation. Cellobiose could not serve as the glucosyl donor in the geraniol transglucosylation by T. citrinoviride β-glucosidase. The glucosyltansferase activity of β-glucosidases holds potential of exploitation in, otherwise difficult, synthesis of higher chain alkyl glucosides. Synthesis of alkyl glucosides having up to C8 (i.e., octylglucosides) has been reported, primarily using almond enzyme in reverse hydrolysis mode [31]. Thus, the synthesis of further higher alkyl (geranyl, C10) observed herein is distinct and complementary to almond like β-glucosidases in this domain of progress.

Transglucosylation of geraniol by β-glucosidase from Trichoderma citrinoviride. Lanes: 1 geraniol; 2 linalool, 3 catalytic reaction with geraniol and pNPG as substrates, 4 catalytic reaction with linalool and pNPG as substrates, 5 catalytic reaction with geraniol and cellobiose as substrates, 6 catalytic reaction with linalool and cellobiose as substrates, 7, 8 and 9 are markers, pNPG, p-nitrophenol and cellobiose, respectively
Proteomic Characterization
The de novo fragment sequences of purified β-glucosidase from T. citrinoviride were compared with other β-glucosidase (family 1 and family 3 glycosylhydrolases) of bacterial or fungal origin using BLAST (Basic Local Alignments Search Tool) service. According to pBLAST results, 11 β-glucosidases from different microbes were selected (considering their e-value and matching score) for comparative sequence alignment using ClustalX 1.81. Thereafter, sequences were manually aligned identifying regions of complete identity (‘*’), semi-conservative change (‘:’), and partially conserved change (‘.’) as presented in Fig. 8. The amino acid residues which were identical with those of T. citrinoviride β-glucosidase have been written in white on black background.

Alignment of the amino acid sequences of Trichoderma citrinoviride β-glucosidase with the amino acid sequences of other microbial β-glucosidases. Amino acids residues are indicated by single letter codes. The regions of complete identity, semi-conservative change and partially conserved change are indicated by ‘*’, ‘:’ and ‘.’, respectively. The amino acid residues which are identical with those of T. citrinoviride β-glucosidase are written in white on black background and those of semi-conservative changes are shown in gray background (Cm, Cmo bgl1; Tr, Trichoderma reesei bgl1; S1, Saccharomycopsis fibuligera bgl1; S2, S. fibuligera bgl2; Cp, Candida pelliculosa bgl1; At, Agrobacterium tumifaciens bgl; Ct, Clostridium thermocellum bgl; Cg, Cellvibrio gilvus; Kf, Kluyveromyces fragilis bgl; Ak, Aspergillus kawachi bgl; Aa, A. aculeantus bgl; Tc, Trichoderma citrinoviride
The comparison indicated very low sequence similarity with family 1 β-glucosidases and relatively higher similarity with family 3 β-glucosidases. Conserved motifs were recognized in the enzyme, DPXL and AKH(L/F)X9R and LX7G in the domain 1, and Xa(G/D/H/A)Xb(Y/F)XXXEEK and GXGL of domain 2, and they were found as signature amino acid landscape to classify as a member of family 3 glycohydrolases. These motifs covered conserved residues of a putative carbohydrate-binding site (XXKHXX), sequence with characteristic N-terminal amino acid landscape of catalytic motif (GXXXXD) and key amino acid residues (GXGL) of the antiparallel/loop (Fig. 8). In the structural model [32], the domain 1 that in a typical glycohydrolase family 3 comprises of a (α/β)8 TIM barrel fold, could be assigned DPXL and AKH(L/F)X9R, and LX7G could be localized to α-helix ‘C2’, junction of β-strand ‘e’ and coil connecting to α-helix ‘E1’ (covering glycosylation site) and α-helix chain ‘F’ and through connecting coil to β-strand ‘g’ (including catalytic site with nucleophile D). The glycine residue in the motif LX7G represents part of the catalytic motif GXXXXD. The motifs Xa(G/D/H/A)Xb(Y/F)XXXEEK and GXGL could be localized in the domain 2 [a (α/β)6 sandwich] at start of the last coil and within the terminal antiparallel loop, respectively. These structural traits of domain 2 indicate ability of the enzyme to attach to insoluble substrates or cell walls, similar to several hydrolytically degradative enzymes. Together with the observed extracellular secretion of the enzyme, this structural feature of domain 2 implies its essential participation in the enzymatic degradation of cell walls as a part of ‘cellulase system’ that could also be symbiotic to the plant in order to augment nutrient acquisition by controlled cell wall loosening in the rhizosphere.
Methods
Chemicals
All biochemicals and reagents unless stated otherwise were purchased from Sigma-Aldrich (USA). Molecular weight markers, chemicals and reagents for running sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were obtained from Bio-Rad Laboratories. Chromatographic media were from GE Healthcare (General Electric, USA) and Sigma-Aldrich (USA). For MALDI analysis chemicals were purchased from Promega, Madison (USA).
Microorganism
Trichoderma citrinoviridae (MTCC No. 2418) was obtained from Microbial Type Culture Collection Center, IMTECH (CSIR) Chandigarh, India and was maintained on potato dextrose agar at 28°C.
Fungal Cultivation for Enzyme Production
An inoculum of 5 × 108 spores/ml was prepared by harvesting 1-week-old culture of T. citrinoviride, and 3 ml of the spore suspension was used as inoculum for 50 mL mineral salt medium [33] in 250-ml Erlenmeyer flask, together with 1% of delignified L. camara. The flasks were incubated at 28°C on rotatory shaker (180 rpm). After 96 h of incubation fungal biomass and media were harvested by filtration and the media was collected.
Isolation and Purification of Enzyme
All steps of enzyme extraction and purification were carried out at 4°C, unless stated otherwise.
Enzyme isolation: The culture supernatant (800 ml) was filtered through glass wool fiber and was used as the crude enzyme source.
Purification: The culture supernatant was subjected to differential (NH4)2SO4 (40–75% saturation) precipitation and the protein precipitated between 40 and 75% saturation was collected by centrifugation at 10,000×g for 20 min in a refrigerated centrifuge (Sarvall RC 5B) and dissolved in 2.0 ml of 50 mM acetate buffer (pH 4.8). The (NH4)2SO4 fraction was loaded on Sephadex G-100 column (1.6 × 91 cm; void volume 50 ml, resin bed volume 183 ml, flux 6 cm/h), pre-equilibrated with acetate buffer (25 mM, pH 4.8). The enzyme was eluted into the same buffer and the fractions were collected and monitored for protein content (A280) as well as for β-glucosidase activity. Active fractions were pooled and precipitated with (NH4)2SO4 (75% saturation). The precipitates were collected by centrifugation at 10,000×g and dissolved in 1.0 ml of acetate buffer (25 mM, pH 4.8), desalted through G-25 column (2.2 × 20 cm) and purified through an anion exchange (Q-Sepharose) resin (bed dimensions 2.0 × 7.0 cm, 22 ml, flux 11 cm/h), pre-equilibrated with acetate buffer (25 mM, pH 4.8) and eluted with using a linear gradient of NaCl (0.1–1.0 M). Fractions (2.0 ml) were assayed for the enzyme activity and protein content (A280). Active fractions were pooled and subjected to hydrophobic interaction chromatography (HIC) through an Octyl Sepharose (CL-4B) column (resin bed 1.5 × 7.0 cm, bed volume 12.5 ml, flux 17 cm/h), pre-equilibrated with acetate buffer (25 mM pH 4.8), containing 1.7 M (NH4)2SO4. The enzyme was eluted with a decreasing gradient (1.7 M to 0.5 M) of (NH4)2SO4. The active fractions were pooled, precipitated with (NH4)2SO4 (75% saturation), the precipitates were dissolved in acetate buffer and loaded on a Sephacryl HR 200 gel filtration column (bed dimensions 1.1 × 45 cm, void volume 20 ml, bed volume 43 ml, flow rate 25 cm/h) pre-equilibrated with acetate buffer (25 mM pH 4.8). Active fractions were pooled and loaded on to an affinity chromatography resin functionalized with concanavalin A (resin bed dimensions 0.9 × 7.0 cm, bed volume 4.5 ml, flow rate 16 cm/h), equilibrated with borate buffer (0.1 M, pH 6.5). The protein was sequentially eluted with borate buffer containing 0.2, 0.3 and 0.4 mM of methyl-α-d-glucopyranoside. The active fractions were pooled and the purified enzyme preparation was analyzed for catalytic enrichment, degree of purity (SDS-PAGE), and for physical-kinetic characteristics and proteomic features.
Enzyme Assay
A priori, assays were optimized for linearity with time and protein concentration. β-Glucosidase was assayed by using a standard reaction mixture (1 ml) containing 5 mM p-nitrophenyl-β-d-glucopyranoside (pNPG), 100 mM acetate buffer (pH 4.8), and appropriately diluted enzyme solution. After incubation at 50°C for 10 min, the reaction was stopped by the addition of 1 M Na2CO3, and the color, developed as a result of p-nitrophenol liberation, was measured at 405 nm in a UV–Visible spectrophotometer. One unit (IU) of β-glucosidase was defined as the amount of enzyme able to release 1 μmol of p-nitrophenol/min in the reaction mixture under the assay condition.
For kinetic characterization assays, the standard assay system was varied as per experimental demand of the analyses.
Transglycosylation
For assaying transglycosylation activity of the enzyme, 1.5 ml reaction was set up with 50 mM primary alcohols (methanol, ethanol, propanol, butanol, pentanol, hexanol, heptanol, octanol, geraniol, linalool) as acceptors, 40 mM cellobiose or 18 mM pNPG as donor, 30% (v/v) dimethyl sulfoxide (DMSO) and 40 μl β-glucosidase in 50 mM acetate buffer (pH 4.8). DMSO was added to create monophasic environment with added alcohols. The reaction mixtures were incubated for 18 h at 30°C. At the end of the reaction, the mixture was extracted with ethyl acetate and the ethyl acetate extract was dried and re-dissolved in minimum volume of ethyl acetate followed by TLC (pre-coated, silica gel) analysis using a mixture of chloroform, ethyl acetate and methanol (70:6:10) as solvent system. The chromatogram was developed using vanillin–sulfuric acid reagent spray followed by heating at 110°C for 10 min. Glycosides were discerned as dark spot of higher polarity (lower R f) than the counterpart substrate (aglycone) substrate.
Denaturing Polyacrylamide Gel Electrophoresis (SDS-PAGE)
Protein quantification was carried out by Lowry’s method using BSA as standard. SDS-PAGE was run in a Mini-PROTEAN-II gel electrophoresis system (Bio-Rad) using standard protocol. The polypeptide pattern in gel lanes was visualized by Coomassie brilliant blue R-250 and silver staining.
Native Molecular Mass Estimation
The molecular weight of the native enzyme was determined by using a calibrated gel filtration on Sephacryl HR-200 column (bed dimensions 1.1 × 45 cm, void volume 20 ml, bed volume 43 ml, flow rate 24 ml/h) using alcohol dehydrogenase (150,000), bovine serum albumin (66,000), and ovalbumin (45,000) as standards.
Mass Spectrometry (MALDI-TOF) Analysis
The in-gel digestion of the separated band of protein (enzyme) on SDS-PAGE and purification of peptides was carried out according to the manufacture’s manual. Briefly, Coomassie Brilliant Blue-stained protein band was excised, sequentially washed with distilled water and 50% (v/v) acetonitrile (ACN) in 25 mM ammonium bicarbonate (pH 8.0), incubated with 5 mM DTE, subjected to S-alkylation by 10 mM iodoacetamide and dehydrated in ACN by vacuum drying. The gel piece was incubated with added 15–20 μl of 10 μg/ml of sequencing grade trypsin (promega, Madison, WI USA) for 15 min followed by the addition of 25 μl of 50 mM ammonium carbonate and allowed to stand overnight at 37°C. The peptide fragments were extracted in 50% ACN/0.3% TFA solution in distilled water by incubating for 15 min and followed by collection of supernatant after centrifugation. The supernatant was dried under vacuum and the residue dissolved in 10 μl of 0.1% TFA. The peptides were purified with C18 RP minicolumn (Millipore, Bedford, MA, USA) before mass spectrometric analysis. The peptide solution was mixed with 2 volumes of α-cyano-4-hydroxycinnamic acid (ABI, Farmingham, USA) of 10 mg/ml in 50% ACN/0.1% TFA and spotted onto a MALDI sample plate.
MS and MS/MS spectra were acquired in the positive ion mode on MALDI-TOF/TOF Mass Spectrometer, Applied Biosystems 4700 Proteomics Analyzer (Framingham, MA, USA). The instrument was operated in the delayed extraction mode with delay of 200 ns. Spectra were obtained by accumulation of 1,000 and 2,000 consecutive laser shots, respectively, in MS and MS/MS mode and laser intensity used were in the range of 3,000–4,000. External mass calibration for MS was performed with 4700 Cal Mix (Applied Biosystems, USA) a standard mixture of six marker peptides. Mass calibration for MS/MS spectra was performed by fragment masses of precursor Glu1-Fibrinopeptide B (1570.6774). Peak harvesting was carried out using 4000 series explorer software (Applied Biosystems). MS/MS spectra were acquired from all major peptide peaks of MS spectrum excluding known tryptic autodigest peaks.
Database search for protein identifications was performed with mass spectrometry data using Global Proteome Server v3.5 software (Applied Biosystems) equipped with MASCOT (Matrix Science) search engine. The database query was made for microbial species against Swiss-Prot and NCBI nr databases using only monoisotopic masses. MS-blast search was performed for sequence homology-based protein identity. As and when required, putative fragment sequences were also searched for conserved and semi-conserved motifs manually. The recognized fragment sequences were compared with amino acid sequences of other glycosidases to discern the homology and assign the glycohydrolase family to the query enzyme.
References
Singhania, R. R., Sukumaran, R. K., Patel, A. K., Larroche, C., & Pandey, A. (2010). Advancement and comparative profiles in the production technologies using solid-state and submerged fermentation for microbial cellulases. Enzyme and Microbial Technology, 46, 541–549.
Chandra, M., Kalra, A., Sharma, P. K., Kumar, H., & Sangwan, R. S. (2010). Optimization of cellulases production by Trichoderma citrinoviride on marc of Artemisia annua and its application for bioconversion process. Biomass and Bioenergy, 34, 805–811.
Chandra, M., Kalra, A., Sharma, P. K., & Sangwan, R. S. (2009). Cellulase production by six Trichoderma spp. fermented on medicinal plant’s processings. Journal of Industrial Microbiology and Biotechnology, 36, 605–667.
Perez-Pons, J. A., Rebordosa, X., & Querol, E. (1995). Properties of a novel glucose-enhanced β-glucosidase purified from Streptomyces sp. (ATCC 11238). Biochimica Biophysica Acta, 1251, 145–153.
Wood, M., McCray, S. I., Wilson, C. A., Bhat, K. M., & Grow, L. A. (1998). Aerobic and anaerobic fungal cellulase, with special reference to their mode of attack on crystalline cellulose. In J. P. Aubert, P. Beguin, & J. Millet (Eds.), Biochemistry and genetics of cellulose degradation. London: Academic Press.
Kohchi, C., Hayashi, M., & Nagai, S. (1985). Purification and properties of β-glucosidase from Candida pelliculosa var. acetaetherius. Agricultural Biology and Chemistry, 49, 779–784.
Umezurike, G. M. (1991). The octameric structure of β-glucosidase from Botryodiplodia theobromae Pat. Biochemical Journal, 275, 721–725.
Peralta, R. M., Kadowaki, M. K., Terenzi, H. F., & Jorge, J. A. (1997). A highly thermostable β-glucosidase activity from the thermophilic fungus Humicola grisea var. thermoidea: Purification and biochemical characterization. FEMS Microbiology Letters, 146, 291–295.
Fabiana, F. Z., de Maria, L., de Teixeira, M. P., Hector, F. T., & Joao, A. J. (2004). β-Glucosidase activity from the thermophilic fungus Scytalidium thermophilum is stimulated by glucose and xylose. FEMS Microbiology Letters, 240, 137–143.
Hu, Y., Luan, H., Hao, D., Xiao, H., Yang, S., & Yang, L. (2007). Purification and characterization of novel ginsenoside-hydrolyzing β-glucosidase from China white jade snail (Achatina fulica). Enzyme and Microbial Technology, 40, 1358–1366.
Yeoh, H. H., & Wee, Y. C. (1989). Kinetic properties of β-glucosidase from cassava. Phytochemistry, 28, 721–724.
Langston, J., Sheehy, N., & Xu, F. (2006). Substrate specificity of Aspergillus oryzae family 3 β-glucosidase. Biochimica et Biophysica Acta, 1764, 972–978.
Teunissen, M. J., Lahayel, D. H. T. P., Veld, H. V., & Vogels, G. D. (1992). Purification and characterization of an extracellular β-glucosidase from the anaerobic fungus Pyromyces sp. strain E2. Archive of Microbiology, 158, 276–281.
Chen, H., Hayn, M., & Esterbauer, H. (1992). Purification and characterization of two extracellular β-glucosidases from Trichoderma reesei. Biochimca et Biophysica Acta, 1121, 54–60.
Langston, J., Sheehy, N., & Xu, F. (2006). Substrate specificity of Aspergillus oryzae family 3 β-glucosidase. Biochimica et Biophysica Acta, 1764, 972–978.
Seidle, H. F., McKenzie, K., Marten, I., Shoseyov, O., & Huber, R. E. (2005). Trp-262 is a key residue for the hydrolytic and transglucosidic reactivity of the Aspergillus niger family 3 β-glucosidase: Substitution results in enzymes with mainly transglucosidic activity. Archives of Biochemistry and Biophysics, 444, 66–75.
Yu, H. L., Xu, J. H., Lu, W. Y., & Lin, G. Q. (2007). Identification, purification and characterization of β-glucosidase from apple seed as a novel catalyst for synthesis of O-glucosides. Enzyme and Microbial Technology, 40, 354–361.
Romeu, A., Montero, M. A., Ruiz, J. A., De Maria, M. J., Jimenez, J. A., Rojas, A., et al. (1994). Effects of glucose, ethanol Hg(II) and Cu (II) on almond β-glucosidase. Biochemistry and Molecular Biology International, 33, 939–946.
Saibi, W., Amouri, B., & Gargouri, A. (2007). Purification and biochemical characterization of a transglucosilating β-glucosidase of Stachybotrys strain. Applied Microbiology and Biotechnology, 77, 293–300.
Amouri, B., & Gargouri, A. (2006). Characterization of a novel β-glucosidase from a Stachybotrys strain. Biochemical Engineering Journal, 32, 191–197.
Zhang, C., Li, D., Yu, H., Zhang, B., & Jin, F. (2007). Purification and characterization of piceid-β-d-glucosidase from Aspergillus oryzae. Process Biochemistry, 42, 83–88.
Karnchanatat, A., Petsom, A., Sanvanich, P., Piaphukiew, J., Whalley, A. J., Reynolds, C. D., et al. (2007). Purification and biochemical characterization of an extracellular β-glucosidase from the wood-decaying fungus Daldinia eschscholzii (Ehrenb.:Fr.)Rehm. FEMS Microbiology Letters, 270, 162–170.
Lau, A. T. Y., & Wong, W. K. R. (2001). Purification and characterization of a major secretory cellobiase, Cba2, from Cellulomonas biazotea. Protein Expression and Purification, 23, 159–166.
Pardo, A. G., & Forchiassin, F. (1999). Influence of temperature and pH on cellulase activity and stability in Nectria catalinensis. Revista Argentina de Microbiología, 31, 3–15.
Zanoelo, F. F., Polizeli Md Mde, L., Terenzi, H. F., & Jorge, J. A. (2004). β-Glucosidase activity from the thermophilic fungus Scytalidium thermophilum is stimulated by glucose and xylose. FEMS Microbiology Letters, 240, 137–143.
Venturi, L. L., Tereni, H. F., Furriel, R. P. M., & Jorge, J. A. (2002). Extracellular β-d-glucosidase from Chaetomium thermophilum var. coprophilum: Production, purification and some biochemical properties. Journal of Basic Microbiology, 42, 55–56.
Nakkharat, P., & Haltrich, D. (2006). Purification and characterisation of an intracellular enzyme with β-glucosidase and β-galactosidase activity from the thermophilic fungus Talaromyces thermophilus CBS 236.58. Journal of Biotechnology, 123, 304–313.
Isorna, P., Polaina, J., Latorre-Garcia, L., Canada, F. J., Gonzalez, B., & Sanz-Aparicio, J. (2007). Crystal structures of Paenibacillus polymyxa β-glucosidase B-complexes reveal the molecular basis of substrate specificity and give new insights into the catalytic machinery of family I glycosidases. Journal of Molecular Biology, 371, 1204–1218.
Ike, M., Park, J., Tabuse, M., & Tokuyasu, K. (2010). Cellulase production on glucose-based media by the UV-irradiated mutants of Trichoderma reesei. Applied Microbiology Biotechnology, 87, 2059–2066.
Christakopoulos, P., Goodenough, P. W., Kekos, D., Macris, B. J., Claeyssens, M., & Bhat, M. K. (1994). Purification and characterisation of an extracellular β-glucosidase with transglycosylation and exo-glucosidase activities from Fusarium oxysporum. European Journal of Biochemistry, 224, 379–385.
Bhatia, Y., Mishra, S., & Bisaria, V. S. (2002). Microbial β-glucosidases: Cloning, properties, and applications. CRC Critical Reviews in Biotechnology, 22, 375–407.
Harvey, J. H., Srinivasan, V. R., Hrmova, M., Varghese, J. N., & Fincher, G. B. (2000). Comparative modeling of the three-dimensional structures of family 3 glycoside hydrolases. Proteins: Structure, Function, and Bioinformatics, 41, 257–269.
Mandels, M., & Weber, J. (1969). Production of cellulase. Advances in Chemistry Series, 95, 391–414.
Acknowledgments
Authors are thankful to the Director CIMAP, Lucknow, for the facilities and encouragement. The financial grant provided by CSIR under NWP-09 network program is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Chandra, M., Kalra, A., Sangwan, N.S. et al. Biochemical and Proteomic Characterization of a Novel Extracellular β-Glucosidase from Trichoderma citrinoviride . Mol Biotechnol 53, 289–299 (2013). https://doi.org/10.1007/s12033-012-9526-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-012-9526-7