
Abstract
Functional expression of lipase from Burkholderia sp. C20 (Lip) in various cellular compartments of Escherichia coli was explored. The poor expression in the cytoplasm of E. coli was improved by several strategies, including coexpression of the cytoplasmic chaperone GroEL/ES, using a mutant E. coli host strain with an oxidative cytoplasm, and protein fusion technology. Fusing Lip with the N-terminal peptide tags of T7PK, DsbA, and DsbC was effective in enhancing the solubility and biological activity. Non-fused Lip or Lip fusions heterologously expressed in the periplasm of E. coli formed insoluble aggregates with a minimum activity. Biologically active and intact Lip was obtained upon the secretion into the extracellular medium using the native signal peptide and the expression performance was further improved by coexpression of the periplasmic chaperon Skp. The extracellular expression was even more effective when Lip was secreted as a Lip–HlyA fusion via the α-hemolysin transporter. Finally, Lip could be functionally displayed on the E. coli cell surface when fused with the carrier EstA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipases are an important biocatalyst for industrial applications [1]. Microbial cells including bacteria, fungi, and yeasts are the major sources of lipases with various enzymatic activities (e.g., hydrolysis, esterification, transesterification, alcoholysis, acidolysis, and chemo, regio, enantio-selective synthesis), enzyme properties (e.g., pH and temperature stability), and substrate specificities [2]. Recent interest in lipases arises particularly from their applications as a potential biocatalyst for biodiesel production [3]. Many microbial lipases have been isolated, sequenced, and expressed [4–8]. Among them, Burkholderia lipases have certain features of thermal stability, alkaline pH and organic solvent tolerance, transesterification activity, and high substrate selectivity, making them an attractive industrial enzyme [9–11]. Hence, there is a motivation in developing an economically feasible bioprocess for their overproduction.
The Gram-negative bacterium Escherichia coli is still recognized as the most popular host for high-level production of recombinant proteins from both prokaryotic and eukaryotic sources [12]. While the major expression steps of transcription and translation have to be effective to achieve high-level production, it is not uncommon that the recombinant protein yield is limited by posttranslational events, such as disulfide bond formation, solubility, misfolding, proteolysis, and even the toxicity to host cells [13]. The expressed recombinant protein can be targeted in various intracellular compartments including the cytoplasm and periplasm, displayed on the cell surface, or secreted into the extracellular medium. The oxidative environment of the extracytoplasmic compartments is conducive for disulfide bond formation [14]. Extracellular secretion often yields recombinant proteins in the culture medium with minimum contamination from host cell proteins [15]. On the other hand, recombinant proteins can be displayed on the E. coli cell surface for specific applications, such as whole-cell biocatalysts and high throughput peptide library screening [16].
E. coli has several folding modulators to assist the folding of nascent polypeptides, particularly during stressful conditions. These folding modulators, including trigger factor, DnaK/J-GrpE, and GroEL/ES in the cytoplasm and Dsb-family chaperones, DegP, FkpA, Skp, and SurA in the periplasm, can be potentially useful for developing their bioactive conformation upon the high-level production of heterologous proteins [17]. Alternatively, fusion tags, originally developed for protein purification with affinity chromatography, are another effective tool to enhance the functional expression, solubility, bioactivity, and stability of the partner protein [18]. The N-terminal tags can potentially enhance the translational initiation and, therefore, are more popular than the C-terminal ones [19]. Several solubility-enhancing tags have been successfully used to produce hard-to-express recombinant proteins in E. coli [20].
Despite the recognized industrial applicability of Burkholderia lipase, there are very few reports associated with its production so far. One of them is the cloning and heterologous expression of the Burkholderia cepacia lipase gene in E. coli [21]. However, the recombinant lipase was predominantly expressed as insoluble inclusion bodies even with the coexpression of lipase-specific foldase (Lif) which is a natural lipase folding modulator. Another study reported that the mutagenesis and screening approach to obtain soluble variants of Burkholderia lipase for functional expression [22]. In this study, we explored various genetic strategies for the functional expression of a lipase from Burkholderia sp. C20 (Lip) in E. coli. The Burkholderia strain was originally isolated from the food waste and could potentially have a novel lipase(s) with the transesterification activity for biodiesel production [23].
Materials and Methods
Bacterial Strains and Plasmids
Bacterial strains, plasmids, and oligonucleotides used in this study are summarized in Table 1. DH5α was used as the host strain for molecular cloning. JM109, BL21(DE3), and Origami B(DE3) were used as the host strain for Lip expression. Molecular cloning was performed according to standard protocols [24]. Restriction enzymes were purchased from New England Biolabs (Beverly, MA, USA). Polymerase chain reaction (PCR) was conducted in an automated thermal cycler (GeneAmp Thermocycler, Applied Biosystems, Foster City, CA, USA). Burkholderia sp. C20 chromosomal DNA was extracted using DNeasy tissue kit (Qiagen, Valencia, CA, USA). Purification of plasmid DNA was performed using a spin-column kit purchased from Qiagen (Valencia, CA, USA). Plasmid transformation was carried out using an electroporator (Bio-Rad, Hercules, CA, USA) or a chemical method according to Chung and Miller [25].
The lip gene was PCR-amplified using the primer pair P1 and P2, and Burkholderia sp. C20 chromosomal DNA as the template. The primer pair P1 and P2 was designed based on the sequence of the lipA gene of Burkholderia multivorans (NCBI Accession number NC_010805-1). The amplified lip gene was cloned to pPCRscriptSK(+) to obtain pPCRscriptLip and the DNA sequencing result is given in Fig. 1a. The lip gene with the native signal peptide was PCR-amplified using P20/P21 primer pairs on pPCRscriptLip template, digested with NcoI/EcoRI and ligated to similarly digested pTrc99A to obtain pTrcLip. The leaderless(ll) lip gene (without the native signal peptide) was PCR-amplified using the primers P58/P21 and pPCRscriptLip template, digested with NcoI/EcoRI and ligated to similarly digested pTrc99A to obtain the plasmid pTrcLipll. In addition, PCR was performed using pPCRscriptLip as the template and the primer pairs of P131/P27, P58/P27, and P58/P59, respectively. The PCR products were digested, respectively, with SfiI, NcoI/SfiI, and NcoI/SacI and ligated to similarly digested pESTYFP, pEHLYA2-SD, and pETM80/pETM82 to construct pESTLipll, pEHLYALipll, and pllDsbCLipll/pDsbCLipll. The NcoI/EcoRI DNA fragment from pTrcLipll was subcloned into pET20b(+) and pENTR4 to obtain pETLipll and pENTRLipll, respectively. The lip-containing DNA in pENTRLipll was subcloned into pGGWA, pMGWA, pHNGWA, pXGWA, pDest-555, pDest-556, pETM-50, and pETM-52 using the Gateway cloning technology (Invitrogen); resulting in pGSTLipll, pMBPLipll, pHisNusALipll, pTRXLipll, pT7PKLipll, pSkpLipll, pDsbALipll, and pllDsbALipll, respectively. The plasmids pAR3GRO, pARDegP, pARDsbA, pARDsbC, pARFkpA, pARllDsbA, pARllDsbC, pARSurA, and pARSkp, respectively, contain the groEL/ES, degP, dsbA, dsbC, fkpA, lldsbA, lldsbC, surA, and skp genes whose expression was under the regulation of the araB promoter. The plasmid pG-KJE8 contains the dnaK/J-grpE and groEL/ES genes, respectively, fused with araB and zt-lp promoters. pG-TF3 contains the tig and groEL/ES genes, respectively, fused with araB and zt-lp promoters. All chaperone gene(s)-expression plasmids contain a chloramphenicol-resistant (CmR) marker and, therefore, are compatible with all Lip-expression vectors containing an ampicillin-resistant (ApR) marker.


Panel a nucleotide sequence analysis of the open reading frame (ORF) of the lip gene amplified by PCR using the genomic DNA of Burkholderia sp. C20 as the template and the primer pair of P1 and P2 (denoted as “1–6”). The sequences of the primers P1 and P2 were determined based on terminal sequences of the 1095-bp lipase gene (lipA) on the chromosome 2 of Burkholderia multivorans ATCC 17616 (NCBI accession number NC_010805, region 699394–700488). The sequences of lip [1–6] and lipA (denoted as “NC_010805”) have been aligned to share a sequence similarity of 87.9%. The stop codon “TGA” at the 3′ end of 1–6 in the figure was originally “GCT” in the amplified DNA, which was replaced with a “STOP” codon for further cloning experiments. Panel b the translated amino acid sequences of the lip gene (denoted as “translation of 1–6 complete”) and lipA gene (depicted as “translation of NC-010805_1”) have been aligned to share a sequence identity of 85.6% and a sequence consensus of 89.5%. The 1–364 amino acid sequence of lip gene [1–6] was subjected to the BLAST query (www.ncbi.nlm.nih.gov). A sequence identity of 99% (362/364) and 100% positive (364/364) was observed with a potential lipase gene (NCBI accession number YP_002233567) identified from the genomic sequence of Burkholderia cenocepacia J2315. Further DNA sequence analysis of the two ORFs showed 100% similarity (4 nucleotides being different and no gap), confirming the missing of a “STOP” codon in the original PCR product of lip [1–6]. Lipase genes from other Burkholderia sp. shared 70–96% sequence identity with lip [1–6]. It may be noted that lip [1–6] shared only 92 and 93% sequence similarity to two other species of Burkholderia cenocepacia. The arrow represents the predicted cleavage site associated with the signal peptide (40 amino acids) using the SignalP 3.0 server (www.cbs.dtu.dk/services/SignalP/). However, the extracellular lipase purified from Burkholderia sp. HY-10 [11] was determined to have the N-terminal amino acid sequence of “ADTYAATRYPIILVHGLTGTDKYAG” which is similar to the underlined amino acid sequence of “ADDYATTRYPIILVHGLTGTDKYAG”. This was determined as the N-terminal sequence of mature Lip for the subsequent cloning of the lip gene without its native signal peptide coding sequence (ll-lip)
Cultivation
Cells were revived by streaking the stock culture stored at −80°C on an LB agar plate (5 g/l NaCl, 5 g/l Bacto yeast extract, 10 g/l Bacto tryptone, and 15 g/l Bacto agar). The plate was incubated at 37°C for approximately 15 h. An isolated single colony was picked to inoculate 25 ml of LB medium, which was then incubated at 37°C in a rotary shaker at 200 rpm for approximately 15 h. The medium was supplemented with 50 μg/ml ampicillin (Ap) when necessary. Erlenmeyer flasks containing 25-ml LB medium were inoculated with the seed culture and were shaken in a rotary shaker at 28°C and 200 rpm. When the cell density reached approximately 0.5 OD600, the culture was supplemented with 0.1-mM isopropyl β-d-thiogalactopyranoside (IPTG) for induction. After induction, the Erlenmeyer flasks were further shaken at the same conditions for another 4 h at 28°C, or 24 h at 15°C. All the shaker-flask cultivations were conducted at least in duplicate.
Analytical Methods
The culture sample was appropriately diluted with saline solution for measuring cell density in OD600 with a spectrophotometer (DU®520, Beckman Coulter, Fullerton, CA, USA). For the preparation of cell extract, cells at an amount of 20 OD600 units (defined as “OD600 × ml”) were centrifuged at 2°C and 6000×g for 10 min. The supernatant containing extracellular proteins was analyzed for secreted Lip. The cell pellet was resuspended in 0.75 ml of sodium phosphate buffer (0.05 M, pH 7.5). The cell suspension was sonicated for 4 min using an ultrasonic processor (Misonix, Farmingdale, NY, USA) and then centrifuged at 4°C and 12,000×g for 15 min. The supernatant containing soluble proteins was assayed for the intracellular Lip activity. The pellet containing insoluble proteins and cell debris was washed with phosphate buffer, resuspended in TE/SDS buffer (10-mM Tris HCl, pH 8.0, 1-mM EDTA, 1% SDS), and heated to 100°C for 5 min. The protein content of the pellet was analyzed as the insoluble fraction.
Lipase activity was qualitatively evaluated by the area and transparency of the halo formed on 1% tributyrin agar plates. To conduct this, soluble fractions and extracellular fractions of 50-μl were loaded on the plate and incubated at 37°C overnight. On the other hand, Lipase enzyme assay was conducted using a pH stat (Brinkman Metrohm 842 Titrando, FL, USA). An appropriate volume of the soluble fraction of cell lysates was added to 5 ml of 2% olive oil emulsion in water at 55°C [23] and the reaction solution was maintained at pH 9.0 by controlled addition of 0.02 N NaOH [26]. One unit of enzyme activity is defined as the amount of enzyme required to liberate 1 μmol of fatty acid per min.
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) was performed in a Mini-PROTEIN®III electrophoresis cell (Bio-Rad) using a 12.5% polyacrylamide separating gel stacked with a 4% polyacrylamide stacking gel. Protein samples of the cell extract from cells of 0.016 OD600 units for the soluble fraction and 0.2 OD600 units for insoluble fractions, respectively, were used. Electrophoresis was conducted under a constant voltage of 120 V for 120 min. The gel was stained with coommassie blue, dried, and scanned.
Results
Seven scenarios (i.e., A1, A2, B, C, D, E, and F summarized in Fig. 2) for the functional expression and targeting of a lipase from Burkholderia sp. C20 (Lip) in E. coli are compared. The culture performance of Lip expression using JM109 (pTrcLip) (i.e., scenario A1) is summarized in Fig. 3. Visible halos were observed on tributyrin plates (data not shown) for the extracellular medium samples of the IPTG-induced culture with the lipase activity up to 55 U/l (expression system 1 in Fig. 3b), but not for the soluble intracellular fraction. The results imply that bioactive Lip was expressed and secreted into the extracellular medium under the direction of the native signal peptide which is recognized by E. coli. The extracellular expression posed no adverse physiological impact on the bacterial cell growth (Fig. 3a). Since Burkholderia lipase is translocated across the inner membrane while being unfolded via the Sec secretion system prior to the secretion into the extracellular medium [27], folding factors in the cell envelope were coexpressed to investigate their effect on Lip expression and the results are summarized in Fig. 3 as well. Among the folding factors investigated, Skp significantly increased the lipase activity by 36% to 75 U/l (#7 in Fig. 3b); whereas DegP had an adverse effect with neither halos being present on tributyrin plates nor a measurable lipase activity (#6 in Fig. 3b).

Various expression strategies adopted in this study for the production and targeting of Lip in E. coli. A1 secretion of Lip into the extracellular medium: The lip gene containing the native signal peptide is expressed under the regulation of the trc promoter. The native signal peptide is responsible for the extracellular secretion of Lip (denoted as LipA1). A2 secretion of Lip into the periplasm: The lip gene without its native signal peptide coding sequence (ll-lip) is fused with the pelB signal sequence and the pelB::ll-lip fusion is expressed under the regulation of the T7 promoter. The PelB signal peptide is responsible for the translocation of Lip to the periplasm (denoted as LipA2). B Periplasmic expression of Lip fusions: Lip is N-terminally fused with various expression enhancing tags (i.e., DsbA, DsbC, and HisperiMBP) and the Fu::ll-lip gene fusions are expressed under the regulation of the T7 or tac promoter. The signal peptides of DsbA, DsbC, and MBP are, respectively, responsible for the translocation of Fu-Lip to the periplasm (denoted as Fu-LipB). C surface display of Lip: Lip is C-terminally fused with an inactive variant of EstA from Pseudomonas aeruginosa and the ll-lip::estA fusion is expressed under the regulation of the lac promoter. The signal peptide of PhoA is responsible for exporting Lip-Fu (denoted as Fu-LipC). D cytoplasmic expression of Lip: The ll-lip gene is expressed in the cytoplasm under the regulation of trc promoter (denoted as LipD). E Cytoplasmic expression of Lip with an N-terminal fusion tag: Lip is N-terminally fused with various expression enhancing tags (i.e., DsbA, DsbC, GST, MBP, NusA, Skp, T7PK, and TRX) and the Fu::ll-lip gene fusions are expressed in the cytoplasm under the regulation of the T7/tac promoter (denoted as Fu-LipE). F secretion of a Lip fusion into the extracellular medium: Lip is C-terminally fused with HlyA and the ll-lip::hlyA gene fusion is expressed under the regulation of the lac promoter. Lip–HlyA is extracellularly secreted via the α-hemolysin transporter (denoted as Lip-FuF)

Culture performance of Lip expression (scenario A1) using (1) JM109 (pTrcLip), (2) JM109 (pTrcLip, pARFkpA), (3) JM109 (pTrcLip, pARDsbA), (4) JM109 (pTrcLip, pARDsbC), (5) JM109 (pTrcLip, pARSurA), (6) JM109 (pTrcLip, pARDegP), (7) JM109 (pTrcLip, pARSkp). Panel a cell density, panel b lipase activity
The culture performance for Lip expression in the cytoplasm of E. coli (i.e., scenario D) is described in Fig. 4. Among the three hosts investigated, i.e., JM109, BL21(DE3), and Origami B(DE3), the halo associated with tributyrin hydrolysis was visualized only for the culture sample of recombinant Origami B(DE3) and the lipase activity appeared to be extremely low (data not shown). However, neither lipase activity was measurable by pH stat nor the protein band corresponding to Lip overexpression was identifiable by SDS-PAGE (data not shown) for all the culture samples grown at 15 or 28°C. The results suggest two possible expression issues, i.e., ineffective translation or protein degradation. Several cytoplasmic chaperones including GroEL/ES, Trigger factor, DnaK/J-GrpE, and the Dsb-chaperones were coexpressed to investigate their possible effect on the cytoplasmic expression of Lip. Visible halos developed on tributyrin agar plates were slightly visible only for BL21(DE3) (pTrcLipll, pAR3GRO) and BL21(DE3) (pTrcLipll, pG-KJE8), but however not for recombinant Origami B(DE3) (data not shown). Again, the lipase activity was not measurable by pH stat and the Lip band was not identifiable by SDS-PAGE (data not shown).

Culture performance of Lip expression (scenario D) using (1) JM109 (pTrcLipll), (2) Origami B(DE3) (pTrcLipll), (3) BL21(DE3) (pTrcLipll), (4) BL21(DE3) (pTrcLipll, pAR3GRO), (5) BL21(DE3) (pTrcLipll, pG-KJE8). Panel cell density
The culture performance for Lip expression in the periplasm of E. coli (i.e., scenario A2) is summarized in Fig. 5. Using the cell lysate of the BL21(DE3) (pETLipll) culture sample with IPTG induction, a halo was visualized on tributyrin agar plates (data not shown) and a lipase activity up to 50 U/l was measured (Fig. 5b). A protein band corresponding to Lip at 35 kDa were visualized upon the SDS-PAGE analysis, but most of the expressed Lip existed in insoluble fraction (Fig. 5c), implying a potential expression issue of protein misfolding. Neither lowering the cultivation temperature to 15°C nor the coexpression of periplasmic chaperones improved the solubility or lipase activity (data not shown).

Culture performance of Lip expression (scenario A2) using BL21(DE3) (pETLipll). Panel a cell density, panel b lipase activity, panel c SDS-PAGE analysis of the soluble and insoluble fractions of culture samples. C and I represent the cultures “without induction” and “with IPTG induction,” respectively, M represents markers
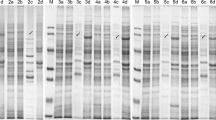
Given the poor culture performance for the cytoplasmic expression of Lip, eight N-terminal fusion tags, i.e., DsbA, DsbC, GST, MBP, HisNusA, Skp, T7PK, and TRX, were used to construct various translational fusions with Lip for their respective expression in the cytoplasm (i.e., scenario E). The culture performance for the expression of these Lip fusions in BL21(DE3) and Origami B(DE3) is summarized in Fig. 6. In general, 28°C appeared to be a better cultivation temperature than 15°C. Halos with a higher intensity were observed for T7PK-Lip, DsbA-Lip, and DsbC-Lip fusions using BL21(DE3) as the expression host and for T7PK-Lip, Skp-Lip, MBP-Lip, and HisNusA-Lip using Origami (DE3) as the expression host. GST-Lip fusion hardly had any lipase activity no matter which host was selected (data not shown). The majority of the overexpressed Lip fusions aggregated as insoluble inclusion bodies (insoluble fraction in Fig. 6c, f). In general, the lipase activity assayed by pH stat in general correlated with the solubility of Lip fusion proteins. Among the eight fusion tags investigated in recombinant BL21(DE3), T7PK, DsbA, and DsbC were effective in solubilizing Lip (lanes 1, 7, and 8 in the soluble fraction of Fig. 6c) and the Lip activities of T7PK-Lip, DsbA-Lip, and DsbC-Lip fusions were assayed to be 105, 135, and 85 U/l, respectively (Fig. 6b). On the other hand, both lipase activity (Fig. 6b) and protein bands corresponding to TRX-Lip, GST-Lip, Skp-Lip, and MBP-Lip (lanes 2, 3, 4, and 5 in Fig. 6c) were minimally detected in the soluble fraction even though halos can be slightly visualized on tributyrin plates for TRX-Lip and Skp-Lip (data not shown). Though HisNusA-Lip fusion had a visible protein band in the soluble fraction (lane 6 in Fig. 6c) and showed a faint halo on the tributyrin plate (data not shown), its lipase activity was hardly detected by pH stat (Fig. 6b). Among the seven fusion tags investigated in recombinant Origami B(DE3), the lipase activities of T7PK-Lip, Skp-Lip, MBP-Lip, HisNusA-Lip fusion proteins were assayed to be 92, 55, 25, and 35 U/l (Fig. 6e), respectively, whereas the other fusions had no detectable lipase activities with faint halos on tributyrin plates. Note that the lipase activity of Skp-Lip was detected by both pH stat and a visible halo on the tributyrin plate though the protein band corresponding to Skp-Lip was hardly detectable (lane 4 in Fig. 6f). In general, the expressed fusions had a higher protein solubility in Origami B(DE3) than BL21(DE3) (Fig. 6c vs. f), implying that the enhancement in the protein solubility could be associated with disulfide bond formation.


Culture performance of Lip expression (scenario E) using an E. coli host harboring (1) pT7PKLipll, (2) pTRXLipll, (3) pGSTLipll, (4) pSkpLipll, (5) pMBPLipll, (6) pHisNusALipll, (7) pllDsbALipll, (8) pllDsbCLipll. Panel a, d cell density, panel b, e lipase activity, panel c, f SDS-PAGE analysis of the soluble and insoluble fractions of culture samples. Various Lip protein fusions have been indicated. Note that panels a–c represent the cultures using BL21(DE3) as the expression host, and panels d–f represent the cultures using Origami B(DE3) as the expression host. C and I represent the cultures “without induction” and “with IPTG induction,” respectively, M represents markers
Similarly, three N-terminal fusion tags, i.e., HisperiMBP, DsbA, and DsbC, were used to construct various translational fusions with Lip for their respective expression in the periplasm (i.e., scenario B). The culture performance for the expression of these Lip fusions in BL21(DE3) is summarized in Fig. 7. Though visible halos developed on tributyrin plates for these Lip fusions (data not shown), their lipase activities were undeterminable by pH stat. Apparently, these fusion tags were incompetent in solubilizing the expressed Lip since almost all the Lip fusions were detected in the insoluble fraction as inclusion bodies (Fig. 7b).

Culture performance of Lip expression (scenario B) using (1) BL21(DE3) (pDsbA-Lipll), (2) BL21(DE3) (pDsbC-Lipll), (3) BL21(DE3) (pHisperiMBP-Lipll). Panel a cell density, panel b lipase activity, panel c SDS-PAGE analysis of the soluble and insoluble fractions of culture samples. Various Lip protein fusions have been indicated. C and I represent the cultures “without induction” and “with IPTG induction,” respectively, M represents markers
Given the above technical limitations associated with intracellular expression in E. coli and the fact that Lip is an extracellular lipase in the original Gram-negative bacterium Burkholderia, extracellular secretion of Lip appears to be a plausible approach. To do this, a translational fusion of Lip with the 23-kDa C-terminal of HlyA was constructed for extracellular secretion of Lip–HlyA via the Hly-transporter (i.e., scenario F). The secretion was assisted by HlyB and HlyD which were coexpressed from the plasmid pVDL9.3. Using JM109 harboring pEHLYALipll and pVDL9.3, the culture performance for the expression and secretion of Lip–HlyA is summarized in Fig. 8. Visible halos were developed on tributyrin agar plates for the extracellular medium samples (data not shown), but not the intracellular fractions; indicating that HlyA can mediate the secretion of bioactive Lip. Lipase activity up to 95 U/l was obtained in the 24-h IPTG-induced culture sample without affecting cell growth (Fig. 8b). Note that the samples of the control cultures without IPTG induction also developed a visible halo owing to the leaky expression associated with the lac promoter, but its lipase activity was very low when compared to that of the IPTG-induced culture (Fig. 8b). The Hly-transporter appeared to be more effective than using the native signal peptide of Lip for achieving functional Lip secretion in the growth medium of E. coli (95 U/l in Fig. 8c vs. 55 U/l in Fig. 3b). However, Lip secreted with the use of the native signal peptide is an intact Lip as opposed to the Lip–HlyA fusion secreted via the Hly-transporter.

Culture performance of Lip expression (scenario F) using JM109 (pEHLYA-Lipll) Panel a cell density, panel b lipase activity. Numbers represent the post-induction times (in hours) at which samples were taken
Finally, displaying Lip on the E. coli cell surface using the carrier protein of EstA*, which is an inactive variant of esterase from Pseudomonas aeruginosa [28], was explored (scenario C) and the culture performance of JM109 harboring pESTLipll is summarized in Fig. 9. Upon the qualitative analysis of the lipase activity on tributyrin agar plates, halos were observed for both the whole cell and the extracellular fraction, whereas no halo was visible for the soluble intracellular lysate (data not shown). The presence of the lipase activity in the extracellular fraction suggests Lip detachment from the cell membrane and/or a potential cell lysis. The whole cells harvested from the IPTG-induced culture yielded a lipase activity up to 25 U/l upon conducting the lipase assay by pH stat (Fig. 9b), confirming the functional display of Lip on the E. coli cell surface.

Culture performance of Lip expression (scenario C) using JM109 (pEST-Lipll) Panel a cell density, panel b lipase activity
Discussion
Functional expression of recombinant proteins in E. coli can be potentially limited by disulfide bond formation. In this study, it appears that Lip expression in the oxidative cytoplasm of Origami B(DE3) slightly developed the lipase activity, as opposed to the cytoplasmic expression of Lip in the reductive cytoplasm of BL21(DE3) which has no lipase activity (Fig. 4). The results suggest certain disulfide bond(s) could be critical for functional expression of Lip. It was reported that the Cys190–Cys269/Cys270 disulfide bridge, which is conserved in lipases from Gram-negative bacteria, is required for protein stabilization against heat denaturation and proteolysis [29–31]. Such conserved cysteine residues of Cys190 and Cys270 exist in the mature Lip. In addition to disulfide bond formation, ineffective translation or proteolysis could also contribute to the poor Lip yield associated with the cytoplasmic expression. Coexpression of GroEL/ES, which assists the folding of de novo proteins and the refolding of misfolded proteins [32, 33], had a slightly positive effect in developing the lipase activity in BL21(DE3). The low expression yield could be partially alleviated with the use of fusion tags and high-level expression was achieved for all the Lip fusions except Skp-Lip. Though lipase activity was detected for several Lip fusions, the majority of the expressed Lip fusions aggregated as insoluble inclusion bodies in the cell, implying another expression issue of protein misfolding. It is interesting to observe that the functional expression of the Lip fusion with DsbA was achieved in BL21(DE3), but not in Origami B(DE3); whereas the functional expression of Lip fusions with MBP, Skp, and HisNusA was achieved in Origami B(DE3), but not in BL21(DE3). T7PK was reported to be effective in producing hard-to-express proteins in E. coli [34] and it was the only fusion tag achieving the functional expression in both BL21(DE3) and Origami B(DE3).
Similarly, the majority of the heterologously expressed Lip, either non-fused Lip or Lip fusions, in the periplasm of E. coli formed inclusion bodies though the periplasm is an oxidative compartment suitable for disulfide bond formation. The first step for the secretion of extracellular lipase in Burkholderia is protein translocation across the cytoplasmic membrane to the periplasm via the Sec secretion system and the export occurs concurrently with the removal of the N-terminal signal sequence while maintaining a partially folded and near native conformation for the secreted lipase [27]. Further transport of proteins across the outer membrane into the extracellular medium can be mediated by the Xcp secreton, which is a complex machinery comprising of at least 12 different proteins and well conserved in the Gram-negative bacteria species such as Pseudomonas and Burkholderia [35]. Though the expressed Lip was targeted to the periplasm of E. coli via the Sec secretion system, the presence of insoluble Lip aggregates, even under the situation with the coexpression of periplasmic folding factors, suggests that proper folding of Lip was prevented in the periplasm. Interestingly, the expression of Lip with its native signal peptide produced active enzyme in the extracellular medium of E. coli, implying that extracellular secretion assists the proper folding of Lip. Apparently, the signal peptide of Burkholderia Lip is recognized by E. coli for the extracellular secretion of Lip even though no Xcp-secreton-like system has been reported for E. coli. Technically, it will be interesting to explore the use of the Lip signal peptide for the extracellular secretion of recombinant proteins since most of the signal peptides adopted in E. coli are primarily for the secretion of recombinant proteins into the periplasm [36–39]. The extracellular expression of Lip was further improved by Skp coexpression. Skp is a cell envelope chaperone that helps maintaining an intermediate folded conformation for outer membrane proteins to be competent for export and insertion onto the outer membrane [40]. It was demonstrated to be effective in improving the folding of recombinant antibody fragments in the periplasm [41] and the display of recombinant proteins on the cell surface [42]. The improved extracellular expression of Lip associated with Skp coexpression might be due to the prevention of premature Lip folding in the periplasm so that the subsequent export across the outer membrane was facilitated.
Since extracellular secretion appears to be an effective approach for the functional expression of Lip, another strategy of using the α-hemolysin transporter for extracellular secretion was successfully explored in this study. The extracellular secretion was conducted through the construction of a Lip–HlyA fusion whose translocation across the inner and outer membranes in E. coli is mediated by an export conduit which is assembled by the Hly-transport component proteins of HlyB, HlyD [43], and the outer membrane protein TolC [44]. No signal peptide is required for such direct secretion of Lip–HlyA from the cytoplasm to the cell exterior without a periplasmic intermediate [45]. It was reported that functionally active single chain Fv antibodies with correct disulfide bonds were secreted via the Hly-transporter [46], whereas the activity was demolished when an E. coli mutant defective in thioredoxin reductase was used as the host for the secretion [47]; suggesting that proper disulfide bonds could be formed during the protein transport through the export conduit or in the oxidative culture medium. Such a feature might be helpful in terms of forming the disulfide bridge of Cys190–Cys270 in Lip–HlyA which is required for stabilizing the active protein [29]. Comparing the two secretion strategies, both of them posed a minimum physiological impact on cell growth, but the one via the Hly-transporter appeared to produce a higher lipase activity. However, a Lip–HlyA fusion was produced and, as a result, an additional protein cleavage step would be required to obtain the native Lip. On the other hand, the native Lip was produced through the secretion based on the native signal peptide.
Finally, Lip was functionally expressed on the E. coli cell surface using EstA*, an inactive variant of EstA from Pseudomonas aeruginosa, as a carrier protein. EstA has been shown to be properly localized on the outer membrane upon heterologous expression in E. coli and, as a result, it can serve as a carrier for the functional display of several lipolytic enzymes from Fusarium solani, Bacillus subtilis, and Serratia marcescens on the E. coli cell surface [28]. Since the N-terminal extracellular domain of EstA is intact in this design, the passenger protein fused to the carrier can be relatively large, fully exposed on top of the carrier protein, and placed far from the lipopolysaccharide layer as compared to other displaying systems. In this study, whole cells with the expressed Lip–EstA* were found to have the lipase activity, implying the functional expression of Lip on the cell surface. However, the lipase activity was also detected in the extracellular medium, suggesting some of the displayed Lip was released. The release of the displayed Lip could be possibly due to the proteolytic attack by certain outer membrane protease, such as OmpT [48], or the release of the whole Lip–EstA* fusion into the medium since EstA is not a native E. coli protein and the integration of EstA with the outer membrane might not be firm enough. The E. coli cells with bioactive Lip being expressed on the outer membrane can be used as a whole-cell biocatalyst though the displaying performance could be further improved.
References
Seitz, E. W. (1973). Industrial application of microbial lipases: A review. Journal of the American Oil Chemists’ Society, 51, 12–16.
Jaeger, K. E., & Eggert, T. (2002). Lipases for biotechnology. Current Opinion in Biotechnology, 13, 390–397.
Nelson, L. A., Foglia, T. A., & Marmer, W. N. (1996). Lipase-catalyzed production of biodiesel. Journal of the American Oil Chemists’ Society, 73, 1191–1195.
Catoni, E., Dannert, C. S., & Schmid, R. D. (1997). Overexpression of lipase A and B of Geotrichum candidum in Pichia pastoris: High-level production and some properties of functional expressed lipase B. Biotechnology Techniques, 9, 689–695.
Dannert, C. S., Rua, M. L., Atomi, H., & Schmid, R. D. (1996). Thermoalkalophilic lipase of Bacillus thermocatenulatus: Molecular cloning, nucleotide sequence, purification, and some properties. Biochimica et Biophysica Acta, 1301, 105–114.
Jensen, B. H., Andreasen, F., Christensen, T., Christensen, M., Thim, L., & Boel, E. (1989). Rhizomucor miehei triglyceride lipase is processed and secreted from transformed Aspergillus oryzae. Lipids, 24, 781–785.
Tsuchiya, A., Nakazawa, H., Toida, J., & Ohnishi, K. (1996). Cloning and nucleotide sequence of the mono- and diacylglycerol lipase gene (mdlB) of Aspergillus oryzae. FEMS Microbiology Letters, 143, 63–67.
Xu, Y., Yasin, A., Tang, R., Scharer, J. M., Moo-Young, M., & Chou, C. P. (2008). Heterologous expression of lipase in Escherichia coli is limited by folding and disulfide bond formation. Applied Microbiology and Biotechnology, 81, 79–87.
Fernandez, V. G., Brieva, R., & Gotor, V. (2006). Lipases: Useful biocatalysts for the preparation of pharmaceuticals. Journal of Molecular Catalysis B: Enzymatic, 40, 111–120.
Noureddini, H., Gao, X., & Philkana, R. S. (2005). Immobilized Pseudomonas cepacia lipase for biodiesel fuel production from soybean oil. Bioresource Technology, 96, 769–777.
Park, D. S., Oh, H. W., Heo, S., Jeong, W. J., Shin, D. H., Bae, K. S., et al. (2007). Characterization of an extracellular lipase in Burkholderia sp. HY-10 isolated from a longicorn beetle. Journal of Microbiology, 45, 409–417.
Marino, M. H. (1989). Expression systems for heterologous protein production. BioPharm, 2, 18–33.
Makrides, S. C. (1996). Strategies for achieving high-level expression of genes in Escherichia coli. Microbiological Reviews, 60, 512–538.
Georgiou, G., & Segatori, L. (2005). Preparative expression of secreted proteins in bacteria: Status report and future prospects. Current Opinion in Biotechnology, 16, 538–545.
Mergulhao, F. J. M., Summers, D. K., & Monteiro, G. A. (2005). Recombinant protein secretion in Escherichia coli. Biotechnol. Advan., 23, 177–202.
Lee, S. Y., Choi, J. H., & Xu, Z. (2003). Microbial cell-surface display. Trends in Biotechnology, 21, 45–52.
Baneyx, F., & Mujacic, M. (2004). Recombinant protein folding and misfolding in Escherichia coli. Nature Biotechnology, 22, 1399–1408.
Terpe, K. (2003). Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Applied Microbiology and Biotechnology, 60, 523–533.
Hammarstrom, M., Woestenenk, E. A., Hellgren, N., Hard, T., & Berglund, H. (2006). Effect of N-terminal solubility enhancing fusion proteins on yield of purified target protein. Journal of Structural and Functional Genomics, 7, 1–14.
Esposito, D., & Chatterjee, D. K. (2006). Enhancement of soluble protein expression through the use of fusion tags. Current Opinion in Biotechnology, 17, 353–358.
Quyen, D. T., Schmidt-Dannert, C., & Schmid, R. D. (1999). High-level formation of active Pseudomonas cepacia lipase after heterologous expression of the encoding gene and its modified chaperone in Escherichia coli and rapid in vitro refolding. Applied and Environmental Microbiology, 65, 787–794.
Guenot, S. P., Lafaquière, V., Guieysse, D., Landric-Burtin, L., Monsan, P., & Remaud-Siméon, M. (2008). Small-scale production of Burkholderia cepacia ATCC21808 lipase adapted to high-throughput screening. J. Biomol. Screen., 13, 72–79.
Liu, C. H., Lu, W. B., & Chang, J. S. (2006). Optimizing lipase production of Burkholderia sp. by response surface methodology. Process Biochemistry, 41, 1940–1944.
Sambrook, J., Fritsch, E. F., & Maniatis, T. (1989). Molecular cloning: A laboratory manual. New York: Cold Spring Harbor Laboratory Press.
Chung, C. T., & Miller, R. H. (1993). Preparation and storage of competent Escherichia coli cells. Methods in Enzymology, 218, 621–627.
Tietz, N. W., & Repique, E. V. (1973). Proposed standard method for measuring lipase activity in serum by a continuous sampling technique. Clinical Chemistry, 19, 1268–1275.
Frenken, L. G. J., Egmont, M. R., Batenburg, A. M., Bos, J. W., Visser, C., & Verrips, T. (1992). Cloning of the Pseudomonas glumae lipase gene and determination of the active site residues. Applied and Environmental Microbiology, 58, 3787–3791.
Becker, S., Theile, S., Heppeler, N., Michalczyk, A., Wentzel, A., Wilhelm, S., et al. (2005). A generic system for the Escherichia coli cell-surface display of lipolytic enzymes. FEBS Letters, 579, 1177–1182.
Kim, K. K., Song, H. K., Shin, D. H., Hwang, K. Y., & Suh, S. W. (1997). The crystal structure of a triacylglycerol lipase from Pseudomonas cepacia reveals a highly open conformation in the absence of a bound inhibitor. Structure, 5, 173–185.
Noble, M. E. M., Cleasby, A., Johnson, L. N., Egmond, M. R., & Frenken, L. G. J. (1994). Analysis of the structure of Pseudomonas glumae lipase. Protein Engineering, 7, 559–562.
Lang, D., Haalck, L., Hofmann, B., Hecht, H. J., Spener, F., Schmid, R. D., et al. (1994). Crystallization and preliminary X-ray analysis of a lipase from Chromobacterium viscosum. Acta Crystallographica Section D: Biological Crystallography, 50, 225–227.
Hartl, F. U., & Hayer-Hartl, M. (2002). Molecular chaperones in the cytosol: From nascent chain to folded protein. Science, 295, 1852–1858.
Ewalt, K. L., Hendrick, J. P., Houry, W. A., & Hartl, F. U. (1997). In vivo observation of polypeptide flux through the bacterial chaperonin system. Cell, 90, 491–500.
Chatterjee, D. K., & Esposito, D. (2006). Enhanced soluble protein expression using two new fusion tags. Protein Expression and Purification, 46, 122–129.
Tomassen, J., Filloux, A., Bally, M., Murgier, M., & Lazdunski, A. (1992). Protein secretion in Pseudomonas aeruginosa. FEMS Microbiology Reviews, 103, 73–90.
Moks, T., Abrahmsen, L., Holmgren, E., Bilich, M., Olsson, A., & Uhlen, M. (1987). Expression of human insulin-like growth factor I in bacteria: use of optimized gene fusion vectors to facilitate protein purification. Biochemistry, 26, 5239–5244.
Lo, A. C., MacKay, R. M., Seligy, V. L., & Willick, G. E. (1988). Bacillus subtilis β-1, 4-endoglucanase products from intact and truncated genes are secreted into the extracellular medium by Escherichia coli. Applied and Environmental Microbiology, 54, 2287–2292.
Khosla, C., & Bailey, J. E. (1988). Heterologous expression of a bacterial hemoglobin improves the growth properties of recombinant Escherichia coli. Nature, 331, 633–635.
Schein, C. H., Boix, E., Haugg, M., Holliger, K. P., Hemmi, S., Frank, G., et al. (1992). Secretion of mammalian ribonucleases from Escherichia coli using the signal sequence of murine spleen ribonuclease. Biochemical Journal, 283, 137–144.
Bulieris, P. V., Behrens, S., Holst, O., & Kleinschmidt, J. H. (2003). Folding and insertion of the outer membrane protein OmpA is assisted by the chaperone Skp and by lipopolysaccharide. The Journal of Biological Chemistry, 278, 9092–9099.
Bothmann, H., & Pluckthun, A. (1998). Selection for a periplasmic factor improving phage display and functional periplasmic expression. Nature Biotechnology, 16, 376–380.
Narayanan, N., & Chou, C. P. (2008). Physiological improvement to enhance Escherichia coli cell-surface display via reducing extracytoplasmic stress. Biotechnology Progress, 24, 293–301.
Gentschev, I., Dietrich, G., & Goebel, W. (2002). The E. coli alpha-hemolysin secretion system and its use in vaccine development. Trends in Microbiology, 10, 39–45.
Andersen, C., Koronakis, E., Bokma, E., Eswaran, J., Humphreys, D., & Hughes, C. (2002). Transition to the open state of the TolC periplasmic tunnel entrance. Proceedings of the National Academy of Sciences of the United States of America, 99, 11103–11108.
Thanabalu, T., Koronakis, E., Hughes, C., & Koronakis, V. (1998). Substrate-induced assembly of a contiguous channel for protein export from E coli: Reversible bridging of an inner-membrane translocase to an outer membrane exit pore. EMBO Journal, 17, 6487–6496.
Fernandez, L. A., Sola, I., Enjuanes, L., & de Lorenzo, V. (2000). Specific secretion of active single-chain Fv antibodies into the supernatants of Escherichia coli cultures by use of the hemolysin system. Applied and Environmental Microbiology, 66, 5024–5029.
Fernandez, L. A., & de Lorenzo, V. (2001). Formation of disulphide bonds during secretion of proteins through the periplasmic-independent type I pathway. Molecular Microbiology, 40, 332–346.
Grodberg, J., & Dunn, J. J. (1988). ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification. Journal of Bacteriology, 170, 1245–1253.
Phillips, T. A., Vanbogelen, R. A., & Neidhardt, F. C. (1984). Lon gene product of Escherichia coli is a heat-shock protein. Journal of Bacteriology, 159, 283–287.
Woodcock, D. M., Crowther, P. J., Doherty, J., Jefferson, S., Decruz, E., Noyerweidner, M., et al. (1989). Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Research, 17, 3469–3478.
Yanisch-Perron, C., Vieira, J., & Messing, J. (1985). Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mp18 and pUC19 vectors. Gene, 33, 103–119.
Perez-Perez, J., & Gutierrez, J. (1995). An arabinose-inducible expression vector, pAR3, compatible with ColE1-derived plasmids. Gene, 158, 141–142.
Pan, K.-L., Hsiao, H.-C., Weng, C.-L., Wu, M.-S., & Chou, C. P. (2003). Roles of DegP in prevention of protein misfolding in the periplasm upon overexpression of penicillin acylase in Escherichia coli. Journal of Bacteriology, 185, 3020–3030.
Xu, Y., Lewis, D., & Chou, C. P. (2008). Effect of folding factors in rescuing unstable heterologous lipase B to enhance its overexpression in the periplasm of Escherichia coli. Applied Microbiology and Biotechnology, 79, 1035–1044.
Wu, M. S., Pan, K. L., & Chou, C. P. (2007). Effect of heat-shock proteins for relieving physiological stress and enhancing the production of penicillin acylase in Escherichia coli. Biotechnology and Bioengineering, 96, 956–966.
Nallamsetty, S., Austin, B. P., Penrose, K. J., & Waugh, D. S. (2005). Gateway vectors for the production of combinatorially-tagged His6-MBP fusion proteins in the cytoplasm and periplasm of Escherichia coli. Protein Science, 14, 2964–2971.
Dummler, A., Lawrence, A. M., & de Marco, A. (2005). Simplified screening for the detection of soluble fusion constructs expressed in E. coli using a modular set of vectors. Microb. Cell Fact., 4, 34–39.
Nishihara, K., Kanemori, M., Yanagi, H., & Yura, T. (2000). Overexpression of trigger factor prevents aggregation of recombinant proteins in Escherichia coli. Applied and Environmental Microbiology, 66, 884–889.
Busso, D., Delagoutte-Busso, B., & Moras, D. (2005). Construction of a set Gateway-based destination vectors for high-throughput cloning and expression screening in Escherichia coli. Analytical Biochemistry, 343, 313–321.
Acknowledgments
This study is supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada and the Canada Research Chair (CRC) program. We greatly appreciate the gift of the strain Burkholderia sp. C20 from Dr. Jo-Shu Chang. We thank Deb K. Chatterjee, Didier Busso, Luis Angel, Nikolay K. Tzvetkov, and EMBL for providing different vectors used in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Narayanan, N., Khan, M. & Chou, C.P. Enhancing Functional Expression of Heterologous Burkholderia Lipase in Escherichia coli . Mol Biotechnol 47, 130–143 (2011). https://doi.org/10.1007/s12033-010-9320-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-010-9320-3