
Abstract
Cyt1Aa is a cytolytic toxin, found together with the delta-endotoxins in Bacillus thuringiensis subsp. israelensis parasporal insecticidal crystals. The latter are used as an environmental friendly insecticide against mosquitoes and black flies. Contrary to Cry delta-endotoxin, the mode of action of Cyt1Aa is not completely understood. In the absence of direct structural data, a novel mutated cyt1Aa gene was used to obtain indirect informations on Cyt1Aa conformation changes in the lipid membrane environment. A mutated cyt1Aa gene named cyt1A97 has been isolated from a B. thuringiensis israelensis strain named BUPM97. The nucleotide sequence predicted a protein of 249 amino acids residues with a calculated molecular mass of 27 kDa. Both nucleotide and amino acid sequences similarity analysis revealed that cyt1A97 presents one amino acid different from the native cyt1Aa gene. This mutation was located in the helix α C corresponding to a substitution of Met115 by a Thr. The heterologous expression of the cyt1A97 and another cyt1Aa-type gene called cyt1A98, not affected by such mutation used as control, was performed in Escherichia coli. It revealed that the mutated Cyt1A97 protein was over produced as inclusion bodies showing a very weak toxicity to E. coli contrarily to Cyt1A98 that stopped E. coli growth. Hence, hydrophobic residue Met at position 115 of Cyt1Aa should play a very important role for the maintenance of the structure and cytolytic functions of Cyt1Aa.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cytolytic (Cyt) endotoxins are produced by a minor group of B. thuringiensis, mostly in subsp. that are toxic to dipteran insects [1 – 3]. Cyt toxins exhibit a broad spectrum of activity against a variety of eukaryotic cells in vitro but are highly specific towards dipteran insects in vivo [4 – 6]. B. thuringiensis subsp. israelensis is the mosquitocidal subspecies most thoroughly studied. One of the major components of its toxin is the cytolytic endotoxin Cyt1Aa. The Cyt1Aa toxin is of particular interest because of its affinity for unsaturated fatty acids in the lipid layer of the membrane, whereas Cry and B. sphaericus Bin proteins target specific receptor molecules. The level of mosquito larvicidal activity of Cyt1Aa by itself is low [7 – 8]. Despite its relatively low mosquitocidal activity, Cyt1Aa was demonstrated to increase Cry protein toxicity in B. thuringiensis subsp. israelensis through synergism [7, 9, 10]. Cyt1A synergizes or suppresses resistance to Cry11Aa toxin by functioning as a membrane-bound receptor. Recent evidences suggested that Cyt synergizes or overcomes resistance to mosquitocidal-Cry proteins by functioning as a Cry membrane-bound receptor. These data represent an example of an insect pathogenic bacterium that carries a toxin and also its functional receptor, promoting toxin binding to target membranes and toxicity. [11 – 14]. Moreover, expression of cloned Cyt1Aa is lethal to Escherichia. coli [15] and to B. thuringiensis subsp. kurstaki (CryB) cells [8). Loss of colony-forming ability without substantial lysis, associated with immediate inhibition of DNA synthesis, was observed after induction of recombinant E. coli cells [15]. The chromosome replication complex has been implicated as the target for Cyt1Aa in E. coli [15] because the membrane fraction attached to DNA is enriched in phosphatidylethanolamine [16], which is among those phospholipids known to preferentially interact with Cyt1Aa [17]. Thus, in B. thuringiensis subsp. israelensis both Orf2 and P20 may act as chaperones to initiate, facilitate or stabilize crystal formation in their original hosts, and protect E. coli and B. thuringiensis strains from lethal action of Cyt1Aa. The detailed mechanism in which the Cyt toxins damage the target cell is still under debate. It could act as a pore former as described previously [18] or kill the cell via a detergent-like mechanism as suggested by Manceva et al. [19]. Prediction of the Cyt1Aa structure became possible only when Li et al. [20] determined the X-ray structure of the related B. thuringiensis toxin Cyt2Aa1 (CytB) based on sequence analysis, hydrophobicity and mutagenesis data. The three-dimensional structure of CytB was determined [20], and found to have a single domain of α/β architecture [20]. Similar to Cyt2Aa1, Cyt1Aa1 appears to consist of two outer α-helix hairpins (helices A-B and C-D) flanking a core of mixed β-sheet (strands 1–7) [21]. Based on this structure, plus the previous work on biochemical characterization of the toxin mutant and the results of proteolysis, Li et al. [20] proposed that it is the β-sheets rather than the α-helix that are responsible for membrane binding and pore formation. Whereas, Gazit and Shai [22] demonstrated that helix-1 and helix-2 are involved in the toxic mechanism of Cyt1Aa toxin of B. thuringiensis subsp. israelensis. Both these hydrophobic helical segments may facilitate pore formation by the toxin, with a predominant contribution by helix-2 [22]. In fact, that helix-2 can permeate phopholipid vesicules better than helix-1, may be due to its higher ability to form aggregates within the membrane, as revealed from their binding isotherm. Characterization of the toxin structure in the membrane-bound state, is however the key to understanding its biological function in a membrane. A conformational change of the toxin upon its binding to the membrane was observed [20]. Current evidence suggests that the helical hairpin, containing α C, is the most likely machinery for membrane insertion and channel formation [20]. Thus, in the present work, we evidenced the importance of the Met115 of Cyt1Aa helix α C of B. thuringiensis israelensis for its cytolytic activity in E. coli.
Materials and Methods
Bacterial Strains, Plasmids and Growth Conditions
Bacillus. thuringiensis subsp. israelensis BUPM97 was isolated from a Tunisian soil in the laboratory according to Jaoua et al. [23]. This strain was selected for its insecticidal activity [24] against larvae of several dipterans insects such as Culexpipiens. B. thuringiensis subsp. israelensis BUPM98 was used as reference strain. For routine use in the laboratory, isolates were grown in Luria-Bertani (LB) medium at 30 °C with shaking at 200 rev min-1, and for long storage, the isolates were stored as glycerol stocks at (80 °C). E. coli strain named pMOSBlue cells (endA1 hsdR17 (rk12−mk12+) supE44thi-1 recA1 gyrA96 relA1 lac[F′ proA+B+ laclqZΔM15: Tn10(TcR)]) was obtained from Amersham (Paris, France) and used as the host organism for recombinant DNA cloning and heterologous expression. Plasmid pMOSBlue (Amersham, Paris, France) and pBADGFPuv (Clontech; GenBank accession no. U62637) were used in this study as cloning vectors and expression vector. pBADGFPuv plasmid is a vector that was used for substituting gfp gene by other genes for expressing downstream arabinose operon promoter (PBAD) [25]. pBADcyt1A98 is a recombinant vector expressing a normal B. thuringiensis israelensiscyt1Aa gene downstream PBAD and used control for Cyt1A97 cytolytic activity assessment.
DNA Extraction and PCR Amplification
Plasmid DNA was extracted from B. thuringiensis strains using the alkali lysis method including a lysosyme treatment step [26]. The oligonucleotides used in this study were synthesized by the “Centre de Génétique Moléculaire, CNRS, GENSET, Orsay, France”. Primers used to detect and to clone cyt-like gene were designed based on the published sequence cyt1A(a) gene [GenBank accession no. AL731825]. DNA extracted from B. thuringiensis strains was used as template for the PCR using a “Gene Amp PCR System 2700” (Applied Biosystems). PCR was accomplished as described by Jaoua et al. [23].
Cloning and Sequencing of cyt1A97 from BUPM97 and Transformation Procedures
The gene cyt1Aa, designed cyt1A97, was amplified by PCR using total DNA isolated from strain BUPM97 as template, Taq DNA polymerase (Amersham, Paris France) and primers D23 (5′GTTGTAAGCTTATGGAAAAT3′) and D24 (5′TTAGAAGCTTCCATTAATA3′) complementary to the regions located at the initiation codon (ATG) and the termination codon (TAA), respectively. The amplified fragment (0.75 kb) corresponding to the Open Reading Frame (ORF) of the cyt-gene was purified from the agarose gel with MiniElute Gel Extraction Kit (Qiagen S.A. France) and cloned in pMOSBlue vector generating a recombinant plasmid pMOScyt1A97. Ligation and transformation of pMOSBlue cells was performed according to the manifacturer’s protocol (Amersham, Paris France). Selection of E. coli pMOSBlue cells transformants was performed on LB medium plates containing ampicillin (100 μg ml−1). The sequencing of the cyt1A97 ORF was carried out using a taq DyeDeoxy Terminator Cycle Sequencing kit and a 3,700 ABI Prism DNA sequencer (Applied Biosystems, Foster City, Calif., USA) according to the instructions of the manufacturer. The cloned fragment sequence of approximately 750 bp was determined using the universal primer. To be certain of the real sequence, three PCR clones were used and the same sequence was obtained.
Sequence Alignments
Sequence comparison with the data bases was performed using BLAST through the NCBI e-mail server. Multiple alignments were carried out using CLUSTALW programme [27] to generate a multiple sequence alignment of the selected protein sequences. The sequence of cyt1A97 has been deposited in the GeneBank data base under the accession No. EF656359.
Heterologous Expression of cyt1A97 in E. coli
Over-expression of cyt1A97 was achieved by cloning its ORF in the pBADGFPuv vector [25]. In this vector, expression is under control of the arabinose operon promoter (PBAD), which can be induced in the presence of L-arabinose [25]. The NheI-EcoRI restriction fragment of this plasmid, containing the green fluorescent protein (GFP) coding sequence (positions 1,340–2,063), was replaced by an XbaI-EcoRI restriction fragment containing cyt1A97 coding sequence obtained from the plasmid pMOScyt1A97. This cloning step positioned the cyt1A97 ORF just downstream of the PBAD promoter and allowed the expression of the latter in pMOSBlue cells. The recombinant plasmid named pBADcyt1A97 (6 kb) was transferred by transformation to pMOSBlue cells. Recombinant E. coli clones carrying the cyt1A97 gene were grown at 37 °C overnight in LB broth supplemented with 100 μg of ampicillin per ml. This preculture was used to inoculate 50 ml of LB medium with an initial absorbance at 660 nm of 0.045. When the optical density reached 0.25 at 660 nm PBAD promoter was induced by L-arabinose (0.125 g l−1 final concentration) and host cells were grown for 3 h post-induction at 37 °C and 200 rev min−1. Then, cells were harvested by centrifugation at 2,400 × g for 10 min. The pellet was washed and suspended in distilled water and then analysed by sodium dodecyl sulphate (10% w/v) polyacrylamide gel electrophoresis (SDS-PAGE).
Protein Modelling
Structure model of Cyt1A97 as well as Cyt1A98 were generated using the structure of the mosquitocidal delta-endotoxin CytB from Bacillus thuringiensis subsp. kyushuensis as template [20]. (pdb-ID:1cby) was determined by X-ray crystallography and solved at 2.6Å. The generated models were performed by the automated protein structure homology-modelling server SWISS-MODEL [28]. Finally, PyMOL [29] was used to render figures.
Concerning the Side chain modelling, the reconstruction of the model side chains is based on the weighted positions of corresponding residues in the template structures. Starting with conserved residues, the model side chains are built by iso-sterically replacing template structure side chains. Possible side chain conformations are selected from a backbone dependent rotamer library, which has been constructed carefully taking into account the quality of the source structures. A scoring function assessing favourable interactions (hydrogen bonds, disulphide bridges) and unfavourably close contacts is applied to select the most likely conformation. So there are no problems with side chain locations and interactions in generated models, and so model built structure will generally provide a useful basis for structure analysis.
Results
Cloning of the cyt1Aa-type Gene from the B. thuringiensis subsp. israelensis Strain BUPM97
Due to the interest provided by the cry genes rearrangement we had evidenced in B. thuringiensis subsp. israelensis strain BUPM97 [24], the investigation of the other components of the crystal, Cyt1Aa, was carried out. A PCR fragment of 750 bp containing the ORF of cyt1A97 was cloned in the vector pMOSBlue. The resultant recombinant plasmid designated pMOScyt1A97 was used for the sequencing of the cyt1A97 gene. The obtained sequence corresponds to an ORF of 750 bp encoding a Cyt1Aa protein of 249 amino acid residues. The predicted molecular mass of this protein was 27 kDa. The search for sequence similarity of cyt1A97 with the up to now known genes, using the Blast programme and multiple alignments using CLUSTALW programme (Fig. 1), showed that when compared with all known cyt1Aa-type genes, there was a substitution at position 342 of T to A (transversion) without any change in the amino acid sequence and another substitution at position 344 of T to C (transition) resulting in the substitution of Met115 for Thr located in helix-2 of Cyt1Aa.

Alignment of the amino acids sequences of Cyt1A97, B. thuringiensis israelensis Cyt1Aa and B. thuringiensis kyushuensis CytB, proteins using CLUSTALW programme. In helix-α-C, mutation Met115 Thr was marked with an arrow
Heterologous Expression of cyt1A97 in E. coli
To study its expression, cyt1A97 was cloned in the pBAD vector. The gene cyt1A97 was cloned downstream of the strong PBAD promoter in the pBADGFPuv vector. Microscopic examination of recombinant transformants E. coli pMOSBlue cells (pBADcyt1A97) revealed the presence of small inclusions (data not shown). The pellet of the recombinant cells was analysed by SDS-polyacrylamide gel electrophoresis (Fig. 2), using as control cyt1Aa-gene of the reference strain B. thuringiensis israelensis BUPM98, cloned and expected in pMOSBlue cells, in the same vector (data not shown). The SDS-PAGE analysis showed the presence of the expected 27 kDa protein (Fig. 2) corresponding to Cyt1A97 protein. These results demonstrate that Cyt1A97 originating from B. thuringiensis subsp. israelensis was well expressed in E. coli, contrary to Cyt1A98 which was not detected in the SDS-PAGE.

Heterologous expression of Cyt1A97 in E. coli: SDS–PAGE analysis. Lanes: 1, molecular weight markers (RPN 800, Amersham, Paris France): 96, 66, 45, 30, 20.1 and 14 kDa; 2, pMOSBlue cells (pBADcyt1A97) induced with arabinose; 3, pMOSBlue cells (pBADcyt1A97) uninduced; 4, pMOSBlue cells (pBADcyt1A98) uninduced; 5, pMOSBlue cells (pBADcyt1A98) induced with arabinose
Effect of a Single Mutation in Cyt1A97 on its Cytotoxicity
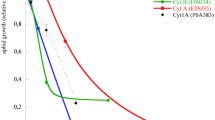
To further investigate the effect of the substitution Met115 Thr of Cyt1A97, the growth kinetics of pMOSBlue cells (pBADcyt1A97), and that of pMOSBlue cells (pBADcyt1A98) used as positive control of cytolytic activity, were performed. The growth kinetics of the strain expressing Cyt1A97 was compared to that expressing the Cyt1A98 that is not affected by the mutation Met115 Thr (Fig. 3). The mutant cyt1A97 gene was highly expressed as inclusion bodies in E. coli cells. However, the recombinant E. coli cells expressing the native non-mutated cyt1A98 gene delayed cell division just after induction. These results reveal that the substitution Met115 Thr affected the cytotoxicity potential of Cyt1A97 in E. coli and demonstrate well that Met115 is essential for structural folding and cytolytic activity of Cyt1Aa.

Growth kinetics curve in shake flask culture of E. coli pMOSBlue cells containing, respectively, cyt1A97 and cyt1A98. △ pMOSBlue cells (pBADcyt1A97) without induction; ▲ pMOSBlue cells (pBADcyt1A97) induced with arabinose; ○ pMOSBlue cells (pBADcyt1A98) without induction; ● pMOSBlue cells (pBADcyt1A98) induced with arabinose
Effect of the Met115 Thr Mutation on the Structure of Cyt1A97
To investigate the importance of the Met115 Thr substitution at a molecular level, a three-dimensional model of Cyt1A97, was constructed on the basis of the crystal structure of CytB of B. thuringiensis subsp. kyushuensis [20] (Fig. 4a). The analysis of the generated model showed that Cyt1A97 and CytB share together the same structure as expected from their sequences identity (39%) [30, 31].

(a) Schematic ribbon diagram of Cyt1A97 structure, (dashed lines): The boundary region between the sheet and helix hairpin C-D, proposed to be the hinge for the conformational change that leaves the helices on the membrane surface. (Broken lines): hydrogen bond between Gln111 and Thr115. (b) Focus on the Gln111/Thr115 region showing the position and side-chain orientation of Met115 in Cyt1A98 (A) and Thr115 in Cyt1A97 (B). Broken lines are hydrogen bonds
In fact, Cyt1A97 is a single domain protein of α/β architecture, with two outer layers of α-helices and a β-sheet in between. However, unlike most proteins of α/β architecture which contain βαβ units, CytB shows an uncommon topology in that its helical layers consist of hairpins, which can be moved relative to the β-sheet without unravelling the sheet [20]. The helices αA, αB, αC and αD have an amphiphilic character, with hydrophobic residues packed against the β-sheet, and polar and charged residues on the molecular surface [20, 32]. Figure 4 illustrates the overall structure of Cyt1A97 and the position of Thr115 in the top of the αC helix. In fact, the comparison between Cyt1A97 and Cyt1A98 focussed on Met115 Thr region showed that Thr115, establishes, by its hydroxyl group, an interaction with the carbonyl group of Gln111 located in the boundary region between the sheet and the helix hairpin C-D (Fig. 4b). This interaction takes more importance when we know that conformational change in this region is important in the cytotoxic activity of both Cyt1A and CytB proteins [20]. Hence, any hindrance that affects this part of the protein will negatively affect its activity.
Discussion
In the present work, we characterized another cyt1Aa-type gene cloned from a newly isolated strain BUPM97 of B. thuringiensis israelensis [33]. Contrary to the published cyt1Aa-type gene of B. thuringiensis israelensis strains, the coding sequence of cyt1A97 is different by a substitution of Met115 by Thr (Fig. 1). Regarding the effect of the reported substitution in cyt1A97, modifications in toxicity and cytolytic activity of the parasporal crystal of BUPM97 were expected.
The predicted model of Cyt1A97 structure, using the server SWISS-MODEL which is based on the X-ray structure of Cyt2Aa1 [20], revealed no modification in the three-dimensional structure. The question thus arises of its possible mode of action in bacteria. The leading hypothesis of Cyt1Aa’s mode of action is that the toxin assembles into cation-selective channels in the membrane, through which ions equilibrate across the membrane, resulting in colloid-osmotic lysis and cell death. The protein is thought to partially insert into the membrane. Initial contact of Cyt1Aa with membranes involves electrostatic interactions of the charged amino acids, situated on its surface, with the lipid head groups. Electrostatic interactions between the charged amino acid residues and the lipid head groups are responsible for binding the protein to the membrane surface, while hydrophobic and/or van der Waals interactions make the membrane binding irreversible [34]. In helix-α C, only the role of three amino acids have been studied. K118 and E121 did not affect toxicity and lipid binding. K124 affects toxicity and lipid binding [22, 33]. Structural investigation of Cyt1A97 shows that Thr115 is located on the top of αC helix, pointing its side chain into the hydrophobic environment, made by the packaging of the hydrophobic residues of the helices against the β-sheet [20, 32]. As described by [20], results of the mutagenesis studies of Cyt1Aa imply a need for a conformational change in the membrane-bound toxin involving a hinge motion along the topological boundary between the β-sheet and the helix pair C-D. They proposed that the helices C-D are lifted off the sheet to lie on the surface of the membrane. This would expose the underlying hydrophobic face of the sheet and cause some part of the sheet to partition into the membrane core. The boundary region between the sheet and helix hairpin C-D is proposed to be the hinge for the conformational change that leaves the helices on the membrane surface while allowing part of the sheet to penetrate the hydrophobic zone of the membrane.
Comparison between Cyt1A97 and Cyt1A98 focussed on Met115 Thr region showed that contrarily to Met115, Thr115 would establish by its hydroxyl group an interaction with the carbonyl group of Gln111 located in the boundary region between the sheet and the helix hairpin C-D (Fig. 4b). Structural investigation of CytB and Cyt1Aa model shows without any doubt that Gln111 (Glu107 in CytB) is not the subject of any effect. This residue is positioned in the loop between β3 and αC, it is a solvent exposed residue, and it participates by its side chain to interact with water and with Ser113 (Ser106 in CytB). Due to steric consideration, the position of Gln111 cannot be altered, and then the interaction of the carbonyl group of Gln111 with the hydroxyl group of Thr115 may occur. This interaction is thought to be the reason of the inhibition of Cyt1A97 toxicity by disturbing C-D hinge motion. The loss of Cyt1A97 cytolytic activity was also confirmed by comparing the cytocidal activity, performed on HeLa cells culture, of the B. thuringiensis israelensis BUPM97 and BUPM98 parasporal crystals containing their respective Cyt proteins. The obtained results (data not shown) showed that BUPM98 solubilized crystals inhibited the HeLa cells growth but those of BUPM97 affected them weekly. These results demonstrate that Met at position 115 could play a key role in facilitating translocation of some parts of the protein through the membrane and is determinant for protein orientation and cytolytic activity on E. coli.
Abbreviations
- B :
-
Bacillus
- E :
-
Escherichia
- PCR:
-
Polymerase Chain Reaction
- Cyt:
-
Cytolytic toxin
- LB:
-
Luria-Bertani medium
References
Promdonkoy, B., Chewawiwat, N., Tanapongpipat, S., Luxananil, P., & Panyim, S. (2003). Cloning and characterization of a cytolytic and mosquito larvicidal d-endotoxin from Bacillus thuringiensis subsp. darmstadiensis. Current Microbiology, 46, 94–98.
Cheong, H., & Gill, S. S. (1997). Cloning and characterization of a cytolytic and mosquitocidal d-endotoxin from Bacillus thuringiensis subsp. jegathesan. Applied and Environmental Microbiology, 63, 3254–3260.
Juarez-Perez, V., Guerchicoff, A., Rubinstein, C., & Delecluse, A. (2002). Characterization of Cyt2Bc toxin from Bacillus thuringiensis subsp. medellin. Applied and Environmental Microbiology, 68, 1228–1231.
Drobniewski, F. A., & Ellar, D. J. (1988) Investigation of the membrane lesion induced in vitro by two mosquitocidal delta-endotoxins of Bacillus thuringiensis. Current Microbiology, 16, 195–199.
Koni, P. A., & Ellar, D. J. (1994) Biochemical characterisation of Bacillus thuringiensis cytolytic delta-endotoxins. Microbiology, 140, 1869–1880.
Schnepf, H. E., Crickmore, N., Van Rie, J., Lereclus, D., Baum, J., Feitelson, J., Zeigler, D.R., & Dean, D. H. (1998) Bacillus thuringiensis and its pesticidal crystal proteins. Microbiology and Molecular Biology Reviews, 62, 775–806.
Crickmore, N., Bone, E. J., Williams, J. A., & Ellar, D. J (1995). Contribution of the individual components of the δ-endotoxin crystal to the mosquitocidal activity of Bacillus thuringiensis subsp. israelensis. FEMS Microbiology Letters, 131, 249–254.
Wu, D., & Federici, B. A. (1993) A 20-kilodalton protein preserves cell viability and promotes CytA crystal formation during sporulation in Bacillus thuringiensis. Journal of Bacteriology, 175, 5276–5280.
Ibarra, J., & Federici, B. A. (1986). Isolation of a relatively non-toxic 65-kilodalton protein inclusion from the parasporal body of Bacillus thuringiensis subsp. israelensis. Journal of Bacteriology, 165, 527–533.
Wu, D., & Chang, F. N. (1985). Synergism in mosquitocidal activity of 26 and 65 kDa proteins from Bacillus thuringiensis subsp. israelensis crystal. FEBS Letters, 190, 232–236.
Georghiou, G. P., & Wirth, M. C. (1997). Influence of exposure to single versus multiple toxins of Bacillus thuringiensis subsp. israelensis on development of resistance in the mosquito Culex quinquefasciatus (Diptera: Culicidae). Applied and Environmental Microbiology, 63, 1095–1101.
Pérez, C., Fernandez, L. E., Sun, J., Folch, J. L., Gill, S. S, Sorberon, M., & Bravo, A. (2005). Bacillus thuringiensis subsp. israelensis Cyt1Aa synergizes Cry11Aa toxin by functioning as a membrane-bound receptor. Proceedings of the National Academy of Sciences of the United States of America, 102, 18303–18308.
Wirth, M. C., & Georghiou, G. P. (1997). Cross-resistance among CryIV toxins of Bacillus thuringiensis subsp. israelensis in Culex quinquefasciatus (Diptera: Culicidae). Journal of Economic Entomology, 90, 1471–1477.
Wirth, M. C., Georghiou, G. P., & Federici, B. A. (1997). CytA enables CryIV endotoxins of Bacillus thuringiensis to overcome high levels of CryIV resistance in the mosquito, Culex quinquefasciatus. Proceedings of the National Academy of Sciences of the United States of America, 94, 10536–10540.
Douek, J., Einav, M., & Zaritsky, A. (1992). Sensitivity to plating of Escherichia coli cells expressing the cytA gene from Bacillus thuringiensis var. israelensis. Molecular & General Genetics, 232, 162–165.
Ballesta, J. P., Cundliffe, E., Daniels, M. J., Silverstein, J. L., Susskind, M. M., & Schaechter, M. (1972). Some unique properties of the deoxyribonucleic acid-bearing portion of the bacterial membrane. Journal of Bacteriology, 112, 195–199.
Thomas, W. E., & Ellar, D. J. (1983). Bacillus thuringiensis var. israelensis crystal delta- endotoxin: Effects on insect and mammalian cells in vitro and in vivo. Journal of Cell Science, 60, 181–197.
Promdonkoy, B., & Ellar, D. J. (2003). Investigation of the pore forming mechanism of a cytolytic d-endotoxin from Bacillus thuringiensis. The Biochemical Journal, 374, 255–259.
Manceva, S. D., Pusztai-Carey, M., Russo, P. S., & Butko, P. (2005). A detergent-like mechanism of action of the cytolytic toxin Cyt1A from Bacillus thuringiensis var. israelensis. Biochemistry, 44, 589–597.
Li, J., Koni, P. A., & Ellar, D. J. (1996). Structure of the mosquitocidal delta-endotoxin CytB from Bacillus thuringiensis sp. kyushuensis and implications for membrane pore formation. Journal of Molecular Biology, 257, 129–152.
Butko, P. (2003). Cytolytic toxin Cyt1A and its mechanism of membrane damage: Data and hypotheses. Applied and Environmental Microbiology, 69, 2415–2422.
Gazit, E., & Shai, Y. (1993). Structural characterization, membrane interaction, and specific assembly within phospholipids membranes of hydrophobic segments from Bacillus thuringiensis var. israelensis cytolytic toxin. Biochemistry, 32, 12363–12371.
Jaoua, S., Zouari, N., Tounsi, S., & Ellouz, R. (1996). Study of the δ-endotoxins produced by three recently isolated strains of Bacillus thuringiensis. FEMS Microbiology Letters, 145, 349–354.
Zghal, R. Z., & Jaoua, S. (2006). Evidence of DNA rearrangements in the 128-kilobase pBtoxis plasmid of Bacillus thuringiensis israelensis. Molecular Biotechnology, 33, 191–198.
Guzman, L. M., Belin, D., Carson, M. J., & Beckwith, J. (1995). Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. Journal of Bacteriology, 177, 4121–4130.
Sambrook, J., Fritsch, E. F., & Maniatis, T. (1989). Molecular cloning. A laboratory manual, 2nd edn. Cold Spring Harbor NY: Cold Spring Harbor Laboratory.
Thompson, J. D., Higgins, D. G., & Gibson, T. J. (1994). The ClustalW: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Research, 22, 4673–4680.
Schwede, T., Kopp, J., Guex, N., & Peitsch, M. C. (2003). SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Research, 31, 3381–3385.
DeLano, W. L. (2002) The PyMOL Molecular Graphics System on World Wide Web http://www.pymol.org.
Chothia, C., & Lesk, A. M. (1986). The relation between the divergence of sequence and structure in proteins. The EMBO Journal, 5, 823–826.
Koni, P. A., & Ellar, D. J. (1993). Cloning and characterization of a novel Bacillus thuringiensis cytolytic delta-endotoxin. Journal of Molecular Biology, 229, 319–327.
Szabo, E., Murvai, J., Fabian, P., Fabian, F., Hollosi, M., Kajtar, J., Buzas, Z., Sajgo, M., Pongor, S., & Asboth, B. (1993). Is an amphiphilic region responsible for the haemolytic activity of Bacillus thuringiensis toxin? International Journal of Peptide and Protein Research, 42, 527–532.
Zghal, R. Z., Tounsi, S., & Jaoua, S. (2006). Characterization of a cry4Ba-type gene of Bacillus thuringiensis israelensis and evidence of the synergistic larvicidal activity of its encoded protein with Cry2A delta-endotoxin of B. thuringiensis kurstaki on Culex pipiens. Biotechnology and Applied Biochemistry, 44, 19–25.
Butko, P., Huang, F., Pusztai-Carey, M., & Surewicz, W. K. (1996) Membrane permeabilization induced by cytolytic delta-endotoxin CytA from Bacillus thuringiensis var. israelensis. Bichemistry, 35, 11355–11360.
Acknowledgements
We thank Mrs. Najeh Belguith-Ben Hassen for her technical assistance. This work was supported by grants from the Tunisian Ministère de l’Enseignement Supérieur de la Recherche Scientifique et de la Technologie et du développement des compétences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zghal, R.Z., Trigui, H., Ali, M.B. et al. Evidence of the Importance of the Met115 for Bacillus thuringiensis subsp. israelensis Cyt1Aa Protein Cytolytic Activity in Escherichia coli . Mol Biotechnol 38, 121–127 (2008). https://doi.org/10.1007/s12033-007-9015-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-007-9015-6