
Abstract
A new lipase gene designated as SlLipA was isolated from Serratia liquefaciens S33 DB-1 by the genomic-walking method. The cloned gene contained an open reading frame (ORF) of 1,845 bp encoding 615 amino acids with a conserved GXSXG motif. Genome sequence analysis showed that an aldo/keto reductase gene closed to the SlLipA gene. The lipase gene was cloned into the expression vector pPICZαA and successfully integrated into the heterologous host, methylotrophic yeast Pichia pastoris GS115. Five transformants could be expressed as secreted recombinant proteins with the high activity on Triglyceride–Agarose plate and as candidates to produce the recombinant enzyme. A C-terminal His tag was used for its purification. The lipase activity of different transformants against substrate para-nitrophenyl laurate (p-NPL) varied from 14 to 16 U ml−1. For the substrates para-nitrophenyl caprate (p-NPC), p-NPL, para-nitrophenyl myristate (p-NPM), para-nitrophenyl palmitate (p-NPP), and para-nitrophenyl stearate (p-NPS), the specific activity was shown to be preferred to long acyl chain length of p-NPS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipase (EC.3.1.1.3) is one of the five international commercial enzymes with maximum output and has broad industrial applications in food agents for modification of milkfat [1], improving flour baking quality [2], and machining oils [3]. Lipase can also be used in synthesis of ideal chemicals in enantioselective transesterification [4] and as carrier for drug delivery [5]. Recently, lipase has become more attractive for biodiesel fuel production by transesterification of oils [6]. Lipases usually display exquisite chemoselectivity, regioselectivity, and stereoselectivity, making them become attractive ideal tools in synthesis of organic chemicals. Thereby, it prompts a demand for diverse lipases from different organisms to expand not only their functionalities, but also their specific applications.
As one of genus Serratia, Serratia liquefaciens is frequently referred to causing the human nosocomial infections [7]. S. liquefaciens strain was studied as a candidate compound for the management of hypertriglyceridemia for producing a pharmacological effect of FR177391 due to its infectivity [8]. Until now, there have been few reports on lipases from S. liquefaciens. In 1978, one test on the lipase and phospholipase revealed that S. liquefaciens exhibited strong lipase, but only a minor phospholipase activity [9]. Yu et al. isolated a strain with the highest lipolytic activity, S. liquefaciens S33 DB-1, from 150 oil-contacted soil samples [10]. The strain showed extracellular alkaline lipase activity of lipolytic olive oil within the pH 7.0–9.0 at a temperature below 45°C [10]. Further investigation elucidated its capability and optimum condition of producing large amount of extracellular alkaline lipase under the induction of oils [11]. To my knowledge, there has not been any report about cloning of lipase gene from S. liquefaciens and expressing of the lipase by heterologous host. So it is a necessity to clone lipase gene from S. liquefaciens and to produce recombinant lipase for large-scale engineering from heterologous host. In this report, we chose S. liquefaciens S33 DB-1 strain to clone and functionally express its lipase gene in P. pastoris.
Materials and Methods
Materials
Restriction enzymes, T4-DNA ligase, Taq polymerase, and LA PCR in vitro Cloning kit were purchased from Takara (Japan). The DNA Gel-Extraction Kit, Mini Plasmid Kit and Yeast Genomic DNA Extraction Kit were from Watson (Shanghai, China). The different lipase substrates including para-nitrophenyl caprate (p-NPC), para-nitrophenyl laurate (p-NPL), para-nitrophenyl myristate (p-NPM), para-nitrophenyl palmitate (p-NPP), para-nitrophenyl stearate (p-NPS), and bovine serum albumin (BSA) as a standard of protein concentration, were all from Sigma (St. Louis, USA). Trypton, peptone, yeast extract, and yeast nitrogen base were from Amresco (Solon, OH, USA). The antibiotic zeocin was supplied by Invitrogen (CA, USA). All reagents were of analytical grade unless otherwise stated.
Strains, Plasmids, and Growth Conditions
Escherichia coli strain TOP10F′ (Invitrogen, CA, USA) was used for vector construction work and Pichia pastoris strain GS115 (his4) (Invitrogen, CA, USA) for lipase expression. E. coli was grown in low salt LB-zeocin medium (10 g tryptone, 5 g yeast extract, 5 g NaCl, and 25 mg zeocin in 1 l water, pH 7.5).
P. pastoris was grown in YPD medium (10 g yeast extract, 20 g peptone, and 20 g dextrose/l) or BMGY medium (10 g yeast extract, 20 g peptone, 13.4 g yeast nitrogen base, 0.4 mg biotin, 10 ml glycerol, and 100 ml 1 M K2HPO4/KH2PO4, pH 6.0 per l). BMMY medium (10 g yeast extract, 20 g peptone, 13.4 g yeast nitrogen base, 0.4 mg biotin, 5 ml methanol, and 100 ml 1 M K2HPO4/KH2PO4, pH 6.0 per l) was used for induction. For selection of transformants, YPDS-zeocin plates were used (182.2 g sorbitol, 20 g agar, 20 g dextrose, 10 g yeast extract, 20 g peptone, 100 mg zeocin/l). MDH (13.4 g yeast nitrogen base, 0.4 mg biotin, 20 g dextrose, 15 g agar, and 4 g histidine/l) and MMH (13.4 g yeast nitrogen base, 0.4 mg biotin, 5 ml methanol, 15 g agar, and 4 g histidine/l) plates were used to confirm the Mut+ (Methanol utilization plus) phenotype.
S. liquefaciens S33 DB-1 was kindly supplied by Dr. Wenxin Zou (Department of Biology, Nanjing University, Nanjing, P R China).
Plasmid pGEM T-easy vector (Promega, Madison, WI, USA) and pPICZαA vector (Invitrogen, CA, USA), were used for cloning and expression, respectively.
Gene Amplification and Sequencing
Genomic DNA was isolated from S. liquefaciens S33 DB-1 with bacterial genomic DNA isolation kit from Watson (Shanghai, China) according to the manufacturer’s protocol and was used as templates for PCR amplification of the conserved region of lipase from S. liquefaciens. Two degenerate oligonucleotide primers, SlLipF (5′-AT(C/T)GC(A/C/T/G)TTTCGCGGCACCAG-3′), SlLipR (5′-TCGTTGAA(G/A)(A/C/T/G)T(G/A)ACGATGTTGT-3′), were designed according to the conserved sequences of other lipase genes and used for the amplification of the core cDNA fragment of SlLipA by standard gradient PCR amplification (from 52 to 60°C) on ThermoHybaid PX2 Thermocycler (Hybaid, UK). Subsequently, amplified at the annealing temperature of 56°C, the core fragment was subcloned into pGEM T-easy vector (Promega, Madison, WI, USA), and transformed into E. coli strain DH5α followed by sequencing. The core fragment was subsequently used to design the gene-specific primers for cloning the full-length DNA of SlLipA by genomic walking.
LA PCR™ in vitro Cloning Kit (TaKaRa, Japan) was used to clone the 3′- and 5′-flanking regions of SlLipA gene. The genomic DNA was completely digested by an appropriate enzyme BamHI, and the digested DNA fragments were ligated to the BamHI cassettes according to the manufacture’s instructions. The ligated DNA fragments with BamHI cassettes were used as the templates for amplification of 3′- and 5′-flanking regions of SlLipA gene.
For the amplification of 3′- and 5′-flanking regions of SlLipA gene, two 3′- and 5′-gene-specific primers were designed. Cassette primers (C1 and C2) were provided by the kit. For the first amplification of 3′-flanking region of SlLipA gene, SlLip3-1 (5′-TGCTGGCCGGTTTCGGCCCGAATGGCTATGCC-3′) and Cassette primer C1 (5′-HOGTACATATTGTCGTTAGAACGCGTAATACGACTCA-3′) were used. For the nested amplification of 3′-flanking region, primers SlLip3-2 (5′-CCAACTATGTCGCCTTTGCTTCTCCTACCCAA-3′) and Cassette primer C2 (5′-HOCGTTAGAACGCGTAATACGACTCACTATAGGGAGA-3′) were used with the diluted products of the first amplification as templates. Two nested specific primers SlLip5-1 (5′-GGCATAGCCATTCGGGCCGAAACCGGCCAGCA-3′) and SlLip5-2 (5′-AGAGATTCGCGCGGGCCGCTGGTGCCGCGAAA-3′) were used for nested PCR amplification of the 5′-flanking region of SLLipA gene with Cassette primer C1 and C2. The first and nested PCRs were carried out under the conditions described in the protocol (LA PCR™ in vitro Cloning Kit, User Manual, TaKaRa): 30 cycles (30 s at 94°C, 2 min at 55°C, 2 min at 72°C). The nested 3′- and 5′-flanking region products were purified and subcloned into pGEM T-easy vector followed by sequencing.
By aligning and assembling the sequences of the core fragment, 3′- and 5′-flanking region products on Contig Express (Vector NTI Suite 8.0), the coding sequence of SlLipA gene was deduced, amplified by using genome DNA as the template and a pair of primers, SlLipF-ORF (5′-ATGGGAATCTTTAATTATCAAGGT-3′) and SlLipR-ORF (5′-GGCCAGTACCACTTGGCCGTCCG-3′). The amplified product was subsequently cloned into pGEM T-easy vector (Promega, Madison, WI, USA), transformed into E. coli strain DH5α followed by sequencing. The PCR amplification and sequencing of the ORF of SlLipA gene were repeated thrice to avoid PCR errors.
Recombinant Vector Construction and Transformation
A pair of primers containing PstI and NotI digesting sites, SlLipA-F-PstI (5′-AACTGCAGAACCAATGGGAATCTTTAATTATCAAGGT-3′) and SlLipA-R-NotI (5′-AAGGAAAAAAGCGGCCGCGGCCAGTACCACTTGGCCGTCCG-3′), were designed and used to amplify the ORF region using the cloned ORF fragment as the template. PCR product was digested with PstI and NotI, and ligated into the vector pPICZαA that was digested with the same enzymes, to generate the expression vector pPICZαA-SlLipA. After transformation of E. coli TOP10F′ with pPICZαA-SlLipA, zeocin-resistant clones were selected and analyzed by sequencing to confirm the correct fusion of SlLipA coding gene to the α-factor secretion signal.
The plasmid was linearized with SacI and electroporated by Bio-Rad MicroPulse (Hercules, CA, USA) into electrocomponent P. pastoris cells (GS115) before the host cells were spread on YPDS-zeocin plates and incubated at 30°C for 2 days. MDH and MMH plates were used to confirm the Mut+ phenotype for metabolizing methanol as the sole carbon source to induce expression of the alcohol oxidase gene (AOX1) that was used to drive expression of the gene of interest encoding the desired heterologous protein [12, 13].
Genomic PCR was used to determine whether the heterologous gene integrated into the yeast genome. The PCR template genomic DNA was isolated according to the procedure of Yeast Genomic DNA Extraction Kit (Watson, Shanghai, China).
Enzyme Screening
Screening of P. pastoris transformants for enzyme secretion was performed using Rhodamine–Triglyceride–Agarose Assay following the procedure [14] to drop 5 μl supernatants after induction by methanol into the Rhodamine–Triglyceride–Agarose plate by a pricher. Enzyme in supernatants hydrolyzed triglyceride to form a clear transparent halo around the pinhole by incubation at 37°C for about 8 h. The SlLipA clones with high hydrolyzing activity toward triglyceride were confirmed according to the size of transparent halo on the agar plate by addition of rhodamine B.
Expression of Recombinant Lipase and Cultivation
Transformants with large hydrolytic cycle were inoculated in 50 ml BMGY medium in a 500 ml baffled flask to an optical density (OD) of 3–4 at 30°C with 250-rpm agitation. The cells were harvested and resuspended in 50 ml BMMY medium to reach OD1.0 for inducing gene expression, growing at 30°C with 250-rpm agitation. Methanol was added to the culture medium to a final concentration of 0.5% every 24 h for the next 3 days to maintain induction. An aliquot of the expression culture was harvested at different time point to analyze expression levels and the optimal time of post-induction.
The culture was chilled rapidly to 4°C, and then the cells were removed by centrifugation. The crude enzyme was precipitated from the culture supernatants by 10% trichloroacetic acid, washed with ice acetone, lyophilized at −50°C and then analyzed by SDS-PAGE [15]. The collected total protein samples of different time courses were analyzed by SDS-PAGE (Bio-Rad, Mini-PROTEAN) on a 10% polyacrylamide gel. The band intensities were determined by Coomassie Blue-stained gel.
Purification of Protein
The purification of protein was carried out using ProBond™ Purification System (Invitrogen, CA, USA) according to the manufacturer’s instructions. The precipitated and lyophilized total protein sample from 50 ml culture supernatants was resuspended in 8 ml Native Binding Buffer (50 mM NaH2PO4, pH 8.0, 0.5 M NaCl), which was then added to the previously equilibrated purification column packed with ProBond™ nickel-chelating resin. The purified protein was eluted with 8 ml Native Elution Buffer (250 mM imidazole, 50 mM NaH2PO4, pH 8.0, and 0.5 M NaCl). The eluted fraction was concentrated to analyze with SDS-PAGE. Protein concentration was determined by the method of Bradford with BSA as a standard [16].
Enzyme Activity and Specificity Assay
Lipase activity was measured by a spectrophotometric method with p-NPL as substrate [17]. The assay mixture contained 44.5 mM Tris–HCl buffer, pH 7.5, and 2.5 mM substrate solution p-NPL that was dissolved in dimethyl sulfoxide (DMSO) before use, and 10 μM of enzyme solution for 10 min at about 25°C room temperature. The enzyme reaction was stopped by adding equal volume of ethanol. The A 405 of liberated p-nitrophenol was measured with p-nitrophenol as a standard. One unit was defined as the mount of enzyme required to release 1 μmol of p-nitrophenol per min under assay conditions [18].
Substrate specificity of SlLipA by a spectrophotometric method toward different p-nitrophenyl esters (pNP-) was determined with p-NPC, p-NPL, p-NPM, p-NPP and p-NPS as substrates. Cleavage of pNP- was measured at room temperature, and reading the corresponding absorbance at 405 nm [19].
Results and Discussion
Cloning and Analysis of Lipase Gene from S. liquefaciens S33 DB-1
On the basis of conserved region of other lipase sequences, two degenerate oligonucleotide primers (SlLipF and SlLipR) were designed and used for gradient PCR amplification of the core cDNA fragment of lipase from S. liquefaciens S33 DB-1. An approximately 500 bp product was amplified and subcloned, followed by sequencing. DNA sequence BLAST search revealed this 461 bp DNA fragment shared over 80% homology with S. marcescens lipase gene, implying that it was probably a partial sequence of lipase gene. Thus this fragment was used to design gene specific primers for both 3′- and 5′-flanking regions of SlLipA.
By 3′- and 5′-genomic walking for amplification, the 1,719 and 627 bp nested PCR products were respectively obtained. Sequencing results revealed that the products were the 562 bp 3′-flanking region and 178 bp 5′-flanking region of SlLipA, respectively. After assembling and analyzing the two sequences and the core fragment on Contig Express (Vector NTI Suite 8.0), ORF Finding analysis on NCBI showed that the genomic fragment contained a 1,845 bp ORF, encoding a protein of 615 amino acids with a calculated molecular mass of 64.85 kDa and the predicted isoelectric point of 4.31 (http://cn.expasy.org/tools/protparam.html). A Shine-Dalgarno sequence, AAGGAA, was found six bases upstream of the ATG start codon (Fig. 1). Confirmed by amplifying and sequencing, ORF of the sequence was analyzed in NCBI for BLAST searching (http://www.ncbi.nlm.nih.gov/BLAST/) and the result showed that SlLipA shared 74% homology with SmLipA (Acc. No. DQ841349). Moreover, the previous research reported that S. proteamaculans and S. liquefaciens had been thought to be synonymous on the basis of DNA relatedness [20]. The SlLipA ORF sequence has been deposited at GenBank under accession number EF202840. In addition, sequence analysis by PSI-BLAST searching (http://www.ncbi.nlm.nih.gov/BLAST/) showed that there was a putative aldo/keto reductase gene in the reverse complemented sequence from 2,085 to 2,588 at 61 bases downstream from the stop codon of 1,845 bp SlLipA ORF.

Nucleotide sequence of a 2.5-kb DNA fragment from S. liquefaciens (Sl) containing both the ORF coding for lipase and the partial CDS for aldo/keto reductase. The start codon (ATG) for lipase was underlined in bold and the stop codon (TGA) was underlined italically in bold. The putative ribosome-binding site was doubly underlined in bold. The reverse complemented sequence was from 2,085 to 2,588 for coding partial aldo/keto reductase. The sequential coding was showed in arrowhead. The sequence data has been deposited at GenBank under the accession number EF202840
Analysis of the deduced amino acid sequence for the reverse complemented sequence from 2,085 to 2,588 by PSI-BLAST searching (http://www.ncbi.nlm.nih.gov/BLAST/) revealed that it coded partial aldo/keto reductase near C-terminus, suggesting that the gene sequence of SlLipA lipase was flanking an aldo/keto reductase gene in the genome of S. liquefaciens (Fig. 1).
Analysis of the deduced amino acid sequence of SlLipA for PSI-BLAST searching (http://www.ncbi.nlm.nih.gov/BLAST/) revealed that SlLipA shared 87% sequence identity with the lipase (Acc. No. ZP_01534977) from S. proteamaculans 568 and about 70% sequence identity with the lipase (Acc. No. ABI83633) from S. marcescens. It suggests that SlLipA belongs to the lipase family (Fig. 2). Simultaneously, SlLipA had 60% lower identity with other lipases, putative lipases from Pseudomonas fluorescens (Acc. No. AAT48728), and other Pseudomonas sp.

Multiple alignment of amino acid sequences of S. liquefaciens lipase (SlLip, Acc. no.: EF202840) and other bacterial lipases. Lipase (Acc. No. ZP_01534977) from Serratia proteamaculans 568 (SPLip), lipase (Acc. No. ABI83633) from Serratia marcescens (SMLip), and lipase (Acc. No. AAT48728) from Pseudomonas fluorescens (PFLip) were compared using CLUSTALW program. The identical amino acids were showed in white with black background and the conserved amino acids were showed in white with gray background. The conserved active-center sequence of lipases, -Gly-X1-Ser-X2-Gly-, was underlined
According to the sequence conservation characteristic, lipases could be divided into GXSXG and GDSL families. The GXSXG lipases are usually named as “classical” lipases, and exist in the majority of bacteria and eukaryotes. Serine residue of the highly conserved Gly-X-Ser-X-Gly motif is a key element of the active site of fatty acid-deesterifying lipases [21, 22]. The amino acid sequence derived from SlLipA nucleotide sequence contained GXSXG motif. On the basis of characteristics possessed by GXSXG family of lipases, SlLipA was predicted to belong to GXSXG family.
Transformation in P. pastoris and Selection of Positive Transformants
For secretive expression, the gene sequence encoding SlLipA was directly fused in frame to the α-factor prepro signal sequence from Saccharomyces cerevisiae to target the protein to the secretory pathway, and placed under the control of methanol inducible alcohol oxidase gene (AOX1) [23, 24]. One hundred positive transformants were selected on solid selective medium (YPD containing zeocin), confirmed by genomic PCR, and induced by BMMY medium. Five transformants of expressing active lipase were determined by transparent halo on Rhodamine–Triglyceride–Agarose plate performed by adding 5 μl induced culture supernatant (Fig. 3). The culture supernatants of positive lipase colony could hydrolyze triglyceride, and a transparent halo appeared in the triglyceride-containing plate.

Rhodamine B-triglyceride agar plate showing different recombinant clones. A 5-μl volume of culture supernatant (sterile filtered) induced by methanol was poured into each preformed hole, and the plate was incubated at 30°C overnight. Transparent halos around the holes (strains labeled A, B, C, D, and E) indicate the production of recombinant lipase
The expression system of recombinant proteins using pPIC and pPICZ in P. pastoris has many advantages for scale-up expression of recombinant proteins [23, 25–29]. P. pastoris, a methylotrophic yeast, could metabolize methanol as its sole carbon source, which was a major advantage to induce the expression of heterologous proteins. There are generally two approaches to add methanol for screening positive colony on plate containing emulsified triglyceride. One was to drop methanol onto the lid of plate, and then invert the plate to induce protein expression of positive colony at 28°C [14, 30, 31]. Another classical approach is to add the culture supernatant after induction by methanol into the hole of plate to observe at 37°C. One disadvantage of the former approach was that the colony could be diluted by volatilization of methanol. In this study, we used the latter approach to screen positive colony using 5 μl of the induced culture supernatant.
Cell Cultivation and SDS-PAGE
Transformants possessing lipolytic activity, as indicated by the formation of a clear halo on triglyceride plates, were selected and grown in shaking flasks containing 50 ml BMGY medium. After the cultures reached an OD600 of 3–4, they were centrifuged and resuspended in 50 ml BMMY medium for induction of lipase expression. The cell density and lipase activity were measured as a function of time after induction with 0.5% methanol (Fig. 4). At the start of induction phage, a high increase of cell density was measured. There is a lag in activity following the rapid increase in cell density, probably due to the remaining glycerol from the cell pellet. After 12 h of post-induction, the lipolytic activity of supernatants was determined. The lipase activity increased steadily, even after 84 h of induction with methanol. The lipase activity of different clones against substrate p-NPL varied between 14–16 U ml−1 in batch after induction of 48 h. No lipase activity was found before induction and in the negative control.

Time course study of SlLipA production. The cell density (●) measured as a function of cultivation time at 600 nm. The lipase activity (Δ) during cultivation in μmol min−1 ml−1 using p-nitrophenyl laurate as substrate. The results are the average of duplicate assays performed in two independent experiments
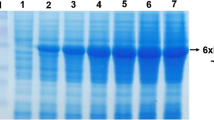
SDS-PAGE analysis of different time-course concentrated samples showed a protein band near the predicted size of 68 kDa with 6× His-tag and c-myc epitope. The protein band became thicker from 48 to 72 h (Fig. 5), demonstrating that the secreted recombinant protein was stable in the medium. The arrow shows the position of the expressed lipase protein.

SDS-PAGE analysis of total secreted protein after induction with 0.5% methanol. Culture supernatants were harvested at various time intervals, precipitated by 10% trichloroacetic acid, washed with acetone, freezingly dried, and then analyzed by SDS-PAGE. The band intensities of Coomassie Blue-stained gel were determined. Lane numbers, 2, 3, 4, and 5 indicate induction time (h) 24, 48, 72, and 96, respectively, after induction by 0.5% of methanol. Lane 1, before induction; Lane M, middle molecular weight protein markers (Watson, Shanghai, China)
Purification of Protein
SlLipA was expressed with 6×His-tag, which was a most convenient way to purify the recombinant protein. ProBond™ nickel-chelating resin used the characteristics of chelating ligand iminodiacetic acid (IDA) that could both bind Ni2+ ions by three coordination sites and highly cross-link agarose matris. On the basis of high affinity and selectivity of ProBond™ nickel-chelating resin for 6× His-tagged recombinant fusion protein, SlLipA recombinant protein was purified using the ProBond™ nickel-chelating resin. The crude protein was loaded onto a Ni2+-IDA column to remove most of the contaminants. The eluted fractions contained almost one single band with a size of approximately 68 kDa, which referred to the size of the cloned SlLipA with fusion of the His tag and c-myc epitope. The molecular mass of purified protein was lower than that of crude protein, which was probably caused by the removal of many modifications from protein in the process of purification. After concentrating the protein preparations, the actual concentration of the purified lipases was determined by measuring the absorbance at 595 nm. The purified protein was analyzed by SDS-PAGE (Fig. 6). The arrow shows the position of the expressed lipase protein.

SDS-PAGE analysis of the purified SlLipA protein (lane L). Lane M showed middle molecular weight protein markers (Watson, Shanghai, China)
Enzymic Specificity Activity Assays
Lipase activity and substrate specificity were usually measured by a spectrophotometric method with p-nitrophenyl esters as substrates [17]. Substrate activity and specificity of recombinant enzyme toward various p-nitrophenyl esters differing in chain length of the acyl moleity from C10 to C18 was studied in this report. As shown in Fig. 7, the recombinant lipase preferred middle acyl group p-NPL (C12) and p-NPM (C14) to p-NPC (C10), but showed the highest activity toward p-NPS (C18). The recombinant lipase shows similarities to Streptococcus and Bacillus lipases, in preference, long chain substrates to short chain substrate [32]. The lipase exhibits preference to long chain substrate, which will make the enzyme an excellent candidate for biocatalytic applications, such as synthesis of specific chemicals and food agents for improving flavor.

Relative specific activity of lipase toward p-nitrophenyl esters. The substrates para-nitrophenyl caprate (p-NPC), para-nitrophenyl laurate (p-NPL), para-nitrophenyl myristate (p-NPM), para-nitrophenyl palmitate (p-NPP), and para-nitrophenyl stearate (p-NPS) were respectively denoted with the acyl chain length of C10, C12, C14, C16, and C18. Each value is the average of three experiments with standard deviation ≤10
With methanol as substrate, the recombinant lipase SlLipA did not have transesterification activity against plant oil in the non-polar organic solvent of n-heptane, suggesting that SlLipA preferred cleavage of esters to transesterification (data not shown).
Conclusions
This is the first report on cloning and expression of a S. liquefaciens lipase in P. pastoris. The useful characteristics of this enzyme are that these show preference to long chain p-NPS substrate, which makes the lipase a specific candidate for biocatalytic applications, such as synthesis of specific chemicals and food agents for improving flavor. Further investigations of SlLipA including methanol feeding rates and non-toxic methanol concentration are required to optimize the product yield.
References
Balcao, V. M., & Malcata, F. X. (1998). Lipase catalyzed modification of milkfat. Biotechnology Advances, 16, 309–341.
Keskin, S. O., Sumnu, G., & Sahin, S. (2004). Usage of enzymes in a novel baking process. Nahrung, 48, 156–160.
Jaeger, K. E., & Eggert, T. (2002). Current opinion in biotechnology. Lipases for Biotechnology, 13, 390–397.
Matsumoto, T., Ito, M., Fukuda, H., & Kondo, A. (2004). Enantioselective transesterification using lipase-displaying yeast whole-cell biocatalyst. Applied Microbiology and Biotechnology, 64, 481–485.
Barbara, F. H. (1995). Liposomes offer hope as medical tools. Information, 6, 793–802.
Fukuda, H., Kondo, A., & Noda, H. (2001). Biodiesel fuel production by transesterification of oils. Journal of Bioscience and Bioengineering, 92, 405–416.
Stock, I., Burak, S., Sherwood, K. J., Grüger, T., & Wiedemann, B. (2003). Natural antimicrobial susceptibilities of strains of ‘unusual’ Serratia species: S. ficaria, S. fonticola, S. odorifera, S. plymuthica and S. rubidaea. Journal of Antimicrobial Chemotherapy, 51, 865–885.
Inami, M., Kawamura, I., Tsujimoto, S., Yasuno, T., Lacey, E., Hirosumi, J., et al. (2005). FR177391, a new anti-hyperlipidemic agent from Serratia. II. Pharmacological activity of FR177391. Journal of Antibiotics (Tokyo), 58, 640–647.
Legakis, N. J., Nicolas, K. J., Xilinas, M., & Papavassiliou, J. (1978). Differentiation of Serratia marcescens and Serratia liquefaciens by tests for lipase and phospholipase production. Journal of Medicinal Microbiology, 11, 225–231.
Zou, W. X., Liu, H., & Yu, W. H. (1996). Isolation of lipase-producing Serratia liquefaciens and characterization of its lipase. Journal of Nanjing University (Natural Science), 32, 713–716.
Liu, H., Zou, W. X., & Yu, W. H. (1995). Studies on the extracellular lipase production of Serratia liquefaciens S33 DB-1. Microbiology (China), 22, 343–346.
Ellis, S. B., Brust, P. F., Koutz, P. J., Waters, A. F., Harpold, M. M., & Gingeras, T. R. (1985). Isolation of alcohol oxidase and two other methanol regulatable genes from the yeast Pichia pastoris. Molecular and Cell Biology, 5, 1111–1121.
Koutz, P. J., Davis, G. R., Stillman, C., Barringer, K., Cregg, J. M., & Thill, G. (1989). Structural comparison of the Pichia pastoris alcohol oxidase genes. Yeast, 5, 167–177.
Jette, J. F., & Ziomek, E. (1994). Determination of lipase activity by a Rhodamine–Triglyceride–Agarose assay. Analytical Biochemistry, 219, 256–260.
Lin, S. F., Chiou, C. M., Yeh, C. M., & Tsai, Y. C. (1996). Purification and partial characterization of an alkaline lipase from Pseudomonas pseudoalcaligenes F-111. Applied and Environment Microbiology, 62, 1093–1095.
Bradford, M. M. (1976). A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye-binding. Analytical Biochemistry, 72, 248–254.
Winkler, U. K., & Stuckmann, M. (1979). Glycogen, hyaluronate, and some other polysaccharides greatly enhance the formation of exolipase by Serratia marcescens. Journal of Bacteriology, 138, 663–670.
Rủa, M. L., Schmidt-Dannert, C., Wahl, S., Sprauer, A., & Schmid, R. D. (1997). Thermoalkalophilic lipase of Bacillus thermocatenulatus: Large scale production, purification and properties, aggregation behaviour and its effect on activity. Journal of Biotechnology, 56, 89–102.
Schmidt-Dannert, C., Sztajer, H., Stocklein, W., Menge, U., & Schmid, R. D. (1994). Screening, purification and properties of a thermophilic lipase from Bacillus thermocatenulatus. Biochimica et Biophysica Acta, 1214, 43–53.
Grimont, P. A. D., Grimont, F., & Irino, K. (1982). Biochemical characterization of Serratia liquefaciens sensu stricto, Serratia proteamaculans, and Serratia grimesii sp. nov. Curr. Microbiology, 7, 69–74.
Brick, D. J., Brumlik, M. J., Buckley, J. T, Cao, J. X., Davies, P. C., Misra, S., et al. (1995). A new family of lipolytic plant enzymes with members in rice, arabidopsis and maize. FEBS Letter, 377, 475–480.
Upton, C., & Buckley, J. T. (1995). A new family of lipolytic enzyme. Trends in Biochemical Sciences, 20, 178–179.
Cregg, J. M., Vedvick, T. S., & Raschke, W. C. (1993). Recent advances in the expression of foreign genes in Pichia pastoris. Bio/Technology, 11, 905–910.
Scorer, C. A., Buckholz, R. G., Clare, J. J., & Romanos, M. A. (1993). The intracellular production and secretion of HIV-1 envelope protein in the methylotrophic yeast Pichia pastoris. Gene, 136, 111–119.
Buckholz, R. G., & Gleeson, M. A. G. (1991). Yeast systems for the commercial production of heterologous protein. Bio/Technology, 9, 1067–1072.
Cregg, J. M., & Higgins, D. R. (1995). Production of foreign proteins in the yeast. Pichia pastoris. Canadian Journal of Botany Supplement, 73, 5981–5987.
Nico-Farber, K., Harder, W., Ab, G., & Veenhuis, M. (1995). Review: Methylotrophic yeasts as factories for the production of foreign proteins. Yeast, 11, 1331–1344.
Sreekrishna, K., Potenz, R. H. B., Cruze, J. A., McCombie, W. R., Parker, K. A., Nelles, L., et al. (1988). High level expression of heterologous proteins in methylotrophic yeast Pichia pastoris. Journal of Basic Microbiology, 28, 265–278.
Wegner, G. H. (1990). Emerging applications of the methylotrophic yeasts. FEMS Microbiology Review, 87, 279–284.
Jiang, Z. B., Luo, Y., Wang, G., Wang, H. P., Ma, Y. S., & Wei, D. Z. (2005). Cloning and expression of a novel lipase gene from Pseudomonas fluorescens B52. Molecular Biotechnology, 31, 95–102.
Li, X. Y., Tetling, S., Winkler, U. K., Jaeger, K. E., & Benedik, M. J. (1995). Gene cloning, sequence analysis, purification, and secretion by Escherichia coli of an extracellular lipase from Serratia marcescens. Applied and Environment Microbiology, 61, 2674–2680.
Tripathi, M. K., Roy, U., Jinwal, U. K., Jain, S. K., & Roy, P. K. (2004). Cloning, sequencing and structural features of a novel Streptococcus lipase. Enzyme Microbial Technology, 34, 437–445.
Acknowledgments
We are grateful to Dr. Wenxin Zou for kindly providing S. liquefaciens S33 DB-1 strain, and we thank China National “863” High-Tech Program, China Ministry of Education and Shanghai Science and Technology Committee for fund support under Project No. 2006AA020202.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yao, H., Yu, S., Zhang, L. et al. Isolation of a Novel Lipase Gene from Serratia liquefaciens S33 DB-1, Functional Expression in Pichia pastoris and its Properties. Mol Biotechnol 38, 99–107 (2008). https://doi.org/10.1007/s12033-007-9007-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-007-9007-6