
Abstract
Ovarian cancer is one of the most lethal gynecological cancers; owning to its late detection and chemoresistance, understanding the pathogenesis of this malignant tumor is much critical. Previous studies have reported that ubiquitin-specific peptidase 39 (USP39) is generally overexpressed in a variety of cancers, including hepatocellular carcinoma, gastric cancer and so forth. Furthermore, USP39 is proved to be associated with the proliferation of malignant tumors. However, the function and mechanism of USP39 in ovarian cancer have not been elucidated. In the present study, we observed that USP39 was frequently overexpressed in human ovarian cancer and was highly correlated with TNM stage. Suppression of USP39 markedly inhibited the growth and migration of ovarian cancer cell lines HO-8910 and SKOV3 and induced cell cycle G2/M arrest. Moreover, knockdown of USP39 inhibited ovarian tumor growth in a xenograft model. In addition, our findings indicated that cell cycle arrest induced by USP39 knockdown might be involved in p53/p21 signaling pathway. Furthermore, we found that the depletion of USP39 inhibited the migration of ovarian cancer cells via blocking epithelial–mesenchymal transition. Taken together, these results suggest that USP39 may play vital roles in the genesis and progression and may serve as a potential biomarker for diagnosis and therapeutic target of ovarian cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As the second most common cancer among gynecological malignancies, ovarian cancer remains one of the most frequent causes of death in female worldwide [1, 2]. Since malignant ovarian tumor normally grows in secluded places and the early symptoms of ovarian malignant tumors are not apparent, most of ovarian cancer patients are diagnosed at an advanced stage with the presentation of metastasis spreading beyond the ovary [3]. Owning to the lack of simple and practical diagnostic method, the treatment effects of ovarian cancer are restricted severely. Therefore, it is necessary to study the molecular mechanisms of ovarian cancer and to seek a reliable diagnostic method. Besides, screening out a novel biomarker for prognostic prediction and looking for a molecular therapeutic target would be more promising in the treatment of advanced ovarian cancer.
Ubiquitin-specific peptidase 39 (USP39), which contains a central zinc finger and two ubiquitin C-terminal hydrolase (UCH) domains, belongs to deubiquitylation family ubiquitin-specific proteases (USPs) [4]. Nevertheless, it is deprived of ubiquitin-specific peptidase activity due to the absence of the catalytic active amino acid residues [5, 6]. Besides, USP39 is well known as a 65-kDa serine/arginine (SR)-related protein, for its N-terminal domain which is rich in arginine, serine and glutamic acids, similar to the RS domain of SR-related proteins, which is essential to recruit the tri-snRNP to the pre-spliceosome to form a mature spliceosome [7]. Previous research showed that SR-related proteins (SRSF1, SRSF3, SRSF5, etc.) were often overexpressed in ovarian cancer and closely related to proliferation, metastasis and prognosis of the malignancy [8,9,10]. Researchers have proved when SRSF1 was stimulated in ovarian cancer cells, it can be transported to the nucleus and affected the expression of p53 by regulating the splicing of p53 pre-mRNA, and the dysfunction of splicing of pre-mRNA thereby leads to the deletion mutation of p53 [9].
Recently, many effective researches have been implemented aiming at the functions of USP39 in the genesis and development of malignancy. Researchers identified that USP39 may act as a new factor to maintain the spindle checkpoint and support successful cytokinesis, probably through regulating splicing of Aurora B and other mRNAs [5]. Wang et al. demonstrated that high level of USP39 was present in human breast tumor cells and regulated the cell survival and proliferation [11]. Pan et al. found that suppression of USP39 inhibited cell proliferation and colony formation of human hepatocellular carcinoma cells along with cell cycle arrested at G2/M phase [12]. Other research discovered that knockdown of USP39 significantly decreased medullary thyroid carcinoma cells’ proliferation and induced cell cycle arrested at G2/M phase, along with the down-regulation of G2/M phase-associated proteins, such as cyclin B1 and CDK1 [13]. In addition, USP39 was proved to be a target of SUMOylation specific protein (SENP), and overexpression of USP39 could enhance the proliferation of prostate cancer PC3 cells [14]. These findings suggested that USP39 may act as an essential oncogenic factor and play important roles in various cancers. However, the potential function and molecular mechanism of USP39 in ovarian cancer remain unclear.
The objection of this project was to explore the biological functions of USP39 in human ovarian cancer both in vivo and in vitro, to identify the molecular target of USP39 in HO-8910 and SKOV3 cell lines and to uncover the potential mechanism of USP39 which promotes ovarian cancer development, in an attempt to provide a novel perspective and theoretical basis for clinical early diagnosis and therapy of ovarian cancer.
Materials and methods
Reagents and antibodies
Roswell Park Memorial Institute-1640 (RPMI-1640), Dulbecco’s modified eagle medium (DMEM), fetal bovine serum (FBS), trypsin/EDTA, and penicillin streptomycin were purchased from GIBCO (Grand Island, NY, USA); human TGF-β-Mammalian was purchased from PeproTech (Rocky Hill, USA). Pierce BCA Protein Assay Kit and RevertAid First Strand cDNA Synthesis kit were purchased from Thermo scientific (Rockford, USA); 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) and propidium iodide (PI) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
The following antibodies were used:
USP39 (ab131244) and CDK1 (ab32094) were purchased from Abcam; β-actin (A1978) was purchased from Sigma; slug (9585), β-catenin (8480) and cyclin B1(12231) were purchased from Cell signaling Technology; P53 (sc47698), P21 (sc-136020), P27(sc-71813), E-cadherin (sc-71009) and N-cadherin (sc-59987) were purchased from Santa Cruz.
Immunohistochemical analysis
Detect the expression level of USP39 protein by immunohistochemistry staining on microarray tissue of human ovarian cancer with scoring procedure according to staining intensity and the extent of straining, to count and analyze the distribution of USP39 in human ovarian cancer. Diaminobenzidine was used to visualize the immunohistochemical reaction, followed by counterstaining with hematoxylin. The immunohistochemistry (IHC) staining on tissue array was analyzed through standard light microscopy. Positive cells showed brown granules in the nucleus or cytoplasm. The positive cell percentage was determined by calculating the percentage of positive cells in total observed cells: 5%, 0; 5–25%, 1; 25–50%, 2; 50–75%, 3; 75–100%, 4. The intensity was decided by comparing the staining of tumor cells: no staining or ambiguous staining, 0; weakly staining, 1; medially staining, 2; strongly staining, 3. The staining was quantified according to the sum of the positive cells and the intensity to categorize: 0–1, negative (−); 2–4, positive (+); ≥ 5, strongly positive (++).
Cell culture
The human ovarian cancer HO-8910 and SKOV3 cell lines (purchased from the Cell Bank of the Chinese Academy of Science, Shanghai, China) were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 0.1 mg/ml streptomycin at 37 °C in 5% CO2. The human embryonic kidney cell line 293T (purchased from the American Type Culture Collection) was cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 0.1 mg/ml streptomycin at 37 °C in 5% CO2.
Lentiviral vector production and infection
ShRNA sequences targeting USP39 (5′-GAATAACATAAAGGCCAAT-3′) was inserted into lentivirus vector GV248 (Hollybio. Inc. Shanghai, China). The shUSP39-expressing plasmid and Lenti-Easy Packaging Mix were co-transfected into 293T cells via the lipofectamine2000TM transfection reagent (Invitrogen) according to the manufacturer’s instruction. Supernatants were collected 48 h after transfection, filtered through a 0.4-μm membrane and used to infect the cells. After 5 days of infection and selection with puromycin, cells were observed under fluorescence microscopy. The USP39 expression levels in the selected stable clones were then verified by real-time PCR and Western blotting.
Quantitative real-time PCR (qRT-PCR)
Total RNA from HO-8910 and SKOV3 cells was extracted using TRIzol reagent (Takara); RNA was quantified using NanoDrop Spectrophotometer. As per manufacturer’s recommendations, 2 μg of RNA was used to generate complementary DNA using the Hifair® II 1st Strand cDNA Synthesis Kit (gDNA digester plus). Then, the measurements were taken using the SYBR Green system (Hieff® qPCR SYBR Green Master Mix (No Rox)) in Light Cycler 96 Detection System (Roche). The primers used were as follows: usp39, 5′-TTGGAAGAGGCGAGATAA-3′and 5′-AGGAGCATCAATCATCATC-3′; β-actin, 5′-GTGGACATTCCGCAAAGAC-3′ and 5′- AAAGGGTGTAACGCAACTA -3′. Fold-changes in mRNA levels were calculated. The β-actin was used as the reference gene; relative mRNA levels were determined with the 2−ΔΔCt. All experiments were repeated at least 3 times. Data shown are normalized to β-actin expression and represent the average of three repeated experiments.
Western blotting analysis
Western blotting was performed as previously described [15]. In short, samples were collected by lysing cells in RIPA lysis buffer. Each sample was size-fractionated using SDS–polyacrylamide gel electrophoresis (PAGE) and electro-transferred onto polyvinylidene difluoride transfer membranes (Dupont, Boston, MA, USA). After bolted with milk, the membranes were incubated with primary antibodies overnight at 4 °C and then blotted with horseradish peroxidase conjugated secondary antibodies. The immunoblots were visualized using ECL (GE Healthcare, Bucks, UK).
Cell growth assay
Cells were seeded at a density of 2000 cells/well for five point-in-time (days 1, 2, 3, 4 and 5) in 96-well plates. Cell growth was assessed via MTT assay. We replace the medium mixed with MTT after 4 h by 150 μl DMSO and vibrate the 96-well plates for 10 min. The absorbance at 570 nm was measured to evaluate cell viability.
Colony formation assay
Cells were seeded evenly in 12-well plates at a concentration of 500 cells/well and cultured at 37 °C for 12 days. The culture medium was changed every 3 days. The cells were washed with phosphate-buffered saline (PBS) twice and fixed in 4% paraformaldehyde for 30 min and stained with crystal violet for 20 min and then washed with PBS three times. The plates were photographed with a digital camera.
Cell cycling distribution assay
The cells were harvested by centrifugation at 1200 rpm for 5 min, resuspended with ice-cold PBS twice, fixed with cold 70% ethanol at 4 °C overnight and then centrifuged at 1500 rpm for 5 min to discard ethanol and resuspended with ice-cold PBS twice. The cell pellets were suspended in propidium iodide staining solution (20 mg/ml propidium iodide and 0.2 mg/ml RNase in PBS) and incubated for 30 min at 37 °C. Cell cycle distribution was finally analyzed by flow cytometry.
Wound-healing assay
Cells were seeded onto 12-well plates, and when they reached over 90% confluence, a scratch was made across the cell monolayer with the tip. Cells were gently washed with PBS for three times and maintained in the fresh medium with FBS. Cells were incubated for 24 h and photographed using an inverted tissue culture microscope. The migration potential between two different cells was compared by relative gap distance.
Xenograft assays in nude mice
For xenograft experiments, stable knockdown HO-8910 cells and its control cell line (2 × 106) were implanted separately and subcutaneously into the right foreleg of the female Balb/c nude mice (n ≥ 3). The mice were monitored every 2 days to measure the tumors volume. After 30 days, the mice were killed and tumor weight was measured. Nude mice were used at 4–6 weeks of age. Tumor volume = 0.5 * a2 * b (a, width; b, length).
Statistical analysis
The results of the experimental studies are expressed as the mean ± S.D. for at least three separate determinations for each group. The differences between the groups were examined for statistical significance using the Student’s t test. p < 0.05 was considered to indicate statistical significance.
Results
USP39 is frequently overexpressed and highly related to TNM stage in human ovarian cancer
To evaluate USP39 protein level and distribution in human ovarian cancer tissues, we tested ovarian cancer tissue microarray by using immunohistochemistry (IHC) staining. The microarray contained 156 ovarian carcinoma patient samples, including 8 normal tissues, 26 benign tissues and 122 malignant tumor tissues. Representative immunohistochemical results and quantitative analysis of ovarian carcinoma tissues are shown in Fig. 1a, b. USP39 was strongly expressed in the ovarian cancer tissues, and positive staining of USP39 was detected mainly in the nucleus and partly distributed in the cytoplasm (Fig. 1a, b). In addition, we also analyzed the expression of USP39 mRNA in human ovarian carcinoma using Oncomine database (https://www.oncomine.org) and GEPIA database (http://gepia.cancer-pku.cn/detail.php). These results also demonstrated that gene USP39 expression was increased in ovarian carcinoma samples (Fig. 1c–e). Then, we analyzed whether USP39 expression was associated with clinical pathological characteristics of ovarian carcinoma. As shown in Table 1, USP39 was significantly increased in malignant tumor (94.26%) compared with normal ovarian tissues (37.5%) and benign ovarian tumor tissues (76.92%) (p < 0.05). Besides, the expression of USP39 was different between I and II/III TNM stage. At the stage I, 11.11% (6/55) cases showed negative expression, 31.48% (17/55) cases showed weak expression, and 59.26% (32/55) cases displayed strong expression; at the stage II/III, USP39 negative expression, weak expression and strong positive expression occupied 1.49% (1/67), 23.88% (16/67) and 76.63% (50/67), respectively. In addition, no evidence showed USP39 expression which was related to tumor histological type and age (p > 0.05). The data above demonstrated that USP39 was overexpressed in ovarian cancer tissues and was related to the TNM stage significantly (p < 0.05).

Expression of USP39 is associated with ovarian carcinoma. A IHC staining of USP39 expression in normal and ovarian cancer tissues. Representative images of USP39 expression in normal ovarian tissues (a, 100 ×; g, 200 ×), struma ovarian tissues (b, 100 ×; h, 200 ×), thecoma tissues (c, 100 ×; i, 200 ×), serous adenocarcinoma tissues (d, 100 ×; j, 200 ×), mucinous adenocarcinoma tissues (e, 100 ×; k, 200 ×) and endometrioid adenocarcinoma tissues (f, 100 ×; l, 200 ×). USP39 mostly located in the nucleus. B Quantitative analysis of IHC assay showed that USP39 protein was strongly expressed in the ovarian cancer tissues compared with normal ovarian tissues. C–E Gene expression data from Oncomine database and GEPIA database showed that USP39 mRNA level was overexpressed in human ovarian carcinoma (**p < 0.01; ***p < 0.001; ****p < 0.0001)
Efficient knockdown of USP39 by lentivirus-mediated shRNA in HO-8910 and SKOV3 cells
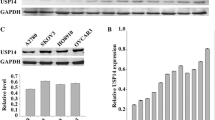
To further determine the role of USP39 in ovarian cancer, we examined the expression level of USP39 in seven ovarian cancer cell lines (TOV-21G, OV2008, HEY, SKOV3, HO-8910, C13 and A2780S) by Western blotting (WB) and found that HO-8910 and SKOV3 possessed the higher expression level of USP39 (Fig. 2a). Therefore, we suppressed USP39 expression in HO-8910 and SKOV3 by USP39 shRNA lentivirus. The infection efficiency of lentivirus was about 90% as represented by the percentage of GFP expression cells, which were subjected to functional analysis (Fig. 2b, c). Real-time PCR and WB indicated that USP39 was reduced about 80% in both the mRNA and protein levels (Fig. 2d–g). These results indicated that USP39 expression was specific and efficient knockdown by lentivirus-mediated shRNA in HO-8910 and SKOV3 cells.

Efficiency of suppression USP39 in HO-8910 and SKOV3 cells. a The protein level of USP39 in ovarian cancer cell lines: TOV-21G, OV2008, HEY, SKOV3, HO-8910, C13 and A2780S. USP39 expression in HO-8910 and SKOV3 cells was extremely higher than in other five cell lines. b, c Lentiviral infection in HO-8910 and SKOV3 cells. Representative GFP expression in control, shCon and shUSP39 in HO-8910 and SKOV3 cells. Upper panel, bright field; lower panel, fluorescence field (200 ×). d–g Real-time PCR and Western blot revealed the expression of USP39 mRNA and protein was efficiently inhibited in HO-8910 and SKOV3 cells (**p < 0.01; ***p < 0.001; ****p < 0.0001)
Knockdown of USP39 inhibits ovarian carcinoma growth in vitro and in vivo
To verify the role of USP39 on cell growth in vitro, MTT assay and colony formation were performed in HO-8910 and SKOV3 cells. The number of USP39 shRNA-transfected cells was reduced significantly compared with negative controls (Fig. 3a, b). Likewise, we demonstrated that USP39 knockdown resulted in significantly reduction in the colony formation compared with the control group (Fig. 3c, d). These results indicated that knockdown of USP39 markedly inhibited the proliferative ability of ovarian cancer in vitro.

Effect of USP39 knockdown on proliferation of ovarian cancer cells in vitro and vivo. a, b USP39-shRNA targeted cells and control cells were observed by Cellomics for 5 days. The growth curve of HO-8910 and SKOV3 cells infection of shUSP39. c, d Images of crystal violet-stained colonies of HO-8910 and SKOV3 cells growing in 6-well plates after 14 days. Corresponding statistics analysis of crystal violet-stained colonies of two groups. e USP39 suppression in HO-8910 cells decreased xenografted tumor growth in nude mice. f, g Tumor weight and size in USP39 knockdown group were significantly reduced compared with the negative control group (*p < 0.05; **p < 0.01; ***p < 0.001)
To further investigate whether USP39 suppression inhibited ovarian tumorigenesis in vivo, we constructed HO-8910 xenograft model. We injected USP39 stable knockdown HO-8910 cell line (shUSP39) and the control cell line (shCon) into BALB/c nude mice separately, and monitored local tumor formation for 30 days. Results showed that the volume and weight of tumors in USP39 knockdown group were significantly lower than in control group (Fig. 3e–g). Taken together, these results indicated that USP39 suppression inhibited ovarian carcinoma growth both in vitro and in vivo, and USP39 is critical for ovarian cancer cell proliferation and tumorigenesis.
Knockdown of USP39 induces G2/M arrest in ovarian cancer cells
To further explore the underlying mechanism of the inhibition of proliferation by reducing the expression of USP39, we employed flow cytometry to detect DNA content in PI-staining cells to analyze whether it was induced by cell cycle abnormality. As depicted in Fig. 4a, b, compared with the control group, knockdown of USP39 induced G2/M arrest in HO-8910 and SKOV3 cells. Further investigation was performed to locate the molecular mechanisms on how USP39 regulates cell cycle arrest in G2/M phase in human ovarian cancer cells. The Western blot analysis showed that knockdown of USP39 down-related the expression of CDK1 and cyclin B1 (Fig. 6a, b). These results implied that proliferation inhibition might be associated with cell cycle arrest and strongly supported that USP39 plays an important role in the malignant growth of ovarian carcinoma.

USP39 silencing induced G2/M cell cycle arrest. Flow cytometric assay was employed to analyze the cell cycle distribution. a, b Knockdown of USP39 in ovarian cancer cells changed cell cycle distribution. The percentage of cells in the G2/M phase was evidently increased, compared with the control group (**p < 0.01; ***p < 0.001)
Silencing USP39 blocks the migration of HO-8910 and SKOV3 cells
To investigate whether USP39 affects the migration ability of ovarian cancer cells, we performed a wound-healing assay. The results showed that the velocity of wound repairing in the shCon group was faster than in the shUSP39 group at 12 h and 24 h (Fig. 5a, b). These results indicated that knockdown of USP39 could block the migration of HO-8910 cells in vitro.

Knockdown of USP39 affected migration of ovarian cancer cells. a, b Representative images of wound healing of HO-8910 and SKOV3 cells infected with shUSP39 and shCon. Silencing of USP39 inhibited the migration of ovarian cancer cells (*p < 0.05; **p < 0.01)
Knockdown of USP39 activates P53 signaling pathway and blocks EMT transition
To investigate the molecular mechanism of USP39 on the proliferation and migration of ovarian cancer, we detected cell cycle and EMT-related regulators by Western blotting assay. We found that USP39 suppression could up-regulate the protein level of p53 and p21 (a downstream target of p53), but had no significant effect on p27 (Fig. 6c, d), suggesting that G2/M arrest induced by USP39 silencing might be associated with p53/p21 signaling pathway. In addition, we tested EMT-related proteins, including β-catenin, N-cadherin, E-cadherin and slug (a downstream target gene of Wnt/β-catenin). As shown in Fig. 6e, f, we found that β-catenin, N-cadherin and slug were decreased and E-cadherin was increased in the wake of USP39 knockdown. Furthermore, cell morphological analysis showed that EMT transition was blocked (Fig. 6g). These findings implied that silencing of USP39 inhibited migration of ovarian cancer cells via blocking EMT transition.

Regulated mechanism of USP39 knockdown in ovarian cancer cells. a, b The total protein was extracted, and the expression of cell cycle-related proteins was analyzed by Western blot assay. c, d Knockdown of USP39 activated the P53 signaling pathway. e–g USP39 suppression blocked the EMT of ovarian cancer cells. Data are reported as mean ± SD of three replicate experiments (*p < 0.05; ***p < 0.001)
Discussion
So far, researchers have discovered and named nearly 100 deubiquitinating enzymes, which are divided into five families; among them, ubiquitin-specific protease (USP) family is the one of the biggest [4]. USPs are involved in many cellular processes, including proliferation of tumor [11], cell cycle [16], cell apoptosis [17] and tumor metastasis [18]. Many reports have indicated that some members of USPs are highly connected with cancers, such as USP2a, USP4, USP5, USP7, USP10, USP13 and USP39 [19,20,21,22,23,24]. Previous studies have reported that USP39 plays important roles in mRNA splicing [6], mitosis processing, and act as an oncogene in several cancers [11, 24]. But up to now, the underlying function and potential mechanism of USP39 in ovarian cancer are still elusive.
In the present study, we observed that USP39 protein was significantly increased in ovarian cancer tissues compared with normal tissues, and the high level of USP39 was related to TNM stage, which is likely to be used as a clinical diagnostic basis of high grade of malignancy and poor prognosis in ovarian cancer. Subsequently, we tested the expression level of USP39 in seven ovarian cancer cell lines and chose HO-8910 and SKOV3 to explore the role of USP39. Lentivirus-delivered short hairpin RNA (shRNA) targeting USP39 was employed to stably down-regulate its endogenous expression. Knockdown of USP39 markedly inhibited cell proliferation and the ability of colony formation. Flow cytometry analysis showed that USP39 knockdown induced G2/M arrest, which could contribute to the inhibition of cell proliferation. Additionally, the suppression of USP39 inhibited the cell migration, which was so distinguished in previous investigates as well. Furthermore, we found that p53/p21 pathway and EMT were involved with the cell cycle processing and cell migration, respectively.
p53 is well known as a tumor suppressor, which regulates the G0/G1 and G2/M cell cycle checkpoints [25], while p27 controls the G1/S cell cycle checkpoint [26]. p21 is a downstream gene of p53, functioning as a link between p53 and cell cycle [27]. In this study, we observed that the human ovarian cancer cells, expressing a wild-type form of p53, were arrested at the G2/M phase of the cell cycle after shUSP39 interference. Then, we found a significant increase in p21, but not in p27, indicating that knockdown of USP39 in ovarian cancer cells resulted in accumulation of p53 and p21, and induced G2/M but not G1/S cell cycle arrest, suggesting that a p53 independent pathway may be involved in inducing the USP39-related changes. It is known that p53 regulates the G2/M checkpoint via modulating the cycle-dependent kinase Cdc2, which is essential for the entry of cells into mitosis [28]. p21 is the transcriptional targets of p53; it can inhibit Cdc2 by anchoring Cdc2 in the cytoplasm where it cannot induce mitosis [29]. A recent research showed that knockdown of USP39 induced the down-regulation of Cdc2 and cyclin B1 in medullary thyroid carcinoma, resulting in G2/M phase arrest [13]. Taken together, we speculate that USP39 regulated G2/M phase mediated by p53/p21/Cdc2/cyclin B1 pathway (described in Fig. 7).

USP39 promotes progression of ovarian cancer cells via blocking P53/P21 pathway and activating β-catenin/LEF/TCF/slug pathway
It is well known that ovarian cancer cells are prone to metastasis; in this process, epithelial-to-mesenchymal transition (EMT) is a necessary step during detachment of tumor cells from the primary tumor site and attachment to metastatic sites [30]. To ascertain the role of USP39 in migration, we observed that knockdown of USP39 in HO-8910 and SKOV3 cells inhibited the migration of ovarian cancer cells, as assessed by scratch-wound assay. Then, we further checked the EMT markers. A switch from E-cadherin to N-cadherin is a key feature of EMT in ovarian cancer [31]. E-cadherin is a transmembrane glycoprotein of the type-I cadherin superfamily. Its cytoplasmic part is linked to the actin cytoskeleton via the catenins [30]. Wnt/β-catenin signaling has a major impact on EMT during cancer progression [32, 33]. Wnt signal activation forced a rapid increase in the level of cytosolic β-catenin and enabled β-catenin to translocate and accumulate in the nucleus where it binds to TCF/LEF transcription factors, and induced the downstream target gene slug transcription [34, 35]. Reduction in E-cadherin levels often released β-catenin from adherens junction, resulting in nuclear β-catenin accumulation, and the consequent transactivation of a panel of target genes [32, 36]. In our experiment, knockdown of USP39 significantly decreased the N-cadherin and increased the E-cadherin expression at the protein levels, and reduced the protein levels of β-catenin and slug. Slug is a transcription inhibitor of E-cadherin; the down-regulation of slug further promotes the expression of E-cadherin and finally leads to EMT inhibition. Therefore, we proposed that USP39 might promote EMT by activating Wnt/β-catenin/TCF/slug/E-cadherin pathway (described in Fig. 7).
Our study provided substantial evidences in supporting that USP39 is overexpressed in most of ovarian cancer cells, and suppression of USP39 reduces proliferation and migration activity. Moreover, we revealed the potential molecular mechanism of USP39 in ovarian cancer cells, which might be involved with p53/p21 pathway and EMT transition. In conclusion, we proposed that USP39 acts as an oncogenic factor, and it has great potential to be a clinical biomarker in diagnosing ovarian cancer, or even be a gene target in treating ovarian cancer patients.
References
Torre LA, Trabert B, DeSantis CE, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68:284–96.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30.
Kuan AS, Teng CJ, Wu HH, et al. Risk of ischemic stroke in patients with ovarian cancer: a nationwide population-based study. BMC Med. 2014;12:53.
Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012;31:2373–88.
van Leuken RJ, Luna-Vargas MP, Sixma TK, Wolthuis RM, Medema RH. Usp39 is essential for mitotic spindle checkpoint integrity and controls mRNA-levels of aurora B. Cell Cycle. 2008;7:2710–9.
Hadjivassiliou H, Rosenberg OS, Guthrie C. The crystal structure of S. cerevisiae Sad1, a catalytically inactive deubiquitinase that is broadly required for pre-mRNA splicing. RNA. 2014;20:656–69.
Makarova OV, Makarov EM, Luhrmann R. The 65 and 110 kDa SR-related proteins of the U4/U6.U5 tri-snRNP are essential for the assembly of mature spliceosomes. EMBO J. 2001;20:2553–63.
He X, Zhang P. Serine/arginine-rich splicing factor 3 (SRSF3) regulates homologous recombination-mediated DNA repair. Mol Cancer. 2015;14:158.
Patwardhan GA, Hosain SB, Liu DX, et al. Ceramide modulates pre-mRNA splicing to restore the expression of wild-type tumor suppressor p53 in deletion-mutant cancer cells. Biochem Biophys Acta. 1841;1571–1580:2014.
Gautrey HL, Tyson-Capper AJ. Regulation of Mcl-1 by SRSF1 and SRSF5 in cancer cells. PLoS ONE. 2012;7:e51497.
Wang H, Ji X, Liu X, et al. Lentivirus-mediated inhibition of USP39 suppresses the growth of breast cancer cells in vitro. Oncol Rep. 2013;30:2871–7.
Pan Z, Pan H, Zhang J, et al. Lentivirus mediated silencing of ubiquitin specific peptidase 39 inhibits cell proliferation of human hepatocellular carcinoma cells in vitro. Biol Res. 2015;48:18.
An Y, Yang S, Guo K, Ma B, Wang Y. Reduced USP39 expression inhibits malignant proliferation of medullary thyroid carcinoma in vitro. World J Surg Oncol. 2015;13:255.
Wen D, Xu Z, Xia L, et al. Important role of SUMOylation of Spliceosome factors in prostate cancer cells. J Proteome Res. 2014;13:3571–82.
Tong Y, Zhang G, Li Y, et al. Corilagin inhibits breast cancer growth via reactive oxygen species-dependent apoptosis and autophagy. J Cell Mol Med. 2018;22:3795–807.
Stegmeier F, Rape M, Draviam VM, et al. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature. 2007;446:876–81.
Chen FZ, Zhao XK. Ubiquitin-proteasome pathway and prostate cancer. Onkologie. 2013;36:592–6.
Yuan X, Sun X, Shi X, et al. USP39 promotes colorectal cancer growth and metastasis through the Wnt/beta-catenin pathway. Oncol Rep. 2017;37:2398–404.
Stevenson LF, Sparks A, Allende-Vega N, Xirodimas DP, Lane DP, Saville MK. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 2007;26:976–86.
Zhang L, Zhou FF, Drabsch Y, et al. USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-beta type I receptor. Nat Cell Biol. 2012;14:717–26.
Dayal S, Sparks A, Jacob J, Allende-Vega N, Lane DP, Saville MK. Suppression of the deubiquitinating enzyme USP5 causes the accumulation of unanchored polyubiquitin and the activation of p53. J Biol Chem. 2009;284:5030–41.
Qian J, Pentz K, Zhu Q, et al. USP7 modulates UV-induced PCNA monoubiquitination by regulating DNA polymerase eta stability. Oncogene. 2015;34:4791–6.
Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–34.
Wang X, Yu Q, Huang L, Yu P. Lentivirus-mediated inhibition of USP39 suppresses the growth of gastric cancer cells via PARP activation. Mol Med Rep. 2016;14:301–6.
Naidu KA, Fang Q, Naidu KA, Cheng JQ, Nicosia SV, Coppola D. P53 enhances ascorbyl stearate-induced G2/M arrest of human ovarian cancer cells. Anticancer Res. 2007;27:3927–34.
Hnit SS, Xie C, Yao M, et al. p27(Kip1) signaling: transcriptional and post-translational regulation. Int J Biochem Cell Biol. 2015;68:9–14.
Horvath V, Soucek K, Svihalkova-Sindlerova L, et al. Different cell cycle modulation following treatment of human ovarian carcinoma cells with a new platinum(IV) complex vs cisplatin. Investig New Drugs. 2007;25:435–43.
Nurse P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344:503–8.
Choi EJ, Kim GH. Apigenin causes G(2)/M arrest associated with the modulation of p21(Cip1) and Cdc2 and activates p53-dependent apoptosis pathway in human breast cancer SK-BR-3 cells. J Nutr Biochem. 2009;20:285–90.
Takai M, Terai Y, Kawaguchi H, et al. The EMT (epithelial-mesenchymal-transition)-related protein expression indicates the metastatic status and prognosis in patients with ovarian cancer. J Ovarian Res. 2014;7:76.
Mao Y, Xu J, Li Z, Zhang N, Yin H, Liu Z. The role of nuclear beta-catenin accumulation in the Twist2-induced ovarian cancer EMT. PLoS ONE. 2013;8:e78200.
Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–66.
Beiter K, Hiendlmeyer E, Brabletz T, et al. beta-Catenin regulates the expression of tenascin-C in human colorectal tumors. Oncogene. 2005;24:8200–4.
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26.
Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281:22429–33.
Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci. 1999;112(Pt 8):1237–45.
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (81872045, 81470793), and the special fund for public welfare research institutes of Fujian Province (2017R1036-1, 2018R1036-4).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Ethical approval
The animal studies were approved by the Institutional Animal Care and Use Committee (IACUC No. XMULAC20180056) of Xiamen University, Fujian. Animal experiments were approved by the Laboratory Animal Ethics Committee of Xiamen University.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yan, C., Yuan, J., Xu, J. et al. Ubiquitin-specific peptidase 39 regulates the process of proliferation and migration of human ovarian cancer via p53/p21 pathway and EMT. Med Oncol 36, 95 (2019). https://doi.org/10.1007/s12032-019-1308-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-019-1308-7