
Abstract
Chimeric antigen receptor (CAR) T cell therapy is genetically engineered tumor antigen-specific anticancer immunotherapy, which after showing great success in hematological malignancies is currently being tried in advanced solid tumors like pancreatic cancer. Immunosuppressive tumor microenvironment and dense fibrous stroma are some of the limitation in the success of this novel therapy. However, genetic modifications and combination therapy is the topic of the research to improve its efficacy. In this article, we summarize the current state of knowledge, limitations, and future prospects for CAR T cell therapy in pancreatic cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer is highly aggressive and lethal malignancy. It is seventh most common cause of cancer-related death worldwide [1] and third most in the United States of America (USA) [2]. The 5 year survival rate is 7.2% and estimated mortality is 40,560 in 2015 in USA [2]. The two main types of tumor are pancreatic ductal adenocarcinoma (PDA) and endocrine tumors. PDA constitutes 90% of all cases, and is characterized by highly aggressive and malignant behavior, delayed diagnosis, resistant to treatment, and high mortality [3, 4]. Generally, at the time of diagnosis, PDA already had local or distant metastasis which make them ineligible for surgical resection, and palliative care is their only option with 5 year survival rate in single digits [5]. Despite recent advances in therapeutic options like FOLFIRINOX (Folinic acid, 5 Fluorouracil, Irinotecan, Oxaliplatin), median overall survival still remains poor [6]. Therefore, novel therapeutic options need to be devised to deal with this highly lethal and dreadful tumor. Immunotherapy recently showed promising results in various tumors and pancreatic cancer can be a potential target for them. Although initial studies of immune checkpoint inhibitors as single agents failed to show therapeutic response in PDA with advancing field and better understanding of immune system in relation to pancreatic cancer, there are still promising chances to have effective immunotherapy for advanced PDA [7].
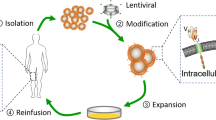
Chimeric antigen receptor (CAR) T cell therapy is a type of Adoptive T cell therapy (ACT), where genetically engineered T cells are used for lysis and degradation of cancer cells. It’s a novel therapeutic modality which has shown outstanding results in hematological malignancies and currently research is going on in solid tumors; PDA can be a potential target for CAR T cell therapy. Immune system generally detects damaged or genetically modified tumor-potential cells and kill them. Cancer cells have the potential to evade these immune mechanisms and can modify immune system to help cancer cells in their proliferation, invasion, and metastasis, known as immune escape. There are various mechanism behind it; some of them are defect in antigen processing and presentation [8, 9], loss of immunogenic cancer antigens, secretion of inhibitory cytokines like IL-10 and TGF-β, genetic modification in tumor oncogenes and tumor suppressor genes, overexpression of CTLA-4, and PD-1 immune checkpoints. Therefore, if we can reverse these immune escape phenomena of cancer cells and activate immune cells against cancer antigens, it can be promising therapy for cancer. Immunotherapy is based on this principle. Conventional activation of T cells is a complex process and is dependent on antigen processing, antigen presentation, and major histocompatibility complex(MHC)- mediated activation to peptide antigens. But CAR T cells uses light and heavy chains of immunoglobulins for antigen binding ectodomain, and therefore, it doesn’t require antigen processing, MHC dependence [10, 11], and can have immunogenic response to non-peptide antigens also [12]. Therefore, CAR T cells can evade various immune escape mechanisms of cancer cells and can have cancer specific immune reactions. There are four main components of CAR. Ectodomain which is extracellular cancer antigen binding domain, consists of single chain variable fragment (scFv) derived from heavy and light chains of antibody. Spacer domain, also known as hinge domain, is formed from IgG4 and CD8 molecule and connects extracellular domain and transmembrane domain. Intracellular domain also known as signaling domain consists of CD3ζ and it initiates and triggers antigen specific immune response. CAR T cell therapy is autologous customized therapy, where T cells are collected from patients’ own peripheral blood with leukapheresis; these T cells are transduced with genetic material for CAR with the help of lentiviruses and retroviruses. Transduced T cells are genetically amplified with in vitro culture systems and each culture system is specific for a particular type of cell line like effector T cell, memory, or naïve T cell. Therefore, we can control the phenotype and can generate required subset of T cells [13]. CAR T cells are then transfused back into patients and they can recognize and bind to tumor specific antigens (TAA). This start a cascade of reaction and lead to phosphorylation of immune receptor tyrosine-based activation motifs (ITAMs) and, therefore, activation of T cells [14]. Cytotoxicity of CAR T cells is mediated by activation of death receptor with binding of fas/fas-ligand and by release of perforin enzyme which induces lysis of tumor cells [15, 16]. On the basis of intracellular domain, CAR T cells are divided into 4 generations. In first generation, CD3ζ is intracellular signaling domain, in second and third generation, there are additions of co-stimulatory molecules like 4-1BB (CD137), CD 28, OX40 which enhances the T cell response; in second generation, there is one co-stimulatory molecule and in third generation, there are two. Fourth generation of CAR T cells have addition of cytokine releasing genes (IL-12 and IL-15) which improves the survival of CAR T cells in immunosuppressive tumor microenvironment [17, 18].
Immunosuppression by PDA
PDA is highly aggressive, malignant, and treatment resistance cancer with extremely high mortality likely due to evasion of immune surveillance by PDA cancer cells. Although PDA is a T cell rich tumor, it induces local or systemic immune dysfunction to prevent cancer cells’ death from cytotoxic immune cells [19, 20]. PDA downregulate the MHC class 1 molecule and inhibit antigen insertion into MHC groove, therefore, inhibit antigen cross-presentation [21]. PDA cells has non-functional Fas receptor to avoid Fas-mediated apoptosis and along with that they express Fas ligand which can induce apoptosis in immune effector cells [22]. PDA secrete IL-10 and transforming growth factor-beta (TGF-β) which are immunosuppressive molecules and inhibit the function of cytotoxic T cells [23, 24]. PDA also secrete Indoleamine 2,3-dioxygenase (IDO) enzyme which catalyzes the breakdown of tryptophan to kynurenine, which is an important nutrient for the survival of lymphocytes, and thereby, suppress antitumor T cell response, along with that, IDO also increase the T regulatory cells (Tregs) in lymph nodes which aggravates immunosuppressive environment [25, 26]. PDA increases the expression of PD-L1 on their surface, which are immune checkpoints; therefore, decrease the activation of T cells [27]. The stromal matrix of PDA comprises of cellular and acellular components like myofibroblasts, pancreatic stellate cells, fibroblasts, blood vessels, extracellular matrix, immune cells, cytokines, and growth factors, and all of them contribute to tumor proliferation, invasion, and metastasis [28]. Along with excessive desmoplasia, PDA also lack vasculature which leads to impaired perfusion and limited drug delivery. The tumor microenvironment of PDA consists of dense fibrotic stroma with variety of inflammatory cells, infiltration with immunosuppressive cells such as myeloid derived suppressive cells (MDSCs), tumor-associated macrophages (TAMs), Tregs, and various other mechanism to avoid tumor cell detection makes tumor microenvironment highly immunosuppressive [29]. As consequence of all the above mentioned mechanisms. As there are multiple steps of immune escape which can be potential targets for immunotherapy also, this makes PDA an immune privilege tumor. Multiple immune escape steps should be targeted simultaneously to have a sustained therapeutic response and single agent immunotherapy should be avoided, as it will lead to failure of immune response due to other immunosuppressive factors, as we have seen in immune checkpoint inhibitors in PDA. Therefore, PDA is a promising candidate for immunotherapy like CAR T cell therapy and these immunosuppressive factors are two edged sword as they can act as obstacles, but with further advances in therapeutic options, they can be excellent targets for different forms of immunotherapy also.
Antigenic targets for CAR T cell therapy
Mesothelin (MSLN)
MSLN is a normal cell surface glycoprotein which is expressed on the surface of mesothelial cells, lining the peritoneum, pericardium, and pleura. It is formed as a precursor protein which is cleaved by the action of endoprotease enzyme to release N-terminal known as megakaryocyte potentiating factor (MPF) and mature MSLN remain behind attached to membrane [30]. As per the distribution of MSLN, it likely functions as a differentiating factor for mesothelial cells but the exact biological function is unknown. MSLN is overexpressed in various cancer cells and its overexpression has been correlated with poor prognosis in various tumors like ovarian cancer [31], PDA [32, 33], cholangiocarcinoma [34, 35], lung adenocarcinoma [36, 37], and triple negative breast cancer [38]. As most of the cancer specific antigens which are used as targets for solid tumors are also present on normal cells, there is nonspecific toxicity. But MSLN is only expressed on dispensable tissue, and therefore, the chances of nonspecific toxicity are less.
As there is overexpression of MSLN in pancreatic cancer, different therapeutic approaches have been evaluated to target MSLN like MSLN vaccine, antibody drug conjugate, chimeric antibody, and CAR T cell therapy. Recent data suggest that new immunotoxins like LMB-100 (old name RG 7787) might have synergistic effects with standard therapy in management of PDA [39]. Initial clinical trials of CRS-207 (Listeria monocytogene-mesothelin vaccine) also have shown promising outcome as compared to standard chemotherapy [40]. Therefore, MSLN can be a potential target for CAR T cell therapy for PDA patients. Other modalities of targeting MSLN are SS1P (Recombinant anti-MSLN immunogenic toxin) [41], BAY 94-9343 (antibody drug targeting MSLN) [40], DMOT4039A (humanized IgG1 anti-MSLN mAb) [42], and BMS-986148 (Anti-MSLN MAb-cytotoxic drug conjugate) [43]. In a clinical trial of MSLN-targeted CAR T cell therapy, a patient with PDA, after receiving 3 weeks of intravenous CAR T cell therapy had a stable disease. After finishing the therapy, FDG PET/CT imaging also showed decrease in uptake value at all sites of disease transiently. Ascitic fluid analysis on day 3 and 15 of CAR T cell therapy have shown decrease in cancerous cell expressing MSLN and c-Met [44]. The transient immune reaction is a promising result and MSLN CAR T cell therapy can be a potential therapeutic tool for management of advanced pancreatic cancer.
CD24
One of the major reasons for highly aggressive and invasive nature of PDA is presence of self-renewable, chemoresistant, and multipotent cells in tumor, known as cancer stem cells (CSC). CSC are known to be responsible for tumor proliferation, invasion, metastasis, and recurrence [45,46,47,48]. Initially, Li et al. found highly aggressive subpopulation of cancer cells in PDA, and transfer of these cells in immunocompromised mice lead to initiation, rapid proliferation, and metastasis of PDA [49]. It has been found that these cells have surface markers like CD44, CD24, and epithelial specific antigen. These surface markers are present only in 0.2–0.8% of cancer cells, but they can increase the tumorigenic potential more than 100 folds as compared to other cancer cells [49, 50]. Therefore, CD24 can be a potential target to control the tumorigenic potential of PDA cancer cells. Maliar et al. [51] successfully eliminated the CD24 + tumor cells with second generation CD24 CAR T cell therapy in human orthotopic pancreatic cancer xenograft model. In this study, total 107 CAR T cells were injected intravenously or intratumorally on 3 or 4 alternating days and then mice were given IL-2 intraperitoneally, two times a day for 10 days. This study showed that as CD24 CAR T cell therapy was more efficacious as compared to HER2 CAR T cell therapy in curing mice, although CD24 expression is markedly less on PDA cancer cells in comparison to HER2. Therefore, this study showed that CD24 can be a potential target for CAR T cell therapy of PDA.
Carcinoembryonic antigen (CEA)
CEA is a 180-kDa GPI-linked glycoprotein which is normally expressed on the surface of colon. However, this oncofetal protein expression increases exponentially during carcinogenesis. Along with that, CEA can be shed into blood stream and can be measured as tumor marker. On a variety of gastrointestinal tumors, expression of CEA is markedly elevated including PDA [52,53,54,55]. Chmielewski et al. [56] did a study regarding efficacy of CEA CAR T cell therapy in mice having orthotopic pancreatic cancer cells. In all, mice size of tumor was reduced to below the limit of detection and long term tumor eradication was seen in 67% of mice. No lymphodepletion was required and there was no destruction of CEA + healthy tissue. CEA targeting CAR T cells therapy trials have not been reported yet, but phase 1 dose escalation trial of CEA CAR T cell therapy with preconditioning chemotherapy and intravenous IL2 in patients with advanced CEA positive tumors have reported that after 6 weeks of treatment, 7 out of 13 patients had stable disease and rest 6 patients progressed [57]. At the second dose escalation level, this trial was discontinued due to pulmonary toxicity in 4 patients [57, 58]. Therefore, CEA can be a promising target for advanced PDA but further research is needed.
MUC1
It is a membrane bound glycoprotein and is commonly overexpressed in various tumors like breast, ovarian, colon, and lung cancer [59]. MUC1 is also overexpressed in around 90% of PDA patients and considered as potential diagnostic, prognostic, and therapeutic marker [60]. Strength of expression is associated with poor prognosis. In various clinical and preclinical trials, MUC1-targeted therapy have been tested [61]. Posey et al [62] developed a CAR that targets MUC1 and it showed target specific cytotoxicity and was able to control growth of tumor cells in xenograft model of pancreatic cancer and T cell leukemia. In this study, MUC-1 CAR T cell therapy led to 100% (6/6) survival of mice as compared to 40% (2/5) survival with untransduced T cells at 16 weeks of therapy, therefore, establishing the role of MUC1 CAR T cell therapy for PDA.
HER2
HER2 is overexpressed in various types of cancers like breast cancer, lung cancer, and gastric cancer [63, 64]. It has been shown in many studies that HER2 can increase the proliferation, invasion, and metastasis of cancer cells, and it is an important biomarker and target of therapy for breast cancer. Presence of HER2 on PDA is controversial [65] but some of the studies have shown its presence on 20–60% of PDA [66, 67]. In the same study as mentioned above for CD24, HER2 CAR T cell therapy has been tested against human orthotopic pancreatic cancer in immunodeficient mice. HER2 CAR T cell therapy was able to reduce the tumor burden even in advanced cases of PDA within 1 week. Sequential therapy with CD24 CAR T cell therapy after 2 months HER2 CAR T cell therapy led 90% survival at 14 weeks [51]. Therefore, HER2 expression was little lower on PDA but it can be a promising therapeutic modality especially in combination.
Toxicity of CAR T cell therapy
CAR T cell therapy have demonstrated positive clinical outcome in hematological malignancy and modest outcome has been seen in various solid tumors. However, CAR T cell therapy also have toxicities like cytokine release syndrome (CRS), on target/off tumor effects, neurological toxicity, and anaphylaxis. The most prevalent and dreadful toxicity of CAR T cell therapy is over immune activation leading to CRS [68]. CRS can present as high grade fever, weakness, malaise, nausea, cardiac dysfunction, renal impairment, tachycardia/hypotension, and disseminated intravascular coagulation with elevation of interferon gamma, IL-10, IL-6, and GM-CSF (granulocyte macrophage colony stimulating factor) [68,69,70]. Generally, for diagnosis of CRS, monitoring of cytokines is technically difficult; therefore, CRP (C-reactive protein) is used as marker for onset and severity of CRS, as it is produced by liver cells in response to IL-6 [69, 70]. The treatment for CRS generally include high dose steroid therapy, ventilator support, vasopressors, and anti-IL-6R antibody (Tocilizumab) which has shown clinical benefit in some of the patients [71]. In various patients receiving CD19 CAR T cell therapy, neurological toxicities has been reported including confusion, aphasia, delirium, myoclonus, and seizure. Cause for neurological toxicity is not identified, likely it is related to elevated cytokine levels [69, 70]. Direct toxic effect of CAR T cells on neurological tissue is another possibility. The ideal target for CAR T cell therapy should be present only on cancer cells, but generally these targets are shared by normal tissues also. Therefore, action of CAR T cells on nonpathogenic tissue lead to on-target/off-tumor toxicity [72]. Targeting of HER2/neu lead to rapid respiratory failure, multi-organ dysfunction, and death in a patient due to reactivity against pulmonary tissue [73]. As there is wide range of expected and unexpected toxicities, integration of suicide gene or elimination gene may be an important component for improvement of this therapy [74,75,76].
Current limitations and future perspectives of CAR T cell therapy
After outstanding success in hematological malignancies, now CAR T cell therapy is one of the most potential therapies for advanced solid tumors like pancreatic cancer. However, solid tumors have additional challenges as compared to hematological malignancies in success of CAR T cell therapy. The main three basic pathophysiological challenges are lack of antigen, hostile tumor microenvironment, and poor trafficking of engineered T cells. These challenges were minimal in liquid cancers as they have one standard antigen, i.e., CD19 which is present on all B cells. Treatment with CD19 CAR T therapy may lead to B cell aplasia but this can be managed by intravenous immunoglobulins and close monitoring for infection. Trafficking of T cells and immunosuppressive microenvironment are also not issue in hematological tumors as CAR T cells are infused directly into blood.
Most common limiting factor for CAR T cell therapy in PDA is hostile tumor microenvironment. As microenvironment of PDA is highly immunosuppressive, it makes survival of engineered T cells difficult, and therefore reduces their efficacy. To deal with this issue, combinational therapies should be tried, like combination of Immune checkpoint inhibitors with CAR T cell therapy [77]. Both are immunotherapies and target cancer cells in different ways, and hence, they can have synergistic effect and improve outcome in PDA. Higher bulk of tumor is associated with poor outcome with CAR T cell therapy has been shown in studies on leukemia patients. Similar response is also expected in PDA; therefore, we should reduce the bulk of tumor before providing CAR T cell therapy with cryoablation, radiation, radiofrequency ablation, or even surgical debulking. Along with reducing bulk, radiation therapy also potentiates immune response which can further potentiate efficacy of CAR T cells [78, 79]. Preconditioning with traditional chemotherapy like cyclophosphamide and fludarabine can also be used as it can also reduce the bulk of tumor along with that it inhibits Tregs, which improves tumor microenvironment for proliferation of CAR T cells [80]. In a research, combination of gemcitabine with rosiglitazone in PDA patients has reduced tumor progression, metastasis, and improved survival also. Rosiglitazone altered the tumor suppressive mediators and made the microenvironment more immunogenic [81]. Similarly, metformin also increased CD8 + TIL in tumor microenvironment [82]. Therefore, if CAR T cell therapy can be combined with rosiglitazone or metformin, immune suppressive nature of tumor microenvironment will reduce and efficacy of CAR T cells will improve.
CAR T cells also need to be modified to improve their efficacy and specificity, like addition of chemokine receptors. Chemokines are molecules released by cancer cells for proper trafficking to tumor site. Receptors of CAR T cells should be complimentary to these molecules for improvement in migration of T cells. Therefore, if CAR T cells have overexpression of these chemokines like CCR2b, CCR4 (CCL17 receptor), and CXCR2 (CXCL1 receptor), migration will improve [83, 84]. In one study on mice migration of MSLN, CAR T cells improved 12.5 times with overexpression of CCR2b in malignant pleural mesothelioma [85]. It can also be tried in PDA. For penetration of extracellular matrix, T cells secrete Heparanase (HPSE) enzyme but [86], while preparing CAR T cells, they become unable to secrete HPSE enzyme due to functional changes. Therefore, researchers developed CAR T cells which can express HPSE and these cells have shown better anticancer response [87]. This strategy can be useful in PDA also.
Conclusion
CAR T cell therapy is promising emerging treatment in cancer care. After its success in hematological malignancies, it is currently tried in Solid tumors. PDA is highly aggressive malignant tumor with high mortality and extremely limited therapeutic option. CAR T cell therapy can be a promising treatment modality for advanced PDA but it has to travel a long distance to come into clinical practice. Currently, there are various limitations and toxicities which are obstructing its way to reality like immunosuppressive tumor microenvironment, nutrient deprivation, hypoxia, dense stoma, poor trafficking of T cells, CRS, on target, off tumor effects, and many more. Active research is going on to deal with these limitations and we are optimistic that in near future, CAR T cell therapy alone or in combination will improve the outcome and survival of PDA patients.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386.
Howlader N, Noone A, Krapcho M, Garshell J, Miller D, Altekruse S, et al. Seer cancer statistics review, 1975–2012. Bethesda: National Cancer Institute; 2015.
Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144(6):1252–61.
Bazhin AV, Shevchenko I, Umansky V, Werner J, Karakhanova S. Two immune faces of pancreatic adenocarcinoma: possible implication for immunotherapy. Cancer Immunol Immunother. 2014;63:59–65. https://doi.org/10.1007/s00262-013-1485-8.
Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039–49. https://doi.org/10.1056/NEJMra1404198.
Vaccaro V, Sperduti I, Milella M. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;365(8):768–9.
Chang JH, Jiang Y, Pillarisetty VG. Role of immune cells in pancreatic cancer from bench to clinical application: an updated review. Medicine. 2016;95:e5541.
Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene. 2008;27:5869–85.
Chang CC, Campoli M, Ferrone S. Classical and nonclassical HLA class I antigen and NK Cell-activating ligand changes in malignant cells: current challenges and future directions. Adv Cancer Res. 2005;93:189–234.
Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejec-tion kinetics and persistence of CAR T cells. Cancer Cell 2015;28(4):415–28. https://doi.org/10.1016/j.ccell.2015.09.0042.
Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–98. https://doi.org/10.1158/2159-8290.
Chmielewski M, Hombach AA, Abken H. Antigen-specific T-cell activation independently of the MHC: chimeric antigen receptor-redirected T cells. Front Immunol. 2013;4:371. https://doi.org/10.3389/fimmu.2013.00371.
Riddell SR, Sommermeyer D, Berger C, Liu LS, Balakrishnan A, Salter A, Hudecek M, Maloney DG, Turtle CJ. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J. 2014;20:141–4. https://doi.org/10.1097/PPO.0000000000000036.
Kershaw MH, Westwood JA, Slaney CY, Darcy PK. Clinical application of genetically modified T cells in cancer therapy. Clin Transl Immunol. 2014;3(5):e16.
Yasukawa M, Ohminami H, Arai J, Kasahara Y, Ishida Y, Fujita S. Granule exocytosis, and not the fas/fas ligand system, is the main pathway of cytotoxicity mediated by alloantigen-specific CD4(+) as well as CD8(+)cytotoxic T lymphocytes in humans. Blood. 2000;95(7):2352–5.
Hombach A, Kohler H, Rappl G, Abken H. Human CD4 + T cells lyse target cells via granzyme/perforin upon circumvention of MHC class II restriction by an antibody-like immunoreceptor. J Immunol. 2006;177(8):5668–75.
Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123(17):2625–35. https://doi.org/10.1182/blood-2013-11-492231 5.
Pegram HJ, Park JH, Brentjens RJ. CD28z CARs and armored CARs. Cancer J. 2014;20(2):127. https://doi.org/10.1097/PPO.0000000000000034.
Nummer D, Suri-Payer E, Schmitz-Winnenthal H, et al. Role of tumor endothelium in CD4 + CD25 + regulatory T cell infiltration of human pancreatic carcinoma. J Natl Cancer Inst. 2007;99:1188–99.
Panni RZ, Sanford DE, Belt BA, et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother. 2014;63:513–28.
Ryschich E, Notzel T, Hinz U, et al. Control of T-cell-mediated immune response by HLA class I in human pancreatic carcinoma. Clin Cancer Res. 2005;11:498–504.
von Bernstorff W, Spanjaard RA, Chan AK, et al. Pancreatic cancer cells can evade immune surveillance via nonfunctional Fas (APO-1/CD95) receptors and aberrant expression of functional Fas ligand. Surgery. 1999;125:73–84.
Bellone G, Turletti A, Artusio E, et al. Tumor-associated transforming growth factor-beta and interleukin-10 contribute to a systemic Th2 immune phenotype in pancreatic carcinoma patients. Am J Pathol. 1999;155:537–47.
Moo-Young TA, Larson JW, Belt BA, et al. Tumor-derived TGF-beta mediates conversion of CD4 + Foxp3 + regulatory T cells in a murine model of pancreas cancer. J Immunother. 2009;32:12–21.
Uyttenhove C, Pilotte L, Theate I, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–74.
Witkiewicz A, Williams TK, Cozzitorto J, et al. Expression of indoleamine 2,3-dioxygenase in metastatic pancreatic ductal adenocarcinoma recruits regulatory T cells to avoid immune detection. J Am Coll Surg. 2008;206:849–54.
Basso D, Fogar P, Falconi M, et al. Pancreatic tumors and immature immunosuppressive myeloid cells in blood and spleen: role of inhibitory co-stimulatory molecules PDL1 and CTLA4. An in vivo and in vitro study. PLoS One. 2013;8:e54824.
Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–76.
Clark CE, Hingorani SR, Mick R, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–27.
Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci USA. 1996;93:136–40.
Cheng WF, Huang CY, Chang MC, et al. High mesothelin correlates with chemoresistance and poor survival in epithelial ovarian carcinoma. Br J Cancer. 2009;100:1144–53.
Winter JM, Tang LH, Klimstra DS, et al. A novel survival-based tissue microarray of pancreatic cancer validates MUC1 and mesothelin as biomarkers. PLoS One. 2012;7:e40157. [Erratum: PLoS One 7, 2012].
Shimizu A, Hirono S, Tani M, et al. Coexpression of MUC16 and mesothelin is related to the invasion process in pancreatic ductal adenocarcinoma. Cancer Sci. 2012;103:739–46.
Kawamata F, Kamachi H, Einama T, et al. Intracellular localization of mesothelin predicts patient prognosis of extrahepatic bile duct cancer. Int J Oncol. 2012;41:2109–18.
Nomura R, Fujii H, Abe M, et al. Mesothelin expression is a prognostic factor in cholangiocellular carcinoma. Int Surg. 2013;98:164–9.
Thomas A, Chen Y, Steinberg SM, et al. High mesothelin expression in advanced lung adenocarcinoma is associated with KRAS mutations and a poor prognosis. Oncotarget. 2015;6:11694–703.
Kachala SS, Bograd AJ, Villena-Vargas J, et al. Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clin Cancer Res. 2014;20:1020–8. (Erratum 20:3896, 2014).
Li YR, Xian RR, Ziober A, et al. Mesothelin expression is associated with poor outcomes in breast cancer. Breast Cancer Res Treat. 2014;147:675–84.
Kolivas E, Rudloff M, Poruchynsky M, et al. Mesothelin-targeted immunotoxin RG7797 has synergistic anti-tumor activity when combined with taxanes. Oncotarget. 2017;8(6):9189–99.
Le DT, Wang-Gillam A, Picozzi V, et al. Safety and survival with GVAX pancreas prime and Listeria monocytogenes–expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. 2015;33:1325–33.
Hassan R, Ebel W, Routhier EL, et al. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immunol. 2007;19:7–20.
Francisco JA, Cerveny CG, Meyer DL, et al. cAC10-vcMMAE, an anti CD30 monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102:1458–65.
Zhao XY, Subramanyam B, Sarapa N, Golfier S, Dinter H. Novel antibody therapeutics targeting mesothelin in solid tumors. Clin Cancer Drugs. 2016;3(2):76–86.
Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–20.
Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121(6):823–35. https://doi.org/10.1016/j.cell.2005.03.032.
O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106–10. https://doi.org/10.1038/nature05372.
Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Can Res. 2005;65(23):10946–51. https://doi.org/10.1158/0008-5472.CAN-05-2018.
Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian inhibiting substance responsiveness. Proc Natl Acad Sci USA. 2006;103(30):11154–9. https://doi.org/10.1073/pnas.0603672103.
Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Can Res. 2007;67(3):1030–7. https://doi.org/10.1158/0008-5472.CAN-06-2030.
Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313–23. https://doi.org/10.1016/j.stem.2007.06.002.
Maliar A, Servais C, Waks T, et al. Redirected T cells that target pancreatic adenocarcinoma antigens eliminate tumors and metastases in mice. Gastroenterology. 2012;143:1375.e1371–1384.e1375.
Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999;9:67–81. https://doi.org/10.1006/scbi.1998.0119.
Albers GH, Fleuren G, Escribano MJ, Nap M. Immunohistochemistry of CEA in the human pancreas during development, in the adult, chronic pancreatitis, and pancreatic adenocarcinoma. Am J Clin Pathol. 1988;90:17–22.
Allum WH, Stokes HJ, Macdonald F, Fielding JW. Demonstration of carcinoembryonic antigen (CEA) expression in normal, chronically inflamed, and malignant pancreatic tissue by immunohistochemistry. J Clin Pathol. 1986;39:610–4. https://doi.org/10.1136/jcp.39.6.610.
Yamaguchi K, Enjoji M, Tsuneyoshi M. Pancreatoduodenal carcinoma: a clinicopathologic study of 304 patients and immunohistochemical observation for CEA and CA19-9. J Surg Oncol. 1991;47:148–54. https://doi.org/10.1002/jso.2930470303.
Chmielewski M, Hahn O, Rappl G, et al. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology. 2012;143:1095.e2–1107.e2.
Thistlethwaite F. A CRUK phase I trial of adoptive transfer of autologous tumour antigen-specific T cells with pre-conditioning chemotherapy and intravenous IL2 in patients with advanced CEA positive tumours. In: NCRI Poster Session; 2010.
Hawkins R. A trial looking at MFEz T cells, chemotherapy and IL-2 (interleukin 2) for cancers that test positive for carcinoembryonic antigen (CEA) (PH1/105). In: Cancer Research UK Clinical Trial Listings; 2010.
Gendler SJ. MUC1, the renaissance molecule. J Mammary Gland Biol Neoplasia. 2001;6:339–53.
Qu CF, Li Y, Song YJ, Rizvi SM, Raja C, Zhang D, et al. MUC1 expression in primary and metastatic pancreatic cancer cells for in vitro treatment by (213)Bi-C595 radioimmunoconjugate. Br J Cancer. 2004;91:2086–93.
Pichinuk E, Benhar I, Jacobi O, Chalik M, Weiss L, Ziv R, et al. Antibody targeting of cell-bound MUC1 SEA domain kills tumor cells. Cancer Res. 2012;72:3324–36. https://doi.org/10.1158/0008-5472.CAN-12-0067.
Posey AD Jr, Schwab RD, Boesteanu AC, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. 2016;44:1444–54.
Bittoni A, Mandolesi A, Andrikou K, Santoni M, Alfonsi S, Lanese A, et al. HER family receptor expression and prognosis in pancreatic cancer. Int J Biol Markers. 2015;30:e327–32.
Rajagopal I, Niveditha SR, Sahadev R, Nagappa PK, Rajendra SG. HER 2 expression in gastric and gastro-esophageal junction (gej) adeno- carcinomas. J Clin Diagn Res. 2015;9:EC06–10.
Te Velde EA, Franke AC, van Hillegersberg R, et al. HER-family gene amplification and expression in resected pancreatic cancer. Eur J Surg Oncol. 2009;35:1098–104.
Safran H, Steinhoff M, Mangray S, et al. Overexpression of the HER-2/neu oncogene in pancreatic adenocarcinoma. Am J Clin Oncol. 2001;24:496–9.
Komoto M, Nakata B, Amano R, et al. HER2 overexpression correlates with survival after curative resection of pancreatic cancer. Cancer Sci. 2009;100:1243–7.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014:124: 188–95.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–28.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18.
Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med. 2012;14:405–15.
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010. 2010;18:843–51.
Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276:1719–24.
Thomis DC, Marktel S, Bonini C, Traversari C, Gilman M, Bordignon C, et al. A Fas-based suicide switch in human T cells for the treatment of graft-versus-host disease. Blood. 2001;97:1249–57.
Serafini M, Manganini M, Borleri G, Bonamino M, Imberti L, Biondi A, et al. Characterization of CD20-transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Hum Gene Ther 2004. 2004;15:63–76.
John LB, Devaud C, Duong CP, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by genemodified T cells. Clin Cancer Res. 2013;19:5636–46.
Vatner RE, Cooper BT, Vanpouille-Box C, et al. Combinations of immunotherapy and radiation in cancer therapy. Front Oncol. 2014;4:325.
Twyman-Saint Victor C, Rech AJ, Maity A, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–7.
Sandin LC, Eriksson F, Ellmark P, et al. Local CTLA4 blockade effectively restrains experimental pancreatic adenocarcinoma growth in vivo. Oncoimmunology. 2014;3:e27614.
Bunt SK, Mohr AM, Bailey JM, et al. Rosiglitazone and gemcitabine in combination reduces immune suppression and modulates T cell populations in pancreatic cancer. Cancer Immunol Immunother. 2013;62:225–36.
Eikawa S, Nishida M, Mizukami S, et al. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci USA. 2015;112:1809–14.
Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. Journal of immunotherapy (Hagerstown:1997). 2010; 33: 780.
Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392 – 402.
Moon EK, Carpenito C, Sun J, Wang L-CS, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin- specific chimeric antibody receptor. Clin Cancer Res. 2011;17(14):4719–30. https://doi.org/10.1158/1078-0432.CCR-11-0351.
Yurchenco PD, Schittny JC. Molecular architecture of basement membranes. FASEB J. 1990;4:1577–90.
Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lym-phocytes. Nat Med. 2015;21(5):524–9. https://doi.org/10.1038/nm.3833.
Acknowledgements
Special thanks to Dr. Manisha Dhananjaya.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest among authors.
Rights and permissions
About this article

Cite this article
Jindal, V., Arora, E., Masab, M. et al. Chimeric antigen receptor T cell therapy in pancreatic cancer: from research to practice. Med Oncol 35, 84 (2018). https://doi.org/10.1007/s12032-018-1145-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-018-1145-0