
Abstract
Breast cancer (BC) is the most common cancer in women worldwide. One in eight women will develop the disease in her lifetime. Notwithstanding the incredible progress made in this field, BC still represents the second most common cause of cancer-related death in women. Targeted drugs have revolutionised breast cancer treatment and improved the prognosis as well as the life expectancy of millions of women. However, the phenomenon of primary and secondary pharmacological resistance is becoming increasingly evident, limiting the efficacy of these agents and calling for a better in-depth knowledge and understanding of the biology as well as the biochemical crosstalk underlying the disease. The advent of laboratory technologies in the clinical setting such as the routine use of next generation sequencing has allowed identification of new genetic alterations as well as providing a precise picture of the molecular landscapes of each tumour. Consequently, new specific therapeutic approaches are becoming available to minimise or delay the occurrence of resistance. In this review, we analyse the latest research and news from the clinical development side for each BC subtype.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
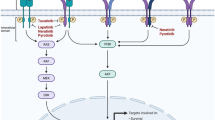
Breast cancer (BC) still remains the most common type of cancer occurring in women, second only to lung cancer as cause of mortality. Compared to the 1990s, the death rate of BC has slowly declined in 10 years, even though this cancer still remains frequent within the population [1]. Metastatic BC (mBC) is treatable but still virtually an incurable disease, with the main goals of care being finalised to the optimisation of length and quality of life. The European biannual Advanced Breast Cancer Conference (ABC) provides evidence-based international and multidisciplinary guidelines for the diagnosis and treatment of MBC confirming how systemic treatment should be tailored to the molecular characteristic of each individual case (Fig. 1).

Signal trasduction and treatments in breast cancer subtypes. Treatments change in accordance to the molecular characteristics of the tumour. In patients that are HR-positive and HER2-negative, the endocrine therapy should be the first-line of treatment. Chemotherapy could always be added during the course of the disease. ER oestrogen receptor, PR progesterone receptor, TDM-1 trastuzumab emtansine. *Conventionally, there is always a combination of endocrine therapy with ovarian suppression in pre-menopausal women. Ovarian suppression is recommended in pre-menopausal women in order to reduce the formation of the breast cancer-feeding hormones [2]
ER/PR+ve metastatic breast cancer
Around 70% of breast tumours express the oestrogen receptor (alpha) and/or the progesterone receptor [3, 4]. These tumours are generally characterised by a favourable prognosis. In fact, since the advent of tamoxifen, anti-oestrogen therapies have completely modified the natural history of the disease, becoming the backbone of hormone-positive BC treatment. However, aside from the cases of de novo resistance displayed by some of these tumours, eventually the majority of ER+ve metastatic breast cancers develop resistance to targeted anti-oestrogen treatment [5, 6]. Several mechanisms underlying the occurrence of pharmacological resistance to targeted therapies have been identified. Accumulation of new genetic mutations (i.e. ESR1) and/or constitutive activation of other signalling pathways represent some of the strategies that tumour cells employ to override oestrogen receptor inhibition [7]. In particular, phosphatidylinositol 3-kinase (PI3K)—mammalian target of rapamycin (mTOR) signalling pathway activation—has been shown to be associated with resistance to endocrine treatment [8–12].
Oestrogen receptor interaction with the ligand induces a biochemical cascade of events culminating, amongst others, with AKT phosphorylation and mTOR activation [13], which translates into cellular proliferation and metabolic signals, and ultimately survival advantage. Constitutive activation of the PI3K signal downstream, mainly mediated by activating mutations of the catalytic subunit of the kinase [13], AKT kinase mutations or PTEN tumour suppressor deletions [14] overcomes oestrogen receptor-targeted inhibition [15]. Moreover, PI3K activation has been shown to cause a decrease in ER levels and therefore a lesser degree of response to anti-oestrogen therapies [16]. It becomes clear how targeting the PI3 kinase pathway has the potential to restore sensitivity to ER inhibition and many studies are investigating this possibility using PI3K and mTOR inhibitors in combination with endocrine therapies.
The BOLERO-2 study evaluated the mTOR-specific inhibitor everolimus (Afinitor, Novartis) in combination with exemestane (Aromasin, Pfizer) in comparison with exemestane monotherapy in patients with metastatic breast cancer, previously treated with a non-steroidal aromatase inhibitor [17, 18]. The study enrolled 724 patients and showed a significant improvement with the combination regime in the progression-free survival (PFS) (10.6 versus 4.1 months) leading to the regulatory approval of the combination for mBC treatment. Very interestingly, in a quest for biomarkers of response to targeted therapies, a parallel translational study run during the BOLERO-2 found that the presence of multiple signalling pathways aberrations is associated with lack of response to everolimus [19]. The data may suggest a multiple targeted combinatorial approach should be reserved to these patients.
Everolimus is undergoing further studies to assess its anti-tumour activity in combination with endocrine therapies in adjuvant and neoadjuvant settings (BOLERO-4, NCT01698918; BOLERO-6, NCT01783444; NCT02291913; NCT02742051 and NCT01805271). Also, a new oral dual mTORC1/2 inhibitor, TAK228, is currently being evaluated in the neoadjuvant setting in combination with tamoxifen (NCT02988986) and letrozole (NCT02619669) in women with ER-positive, PR-positive high-risk early breast cancer.
Other PI3K signalling inhibitors have shown significant anti-tumour activity in preclinical studies and have therefore reached clinical development. AZD5363 a specific AKT inhibitor is being tested in monotherapy (NCT0277569 and NCT01226316) as well as in combination with fulvestrant (Faslodex, AstraZeneca) in metastatic BC previously progressed on aromatase inhibitors (NCT01992952). New PI3K-targeted therapies BYL719 (alpelisib) (specifically designed to target PI3KCA mutations) (NCT01219699 and NCT02437318) and taselisib (NCT02340221) are also being evaluated in the same combinatorial regime with fulvestrant, as well as with letrozole (NCT01923168, NCT01791478, NCT02273973). Buparlisib is also being evaluated in two Phase III trials in combination with fulvestrant in patients previously treated with AIs (BELLE-2, NCT01610284) and after resistance to mTOR inhibitors developed (BELLE-3, NCT01633060). Of note, the BELLE-4 trial evaluated buparlisib in combination with paclitaxel in advanced and metastatic BC with or without PI3K mutation (NCT01572727) and was terminated due to the absence of activity.
The FERGI study is a Phase II trial comparing fulvestrant versus fulvestrant plus pan-PI3K inhibitor pictilisib (GDC-0941) in post-menopausal patients with ER-positive metastatic BC [20, 21]. Results showed no improvement in the median progression-free survival (PFS) with the combination. Also, the study failed to show any correlation between the PI3K mutational status and the combination efficacy [20]. These data are in contrast with the report from the BELLE-2 and BELLE-3 with buparlisib; the mutation status of PI3KCa was predictive of response/outcome in those patients receiving the PI3Kca inhibitor [22]. Taken altogether, these data suggest more translational studies are required in these settings to understand the clinical role of the PI3Kca status with regard to the PI3KCa or/and mTOR inhibitors in mBC.
About the importance of the PI3KCa status, it will also be interesting to evaluate whether specific PI3K isoforms inhibitors in development are more effective than the pan inhibitors [23].
Moreover, inhibition of PI3K or mTOR signalling seems to induce pharmacologic resistance via activation of parallel pathways. Therefore, inhibition of AKT kinase or PI3K upstream of mTOR or combined inhibition of PI3K and mTOR together seems to be the preferred strategies [24]. In fact, dual inhibition of PI3K and mTOR is currently being assessed in a Phase II trial looking at the combination of BYL719 and everolimus plus exemestane (NCT02077933).
Of note, the association of metformin to the above-mentioned combinations (NCT01589367 and NCT01042379) appears to add to the anti-tumour activity, perhaps decreasing the insulin receptor (IR) expression as well as AKT phosphorylation [25] and avoiding the occurrence of hyperglycaemia (the major drug-related toxicity associated with PI3K inhibitors, due to mTOR activation and inhibitory feedback to IGF) [26–28] and insulin resistance.
Disruption of the cyclin D/CDK/pRB pathway occurs in 50–70% of breast cancers [3]. Cyclin D1 amplification is the most frequent alteration and is found in around 70% of BCs [29], in particular in luminal B and HER2-positive BCs [3]. A growing body of evidence suggests the existence of a crosstalk between oestrogen receptor and the cyclin D1/CDK4-6/RB pathway, via downstream signals such as PI3K and mTORC or the convergence of other growth factors intracellular signals [30]. This crosstalk overrides the inhibition of the oestrogen receptor upstream, therefore being responsible for resistance to anti-oestrogen therapies [31] as well as to PI3K-targeted therapies. Also, retinoblastoma tumour suppressor gene-negative breast tumours have shown resistance to tamoxifen in xenografts models and in the clinic, further confirming the convergence of the oestrogen and cyclin D/CDK4/6/RB pathways [32, 33].
Palbociclib (Ibrance, Pfizer Inc.) as a single agent showed minimal activity in a Phase II study in patients with RB-positive breast cancer, with 7% partial responses and 14% rate of stable disease lasting for more than 6 months [34]. Although “partial”, these results indicated the potential for synergistic anti-tumour activity in combination with other targeted drugs and the available data pointed towards a combination with hormone therapies. PALOMA-1, a randomised Phase I/II trial, investigated the combination of palbociclib and letrozole in comparison with letrozole alone, for the treatment of advanced breast cancers in post-menopausal women [35]. The following Phase 2 study showed superiority of the combination of palbociclib and letrozole in comparison with letrozole with a marked increase in the progression-free survival from 7.5 to 26.1 months (HR 0.37, p < 0.001) [36]. The combination was very well tolerated with neutropenia, leukopenia, anaemia and fatigue being the main toxicities detected. These outstanding results gained palbociclib the designation of “breakthrough therapy” and subsequent approval by the Food and Drug Administration (FDA) as new first-line therapy for patients with advanced or metastatic ER-positive, HER2-negative tumours. Further results for the overall survival analysis will be gained from an ongoing randomised Phase III study (PALOMA-2, NCT01942135). Palbociclib was also granted FDA approval in combination with fulvestrant, as first-line treatment of metastatic ER-positive, HER2-negative breast tumours which have progressed after prior endocrine therapy, irrespective of menopausal status, on the basis of the results obtained in the PALOMA-3 study (palbociclib combined with fulvestrant in hormone receptor-positive HER2-negative metastatic breast cancer after endocrine failure [37]).
The other CDK4/6 inhibitors have also yielded extremely clinically relevant results. Ribociclib (LEE011, Novartis) was granted “breakthrough therapy designation” by the FDA in view of the results of the Phase 3 MONALEESA-2 trial. In this study, the combination of ribociclib and letrozole significantly improved progression-free survival (PFS) from 14.7 months in the placebo group to 19.3 months to “not reached” in the ribociclib group (HR 0.59; p = 0.002) in first-line treatment of post-menopausal women with ER-positive/HER2-negative advanced breast cancer [38]. At 18 months, PFS rate in the experimental arm was 63% (95% CI, 54.6–70.3) versus 42.2% in the letrozole arm (95% CI, 34.8–49.5). Other ribociclib-containing combinations are currently being evaluated [39]. Abemaciclib (LY283519, Eli Lilly), another CDK4/6 inhibitor in clinical development, was also granted “breakthrough therapy designation” as a single agent for the treatment of patients with hormone receptor-positive advanced or metastatic breast cancer on the basis of the MONARCH 1 clinical trial results. [40] The drug is currently being evaluated in two Phase 3 trials in comparison with fulvestrant (MONARCH 2) and with a non-steroidal aromatase inhibitor (MONARCH 3) in post-menopausal patients with advanced or metastatic ER-positive/HER2-negative breast cancer [40, 41].
Several trials are investigating multiple combinations of cell cycle inhibitors, PI3K inhibitors and endocrine therapies (NCT02088684 and NCT01872260) [42, 43].
ESR1 mutation in ER+/PR+ metastatic breast cancer and new target tehrapies
The occurrence of new somatic mutations is one of the well-recognised mechanisms of acquired resistance to endocrine therapies in metastatic BC. Recently, two different research groups independently reported relatively high prevalence of previously described mutations of the oestrogen receptor 1 (ESR1) in patients with metastatic BC [42, 43]. Single nucleotide mutations in the ligand-binding domain of the ESR1 gene give rise to constitutively active mutated variants of the receptor, which retain ligand-independent activity [44, 45]. Toy et al. [43] identified ESR1 mutations in 9/36 (25%) metastatic tissue samples from women with ER+ metastatic BC previously treated with multiple endocrine therapies regimes. In the same fashion, further studies found ESR1 mutations in 55 and 38 and 12% of metastatic biopsies from patients with ER+ metastatic BC pre-treated with multiple endocrine therapies [42, 46, 47]. Whilst the first reports linked the occurrence of ESR1 mutations to previous aromatase inhibitors containing regimens, further studies showed the mutations in treatment-naïve and tamoxifen-treated patients [46, 48, 49]. With the advent of “liquid biopsies”, it has become much easier and less invasive for the patient to collect information on the genetic status of the metastases using circulating tumour cells (CTCs) and free circulating DNA (cfDNA). In fact, both of these parameters are thought to provide reliable “indirect pictures” of metastatic disease [50]. Therefore, larger cohorts of patients have been investigated and the ESR1 mutation impact on treatment and prognostic significance looked at [51].
Cell-free DNA in the blood of women enrolled in the BOLERO-2 trial showed a 29% prevalence of ESR1 mutations in this patient cohort, with 6% of the patients carrying double mutations. When the authors looked at progression-free survival (PFS) in the exemestane arm of the trial, patients with ESR1 mutation (D538G) showed a decreased PFS in comparison with the wild-type receptor patients (2.7 vs. 3.9 months, respectively). In the everolimus plus exemestane arm, the PFS increased consistently irrespective of the mutational status of the receptor [51]. When the overall survival (OS) was examined, patients with ESR1 mutations did worse than wild-type patients (median OS 22 vs. 32 months, respectively) and it seems that the type of genetic mutation influenced the OS figures.
In another study (PALOMA-3), patients with advanced or metastatic BC were randomised to receive fulvestrant alone or in combination with palbociclib [52]. In this trial, analysis of the ESR1 status revealed a prevalence of 25% mutations. The benefit of the combination therapy in terms of PFS was seen in the ESR1-mutated population as well as in the wild-type population (with an increase in the PFS from 3.6–9.4 in patients treated with the combination regime).
The intrinsic characteristics of the mutations imply that only drugs that interact directly with the oestrogen receptor, such as SERMs and SERDs, are able to retain their activity against the mutated cells, whereas aromatase inhibitors are not effective in these patients. In fact, as demonstrated in the FERGI study, fulvestrant is active in the patients who carry the ESR1 mutation (37% of the total number of patients in this cohort), with a similar PFS in wild-type and mutated patients in both arms of the trial irrespective of the presence of the PI3K inhibitor [53].
New SERDs are in different stages of clinical development and eagerly waited for. The GDC-810 is a new orally available selective oestrogen receptor degrader currently being evaluated in patients with metastatic breast cancer with or without ESR1 mutations (NCT01823835), after the encouraging results showed in a Phase I trial [54]. Other SERDs in clinical development are Rad-1901, NCT02338349; AZD-9456, NCT02248090; and LCZ-102, NCT02734615.
It is noteworthy that these new drugs are orally available. Their better bioavailability characteristics will likely overcome the limits of fulvestrant and perhaps favour a more extensive use of this very effective family of oestrogen receptor inhibitors.
The main ongoing clinical trials studying safety and anti-tumour activity of experimental compounds or combinations of targeted drugs in ER/PR+ve metastatic BC are detailed in Table 1.
HER2-positive metastatic breast cancer
Human epidermal growth factor receptor 2 gene amplification and/or protein overexpression are found in 15–20% primary breast tumours [55–57] and are associated with aggressive biological behaviour and poor prognosis [55]. Notably, in inflammatory BC, Zell et al. [58] reported up to 40% HER2 protein overexpression. Other epithelial-derived tumours aside from breast cancer have been shown to carry protein overexpression or gene amplification of HER2 (also reviewed by Yan et al. 2014).
HER2-positive BC effectively illustrates the concept of “oncogene addiction”: tumour cells of this subtype depend on activation and downstream signalling of the main “driver”, HER2, for proliferation and survival. Inhibition of the signalling cascade induces cell cycle arrest, apoptosis and tumour shrinkage in vivo. This phenomenon, together with the transmembrane position and the tyrosine kinase activity of the receptor, made HER2 an extremely attractive therapeutic target. Trastuzumab (Herceptin, Genentech) was the first monoclonal antibody specifically directed against the juxtamembrane portion of the HER2 receptor [59]. Its advent, as well as profoundly changing the approach to cancer therapy, has greatly improved the prognosis of HER2-positive BC [60], not only in the early stages, but also in patients with advanced or metastatic disease [61]. The combination of trastuzumab with chemotherapy in the adjuvant and neoadjuvant settings represents now the standard treatment for this BC subtype, after several studies demonstrated the significant superiority of the combination over chemotherapy alone [60, 62, 63]. Notwithstanding the incredible results achieved with trastuzumab, the phenomenon of primary and secondary pharmacological resistance has become increasingly frequent, ultimately limiting treatment outcome [64].
Most of the molecular changes investigated to explain de novo and/or acquired resistance to HER2-targeted therapies are to be attributed to the phenomenon of the “adaptive response”: when the primary oncogenic driver is inhibited, tumour cells are able to survive by activating other “secondary” signalling pathways, ultimately overcoming their “oncogenic dependency” on the main mutation.
Several signalling pathways (either downstream of or “parallel” to HER2) are activated in response to anti-HER2 therapies and therefore responsible for pharmacologic resistance. Phosphoinositide 3-Kinase (PI3K-Akt), EGFR, IGFR, mTOR and MAPK/ERK pathways are the most commonly involved [65–69].
Loss of PTEN or activation of mTOR is able to overcome HER2 signal blockade [69, 70]. Other molecular aberrations involved with trastuzumab resistance include gene mutations and expression of a truncated form of the HER2 protein [71].
More recently, after several reports highlighted the presence of cell cycle molecular aberrations in HER2-positive BC, as well as pointing out the requirement for cyclin D1 and CDK4/6 in HER2 models of tumourigenesis [3, 72, 73], Goel et al. [74] provided stronger evidence of the involvement of the cell cycle in pharmacologic resistance to targeted anti-HER2 therapies. Hyper-activation of the cyclin D1/CDK4/6/pRB pathway has been linked to acquired resistance to anti-HER2 targeted therapies. A small group of tumour cells were shown to be able to survive HER2 targeted inhibition: nuclear overexpression of cyclin D1, the common molecular feature amongst these cells, represents direct indication of cell cycle activation [74].
These data altogether provide enough evidence to highlight the need for new more effective anti-HER2 therapies, specifically in a combinatorial approach, to block the crosstalk between pathways, to achieve simultaneous inhibition of multiple signalling pathways or to obtain complete HER2 signal blockade (using more than one targeted therapy when the mechanisms of action of each compound differ), therefore preventing pharmacologic resistance [75–77].
There is increasing evidence that some of these approaches are paying off.
So far, four anti-HER2 targeted therapies have become available: trastuzumab, pertuzumab, lapatinib and ado-trastuzumab emtansine. Review of each of these agents is beyond the scope of this article. More in-depth information can be found elsewhere [78–80].
Lapatinib (Tykerb, GlaxoSmithKline) is a small molecule with tyrosine kinase activity, which therefore displays totally different characteristics in terms of mechanism of action, safety and toxicity profile from those of trastuzumab. In particular, lapatinib is effective in patients whose tumours express the truncated HER2 protein [81] or lack the tumour suppressor PTEN [82]. Instead, both these aberrations induce resistance to trastuzumab. This is just one example reinforcing the concept of a dual blockade of the epidermal growth factor receptor 2. In fact, two recent trials NeoALTTO and NeoSphere have confirmed superior efficacy of the dual blockade/combination of anti-HER2 agents in comparison with single therapy with each agent alone in early BC [83, 84]. Both studies showed much higher rates of pathologic complete remission when the patients received either trastuzumab plus lapatinib or trastuzumab plus pertuzumab, plus chemotherapy, rather than anti-HER2 monotherapy plus chemotherapy. Lapatinib is currently approved in combination with letrozole or capecitabine for HER2-positive metastatic BC resistant to trastuzumab, when hormonal therapy is indicated.
Pertuzumab (Perjeta, Genentech) inhibits the dimerisation of HER2 and therefore signalling through the receptor. The CLEOPATRA study evaluated the combination of trastuzumab plus chemotherapy plus or minus pertuzumab as first-line therapy for patients with metastatic breast cancer (CLEOPATRA, NCT00567190) [85]. The study showed significant PFS (6.3 months difference) and OS (15.7 months difference) improvements in patients treated with the combination of pertuzumab, trastuzumab and docetaxel versus patients randomised to receive only trastuzumab plus chemotherapy. These data contributed to the FDA approval of pertuzumab in combination with trastuzumab plus chemotherapy (docetaxel) in patients with HER2-positive metastatic BC. In 2013, the antibody was also approved for the neoadjuvant treatment of HER2-positive BC [86].
Pertuzumab is currently being tested in combination with other approved or experimental drugs in the neoadjuvant setting in patients with BC (I-SPY 2 trial, NCT01042379).
Neratinib and afatinib are the second-generation small molecule inhibitors of HER1, HER2 and HER4 (only neratinib) [78, 87]. These agents are currently being evaluated in the clinic in monotherapy or in combination with chemotherapy as well as with other targeted drugs, in the adjuvant and neoadjuvant settings (NCT00915018, NCT00878709, NCT01271725, NCT01441596, NCT01125566) [88, 89]. A Phase II randomised trial of neratinib monotherapy versus the combination of lapatinib plus capecitabine (approved by the FDA) in HER2-positive advanced BC showed significant activity of the second-generation HER2 inhibitor (neratinib) as single-agent therapy, but failed to demonstrate the superiority or inferiority of this molecule to lapatinib plus capecitabine [90].
The LUX-Breast 1 trial (NCT01125566) also compared trastuzumab plus vinorelbine to afatinib plus vinorelbine in patients with metastatic HER2 BC previously treated with at least one trastuzumab-based regime, but failed to show the superiority of the afatinib/vinorelbine combination.
Tucatinib (ONT-380) is also being evaluated in combination with other HER2 inhibitors in Phase I clinical trials (NCT01921335, NCT0198501 and NCT02025192).
Other trials are underway to study the anti-tumour effect of these new agents, as well as to find markers of response to treatment, in breast cancer (NCT01670877 and NCT01042379) and other solid tumours overexpressing HERI or HER2 (mainly lung and gastric cancer) (NCT01522768). An area of great interest and possible application for these second-generation inhibitors, as well as for lapatinib, are the treatment of central nervous system metastatic disease (NCT02650752 and NCT01921335) [91]. The incidence of brain metastases in patients with advanced HER2-positive breast cancer is in fact relatively high, and successful treatment of distant disease in this area would tremendously improve the prognosis for this patient cohort [92, 93].
T-DM1 (Kadcyla, Genentech), a conjugate drug coupling trastuzumab with a cytotoxic anti-microtubule drug called maytansinoid, has shown good results in the early phases of clinical development [94, 95] and was approved by the FDA in February 2013 for treatment of HER2-positive metastatic BC previously treated with trastuzumab and taxanes, after the encouraging results of the EMILIA trial [96]. This agent is currently being tested in a Phase Ib/II trial in combination with the anti-CDK4/6 agent ribociclib compared to trastuzumab plus ribociclib in women with metastatic HER2-positive BC (NCT02657343). New antibody–drug coupled compounds are being evaluated in the clinic: MM-302 uses nanoparticle technology to deliver anthracyclines to HER2-positive tumours cells. The HERMIONE trial looked at the combination of MM-302 with trastuzumab in comparison with chemotherapy plus trastuzumab in patients with advanced/metastatic HER2-positive breast cancer never treated with anthracyclines, who progressed under trastuzumab or TDM-1 therapy (NCT02213744) [97]. The study was prematurely terminated as the experimental combination failed to show any benefit.
Data from BOLERO-3 [98] support the combinatorial approach with anti-mTOR and trastuzumab for women with trastuzumab-resistant, HER2-positive advanced BC.
Other PI3K inhibitors are being evaluated in the clinic for this type of metastatic BC: the PANTHER study, a Phase Ib/II single-arm trial, is currently evaluating the safety, pharmacokinetic profile as well as the anti-tumour activity of copanlisib (PI3Kinase inhibitor) in combination with trastuzumab in recurrent or metastatic HER2-positive BC previously pre-treated with anti-HER2 targeted therapies (NCT02705859).
The first clinical studies testing the combination of trastuzumab and palbociclib, in advanced or metastatic cancers, as well as other combinations of CDK4/6 inhibitors with targeted anti-cancer therapies are also underway (Table 2). The NA-PHER2 trial (NCT02530424) investigates the combination of trastuzumab, pertuzumab, palbociclib and fulvestrant for neoadjuvant treatment of ER-positive, HER2-positive invasive breast cancer. The PATRICIA study (NCT02448420) is a Phase 2 trial designed to compare the combination of palbociclib and trastuzumab plus or minus letrozole for the treatment of HER2-positive, ER+ or ER−, locally advanced or metastatic breast cancer in post-menopausal women previously treated with chemotherapy and trastuzumab.
Lastly, immunotherapies are currently being tested in this molecular subtype of cancer (Table 2): atezolizumab is being evaluated in combination with trastuzumab emtansine (T-DM1) or with trastuzumab plus pertuzumab in patients with HER2-positive BC (NCT02605915), whilst PANACEA is a Phase Ib/II trial investigating the efficacy of pembrolizumab in combination with trastuzumab in women with HER2-positive, metastatic breast cancer, who progressed whilst on trastuzumab (NCT02129556).
As it becomes clear from the type and number of ongoing studies, the current issues regarding the therapeutic approach to HER2-positive breast cancer are being addressed mainly by designing multiple combinations of agents that target different features of transformed cells at the same time. This strategy weakens the very ability of cancer to “adapt” to targeted therapies, depriving cells of the biochemical signals necessary for survival and proliferation. However, more clinical trials are needed to identify reliable biomarkers with the aim to define and target subgroups of patients that are most likely to benefit from specific combinations of therapies. Such achievement will really start the era of “personalised” medicine.
New therapeutical strategies in triple-negative metastatic breast cancer
Historically the idea of immunotherapy against cancer goes back to the early 1910s when William Coley proved that injection of Streptococcus and Serratia into tumours (specifically round-cell sarcoma), caused them to shrink [99]. The bacteria evoked an immune response that ultimately led to shrinkage of the cancer. Today various immunotherapies have been developed based on four different strategies: (1) non-specific immune stimulation, (2) adaptive cell transfer, (3) vaccination and (4) immune checkpoint blockade. The latter represents the most promising approach, in particular after the outstanding results obtained in melanoma and NSCLC [100, 101].
Breast cancer tissue immune profiling exposed how T-lymphocytes represent the main population of immune cells found in the context of the tumour (70–80%), with the rest of the immune cells composed of B-lymphocytes, macrophages, antigen-presenting cells (APC) and natural killer cells (NKC) [102, 103]. A portion of TNBC has been proved to be highly immunogenic, showing relatively high percentage of tumour infiltrating lymphocytes (TILs) [104], higher levels of B7 gene family member type I [105] programmed cell death 1 ligand 1 (PD–L1) protein [106, 107] and mRNA [108, 109] expression levels in comparison with other breast cancer subtypes. It has been shown that there is a strong and consistent correlation between the presence of immune markers and/or TILs and the likelihood of achieving a pCR after neoadjuvant chemotherapy in TNBC [110]. High expression of tumour markers and high TIL density are associated with benefit from chemotherapy in TNBC [111–114]. These data suggest that chemotherapy regimens elicit better results in the presence of a proficient immune system, as initially proposed by preclinical data [115, 116]. Moreover, many research groups have found a strong association between high levels of immune markers or TILs and low risk of relapse and/or death of TNBC patients early treated with systemic chemotherapy [104, 108, 117–126]. These data altogether provide a strong rationale for testing immunotherapies in a highly immunogenic cohort of TNBC.
Many immunotherapy-based studies on breast cancer [126–130] suggest an effective engagement of the immune system, although insufficient to entirely eradicate the tumour, can help reduce the risk of metastasis or maintain tumour dormancy [131].
A new class of immunotherapies targeting the so-called immune checkpoints (mainly PD-1 and its ligand, PDL-1 and CTLA-4) has shown promising results.
Two Phase I trials evaluated immune checkpoint inhibitors in advanced-stage TNBC [132, 133]. In one of them (KEYNOTE-012), anti-PD-1 pembrolizumab induced a response in 18.5% of ER-positive breast cancer patients enrolled in the study [132]. Expression of PD-L1 measured by immunohistochemistry (IHC) was used as criteria for selecting patients for these two trials [132, 133]. However, the validity of PD-L1 as a selection marker remains still controversial from data coming from studies on other solid tumours, where substantial survival benefit was shown when anti-PD-L1 antibodies were used in PD-L1-negative patients [134, 135]. Table 1 summarises key ongoing clinical trials evaluating immune checkpoint inhibitors in combination with chemotherapy in patients with metastatic TNBC. In the Phase III trial NeoTRIPaPDL1 (NCT02620280), patients with metastatic TNBC will be randomly assigned to receive nab-paclitaxel and carboplatin with or without PD-L1 inhibitor (atezolizumab). In another Phase II trial (NCT02530489), atezolizumab will be evaluated in combination with nab-paclitaxel. Also, the safety and efficacy of anti-PD-L1 inhibitor durvalumab will be tested in combination with nab-paclitaxel followed by dose-dense chemotherapy containing cyclophosphamide and doxorubicin in a Phase I/II trial (NCT02489448). Overall, the monoclonal antibodies anti-PD-1 pembrolizumab (Keytruda), nivolumab (Opdivo), PDR001 and immunotherapy drugs, the anti-CTLA4 tremelimumab and anti-PD-L1 atezolizumab (MPDL3280a), MEDI4736 and durvalumab have been tested in patients with metastatic TNBC in the ongoing clinical trials in Table 3.
Other noteworthy molecular targets under clinical investigation for metastatic TNBC are PARP inhibitors, exploited for their “synthetic lethality” [136] PI3K inhibitors [137], histone deacetylase (HDAC) inhibitors [138, 139], MEK inhibitors [140], heat shock protein 90 (HSP90) inhibitors [141, 142], EGFR inhibitors [143], FGFR inhibitors [144, 145] and angiogenic pathway inhibitors [143–145]. There are several PARP inhibitors currently being tested in clinical trials, such as veliparib (Phase III study; NCT02163694), talazoparib (Phase III study; NCT01945775), niraparib (Phase III study; NCT01905592) and rucaparib (Phase I/II study; NCT01074970) to treat patients with MBC, including TNBC; buparlisib a pan-PI3K inhibitor (buparlisib, BKM120, Novartis) is being evaluated in combination with olaparib to treat patients with solid tumours that include met TNBC (Phase I study; NCT01623349); lastly, HDAC inhibitor KHK2375 is being tested either as a monotherapy (Phase I study; NCT02623751) or in combination with cisplatin, in the treatment of patients with met TNBC (Phase I/II study; NCT02393794).
Conclusions
In this review, we have highlighted the importance of integrating biological and clinical data for the clinical development of new drugs in metastatic breast cancer. The new drugs, recently approved in the different settings of mBC, provide concrete clinical opportunities to induce “chronicity” of disease with a respect to the patient’s quality of life. Careful selection of patients and growing opportunities for enrolment in clinical trials represents a great chance to expand treatment options whilst deepening our understanding of cancer biology. Indeed, a molecular stratification of breast cancer patients would be the key for future research in the field helping into the identification of the proper drug for any single patient. In this scenario, various ongoing Phase III clinical trials are testing the efficacy of new molecules according to the hypothesised timing of the single BC subset driver, such as ER or HER2+ or immune phenotype. Novel molecular targets are also emerging in subtypes of breast cancer traditionally lacking actionable mutations. Furthermore, biomarker studies in the metastatic setting should be conducted in the upcoming future and new tools for molecular diagnosis (such as CNV or protein gene expression profile) should become more readily available to move to the concept of precision medicine.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA. Cancer J Clin. 2016. http://www.ncbi.nlm.nih.gov/pubmed/28055103.
Klijn JG, Blamey RW, Boccardo F, Tominaga T, Duchateau L, Sylvester R, et al. Combined tamoxifen and luteinizing hormone-releasing hormone (LHRH) agonist versus LHRH agonist alone in premenopausal advanced breast cancer: a meta-analysis of four randomized trials. J Clin Oncol. 2001;19:343–53. http://www.ncbi.nlm.nih.gov/pubmed/11208825.
Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. Nature Research; 2012;490:61–70. http://www.nature.com/doifinder/10.1038/nature11412.
Dunnwald LK, Rossing M, Li CI. Hormone receptor status, tumor characteristics, and prognosis: a prospective cohort of breast cancer patients. Breast Cancer Res. 2007;9:R6. http://www.ncbi.nlm.nih.gov/pubmed/17239243.
Buzdar AU. Phase III study of letrozole versus tamoxifen as first-line therapy of advanced breast cancer in postmenopausal women: analysis of survival and update of efficacy from the international letrozole breast cancer group. J Clin Oncol. 2004;22:3199-200-1. http://www.jco.org/cgi/doi/10.1200/JCO.2004.99.058.
Dalmau E, Armengol-Alonso A, Muñoz M, Seguí-Palmer MÁ. Current status of hormone therapy in patients with hormone receptor positive (HR+) advanced breast cancer. The Breast. 2014;23:710–20. http://www.ncbi.nlm.nih.gov/pubmed/25311296.
Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011; 62:233–47. http://www.ncbi.nlm.nih.gov/pubmed/20887199.
Miller TW, Hennessy BT, González-Angulo AM, Fox EM, Mills GB, Chen H, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor–positive human breast cancer. J Clin Invest. 2010;120:2406–13. http://www.ncbi.nlm.nih.gov/pubmed/20530877.
Paplomata E, O’Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol. 2014;6:154–66. http://www.ncbi.nlm.nih.gov/pubmed/25057302.
deGraffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, et al. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt activity. Clin Cancer Res. 2004;10:8059–67. http://www.ncbi.nlm.nih.gov/pubmed/15585641.
Bhattacharvva GS, Biswas J, Singh JK, Singh M, Govindbabu K, Ranade AA, et al. Reversal of tamoxifen resistance (hormone resistance) by addition of sirolimus (mTOR inhibitor) in metastatic breast cancer. Eur J Cancer. 2011;47:9. http://linkinghub.elsevier.com/retrieve/pii/S0959804911701150.
Generali D, Fox SB, Brizzi MP, Allevi G, Bonardi S, Aguggini S, et al. Down-regulation of phosphatidylinositol 3′-Kinase/AKT/molecular target of rapamycin metabolic pathway by primary letrozole-based therapy in human breast cancer. Clin Cancer Res. 2008;May 1;14(9):2673-80
Liao JK, Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–41. http://www.ncbi.nlm.nih.gov/pubmed/11029009.
Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo W-L, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT Mutations in breast cancer. Cancer Res. 2008;68:6084–91. http://www.ncbi.nlm.nih.gov/pubmed/18676830.
Creighton CJ, Fu X, Hennessy BT, Casa AJ, Zhang Y, Gonzalez-Angulo AM, et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010;12:R40. http://www.ncbi.nlm.nih.gov/pubmed/20569503.
Fu X, Osborne CK, Schiff R. Biology and therapeutic potential of PI3K signaling in ER+/HER2-negative breast cancer. The Breast. 2013; 22:S12–8. http://www.ncbi.nlm.nih.gov/pubmed/24011769.
Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. N Engl J Med. 2012;366:520–9. http://www.nejm.org/doi/10.1056/NEJMoa1109653.
Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2. Ann Oncol. 2014;25:2357–62. http://www.ncbi.nlm.nih.gov/pubmed/25231953.
Hortobagyi GN, Piccart-Gebhart MJ, Burris HA, Campone M, Noguchi S, Perez AT, et al. Correlation of molecular alterations with efficacy of everolimus in hormone-receptor-positive, HER2-negative advanced breast cancer: Results from BOLERO-2. J Clin Oncol. 2013;31:142. http://www.ncbi.nlm.nih.gov/pubmed/28136528.
Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17:811–21. http://linkinghub.elsevier.com/retrieve/pii/S1470204516001066.
Krop I, Johnston S, Mayer IA, Dickler M, Ganju V, Forero-Torres A, et al. Abstract S2-02: The FERGI phase II study of the PI3K inhibitor pictilisib (GDC-0941) plus fulvestrant vs fulvestrant plus placebo in patients with ER+ , aromatase inhibitor (AI)-resistant advanced or metastatic breast cancer—Part I results. Cancer Res. 2015;75:S2-2-S2-2. http://cancerres.aacrjournals.org/lookup/doi/10.1158/1538-7445.SABCS14-S2-02.
Baselga J, Im S-A, Iwata H, Clemons M, Ito Y, Awada A, et al. PIK3CA status in circulating tumor DNA predicts efficacy of buparlisib plus fulvestrant in postmenopausal women with endocrine-resistant HR+/HER2—advanced breast cancer: first results from the randomized, Phase III BELLE-2 Trial. Emmanuelle Di Tomaso. Patrick Urban; 2015;18.
Juric D, Rodon J, Gonzalez-Angulo AM, Burris HA, Bendell J, Berlin JD, et al. Abstract CT-01: BYL719, a next generation PI3K alpha specific inhibitor: preliminary safety, PK, and efficacy results from the first-in-human study. Cancer Res. 2014;72.
Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6 K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–6. http://www.ncbi.nlm.nih.gov/pubmed/15380067.
Dowling RJ, Niraula S, Chang MC, Done SJ, Ennis M, McCready DR, et al. Changes in insulin receptor signaling underlie neoadjuvant metformin administration in breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res. 2015;17:32. http://www.ncbi.nlm.nih.gov/pubmed/25849721.
Bonanni B, Puntoni M, Cazzaniga M, Pruneri G, Serrano D, Guerrieri-Gonzaga A, et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J Clin Oncol. 2012;30:2593–600. http://www.ncbi.nlm.nih.gov/pubmed/22564993.
Cazzaniga M, DeCensi A, Pruneri G, Puntoni M, Bottiglieri L, Varricchio C, et al. The effect of metformin on apoptosis in a breast cancer presurgical trial. Br J Cancer. 2013;109:2792–7. http://www.ncbi.nlm.nih.gov/pubmed/24157825.
DeCensi A, Puntoni M, Gandini S, Guerrieri-Gonzaga A, Johansson HA, Cazzaniga M, et al. Differential effects of metformin on breast cancer proliferation according to markers of insulin resistance and tumor subtype in a randomized presurgical trial. Breast Cancer Res Treat. 2014;148:81–90. http://www.ncbi.nlm.nih.gov/pubmed/25253174.
Ehab M, Elbaz M. Profile of palbociclib in the treatment of metastatic breast cancer. Breast Cancer. 2016;8:83–91. http://www.ncbi.nlm.nih.gov/pubmed/27274308.
Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136–49. http://www.ncbi.nlm.nih.gov/pubmed/25002028.
Stendahl M, Kronblad Å, Rydén L, Emdin S, Bengtsson NO, Landberg G. Cyclin D1 overexpression is a negative predictive factor for tamoxifen response in postmenopausal breast cancer patients. Br J Cancer. 2004;90:1942–8. http://www.ncbi.nlm.nih.gov/pubmed/15138475.
Lehn S, Fernö M, Jirström K, Rydén L, Landberg G. A non-functional retinoblastoma tumor suppressor (RB) pathway in premenopausal breast cancer is associated with resistance to tamoxifen. Cell Cycle. 2011;10:956–62. http://www.ncbi.nlm.nih.gov/pubmed/21358261.
Bosco EE, Wang Y, Xu H, Zilfou JT, Knudsen KE, Aronow BJ, et al. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest. 2007;117:218–28. http://www.ncbi.nlm.nih.gov/pubmed/17160137.
DeMichele A, Clark AS, Heitjan D, Randolph S, Gallagher M, Lal P, et al. Phase I study of PD 0332991, cyclin-D kinase (CDK) 4/6 inhibitor in combination with letrozole for first-line treatment of patients with ER-positive, HER2-negative breast cancer. 2010 ASCO Annual Meeting, Abstracts, Meeting Library. J Clin Oncol. 31 (suppl; abstr 519). 2013. http://meetinglibrary.asco.org/content/50680-74.
Slamon DJ, Hurvitz SA, Applebaum S, Glaspy JA, Allison MK, DiCarlo BA, et al. A phase II trial of an oral CDK 4/6 inhibitor, PD0332991, in advanced breast cancer. 2013 ASCO Annual Meeting, Abstracts, Meeting Library. J Clin Oncol 2815 s, 2010 (suppl; abstr 3060). 2010. http://meetinglibrary.asco.org/content/113257-132.
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35. http://linkinghub.elsevier.com/retrieve/pii/S1470204514711593.
Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, et al. Palbociclib in hormone-receptor–positive advanced breast cancer. N Engl J Med. 2015;373:209–19. http://www.ncbi.nlm.nih.gov/pubmed/26030518.
Hortobagyi GN, Stemmer SM, Burris HA, Yap Y-S, Sonke GS, Paluch-Shimon S, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738–48. http://www.nejm.org/doi/10.1056/NEJMoa1609709.
Munster PN, Hamilton EP, Franklin C, Bhansali S, Wan K, Hewes B, et al. Phase lb study of LEE011 and BYL719 in combination with letrozole in estrogen receptor-positive, HER2-negative breast cancer (ER+ , HER2− BC). 2014 ASCO Annual Meeting, Abstracts, Meeting Library. J Clin Oncol. 325 s, 2014 (suppl; abstr 533). 2014. http://meetinglibrary.asco.org/content/127461-144.
Dickler MN, Tolaney SM, Rugo HS, Cortes J, Diéras V, Patt DA, et al. MONARCH1: Results from a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as monotherapy, in patients with HR+/HER2- breast cancer, after chemotherapy for advanced disease.2016 ASCO Annual Meeting, Abstracts, Meeting Library. 2016 ASCO Annu. Meet. 2016. http://meetinglibrary.asco.org/content/164546-176.
Tolaney SM, Rosen LS, Beeram M, Goldman JW, Gandhi AW, Papadopoulos KP, et al. Clinical activity of abemaciclib, an oral cell cycle inhibitor, in metastatic breast cancer. Cancer Res. 2015;75:P5-19-13. doi: 10.1158/1538-7445.SABCS14-P5-19-13
Robinson DR, Wu Y-M, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–51. http://www.ncbi.nlm.nih.gov/pubmed/24185510.
Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–45. http://www.ncbi.nlm.nih.gov/pubmed/24185512.
Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol. 1996;10:1388–98. http://www.ncbi.nlm.nih.gov/pubmed/8923465.
Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 1997;57:1244–9. http://www.ncbi.nlm.nih.gov/pubmed/9102207.
Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor- mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757–67. http://www.ncbi.nlm.nih.gov/pubmed/24398047.
Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013;73:6856–64. http://www.ncbi.nlm.nih.gov/pubmed/24217577.
Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Inao T, Sueta A, Fujiwara S, et al. Droplet digital polymerase chain reaction assay for screening of ESR1 mutations in 325 breast cancer specimens. Transl Res. 2015;166:540–553.e2. http://www.ncbi.nlm.nih.gov/pubmed/26434753.
Wang P, Bahreini A, Gyanchandani R, Lucas PC, Hartmaier RJ, Watters RJ, et al. Sensitive detection of mono- and polyclonal ESR1 mutations in primary tumors, metastatic lesions, and cell-free DNA of breast cancer patients. Clin Cancer Res. 2016;22:1130–7. http://clincancerres.aacrjournals.org/cgi/doi/10.1158/1078-0432.CCR-15-1534.
Haber DA, Velculescu VE. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014;4:650–61. http://www.ncbi.nlm.nih.gov/pubmed/24801577.
Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D, et al. Prevalence of ESR1 Mutations in cell-free DNA and outcomes in metastatic breast cancer. JAMA Oncol. 2016;2:1310. http://www.ncbi.nlm.nih.gov/pubmed/27532364.
Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im S-A, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phas. Lancet Oncol. 2016;17:425–39. http://www.ncbi.nlm.nih.gov/pubmed/28153873.
Fribbens C, OLeary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J Clin Oncol. 2016;34:2961–8. http://www.ncbi.nlm.nih.gov/pubmed/27269946.
Mayer I, Bardia A, Dickler M, Manning H, Mahmood U, Ulaner G, et al. Abstract OT3-2-07: Phase I study of ARN-810, a novel selective estrogen receptor degrader, in post-menopausal women with locally advanced or metastatic estrogen receptor positive breast cancer. Cancer Res. 2014;73:OT3-2-07-OT3-2-07. http://cancerres.aacrjournals.org/lookup/doi/10.1158/0008-5472.SABCS13-OT3-2-07.
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. http://www.ncbi.nlm.nih.gov/pubmed/3798106.
Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–12. http://www.ncbi.nlm.nih.gov/pubmed/2470152.
Yan M, Parker BA, Schwab R, Kurzrock R. HER2 aberrations in cancer: Implications for therapy. Cancer Treat Rev. 2014;40:770–80. http://www.ncbi.nlm.nih.gov/pubmed/24656976.
Zell JA, Tsang WY, Taylor TH, Mehta RS, Anton-Culver H. Prognostic impact of human epidermal growth factor-like receptor 2 and hormone receptor status in inflammatory breast cancer (IBC): analysis of 2,014 IBC patient cases from the California Cancer Registry. Breast Cancer Res. 2009;11:R9. http://breast-cancer-research.biomedcentral.com/articles/10.1186/bcr2225.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. http://www.ncbi.nlm.nih.gov/pubmed/11248153.
Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–72. http://www.ncbi.nlm.nih.gov/pubmed/16236737.
Witzel I, Müller V, Abenhardt W, Kaufmann M, Schoenegg W, Schneeweis A, et al. Long-term tumor remission under trastuzumab treatment for HER2 positive metastatic breast cancer—results from the HER-OS patient registry. BMC Cancer. 2014;14:806. http://www.ncbi.nlm.nih.gov/pubmed/25371387.
Smith I, Procter M, Gelber RD, Guillaume S, Feyereislova A, Dowsett M, et al. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet. 2007;369:29–36. http://www.ncbi.nlm.nih.gov/pubmed/17208639.
Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE, Davidson NE, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673–84. http://www.ncbi.nlm.nih.gov/pubmed/16236738.
Pohlmann PR, Mayer IA, Mernaugh R. Resistance to trastuzumab in breast cancer. Clin Cancer Res. 2009;15:7479–91. http://www.ncbi.nlm.nih.gov/pubmed/20008848.
Serra V, Scaltriti M, Prudkin L, Eichhorn PJA, Ibrahim YH, Chandarlapaty S, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30:2547–57. http://www.ncbi.nlm.nih.gov/pubmed/21278786.
Miller TW, Forbes JT, Shah C, Wyatt SK, Manning HC, Olivares MG, et al. Inhibition of mammalian target of rapamycin is required for optimal antitumor effect of HER2 inhibitors against HER2-overexpressing cancer cells. Clin Cancer Res. 2009;15:7266–76. http://www.ncbi.nlm.nih.gov/pubmed/19934303.
Wang Q, Liu P, Spangle JM, Von T, Roberts TM, Lin NU, et al. PI3K-p110α mediates resistance to HER2-targeted therapy in HER2+ , PTEN-deficient breast cancers. Oncogene. 2016;35:3607–12. http://www.nature.com/doifinder/10.1038/onc.2015.406.
Nagata Y, Lan K-H, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27. http://www.ncbi.nlm.nih.gov/pubmed/15324695.
Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K Pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. http://www.ncbi.nlm.nih.gov/pubmed/17936563.
Keck S, Glencer AC, Rugo HS. Everolimus and its role in hormone-resistant and trastuzumab-resistant metastatic breast cancer. Future Oncol. 2012;8:1383–96. http://www.ncbi.nlm.nih.gov/pubmed/23148612.
Scaltriti M, Rojo F, Ocana A, Anido J, Guzman M, Cortes J, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. JNCI J Natl Cancer Inst. 2007;99:628–38. http://www.ncbi.nlm.nih.gov/pubmed/17440164.
Yu Q, Sicinska E, Geng Y, Ahnström M, Zagozdzon A, Kong Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32. http://www.ncbi.nlm.nih.gov/pubmed/16413469.
Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9:13–22. http://linkinghub.elsevier.com/retrieve/pii/S153561080500396X.
Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 Inhibitors. Cancer Cell. 2016;29:255–69. http://linkinghub.elsevier.com/retrieve/pii/S153561081630040X.
Baselga J, Gelmon KA, Verma S, Wardley A, Conte P, Miles D, et al. Phase II Trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol. 2010;28:1138–44. http://www.ncbi.nlm.nih.gov/pubmed/20124182.
Portera CC, Walshe JM, Rosing DR, Denduluri N, Berman AW, Vatas U, et al. Cardiac toxicity and efficacy of trastuzumab combined with pertuzumab in patients with trastuzumab-insensitive human epidermal growth factor receptor 2-positive metastatic breast cancer. Clin Cancer Res. 2008;14:2710–6. http://www.ncbi.nlm.nih.gov/pubmed/18451236.
Nahta R, Hung M-C, Esteva FJ. The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res. 2004;64:2343–6. http://www.ncbi.nlm.nih.gov/pubmed/15059883.
Zhang X, Munster PN. New protein kinase inhibitors in breast cancer: afatinib and neratinib. Expert Opin Pharmacother. 2014;15:1277–88. http://www.tandfonline.com/doi/full/10.1517/14656566.2014.913570.
Chung A, Cui X, Audeh W, Giuliano A. Current status of anti-human epidermal growth factor receptor 2 therapies: predicting and overcoming herceptin resistance. Clin Breast Cancer. 2013;13:223–32. http://linkinghub.elsevier.com/retrieve/pii/S1526820913001006.
Saini KS, Azim HA, Metzger-Filho O, Loi S, Sotiriou C, de Azambuja E, et al. Beyond trastuzumab: new treatment options for HER2-positive breast cancer. The Breast. 2011;20:S20–7. http://www.ncbi.nlm.nih.gov/pubmed/22015288.
Scaltriti M, Chandarlapaty S, Prudkin L, Aura C, Jimenez J, Angelini PD, et al. Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2-positive breast tumors coexpressing the truncated p95HER2 receptor. Clin Cancer Res. 2010;16:2688–95. http://clincancerres.aacrjournals.org/cgi/doi/10.1158/1078-0432.CCR-09-3407.
Dave B, Migliaccio I, Gutierrez MC, Wu M-F, Chamness GC, Wong H, et al. Loss of phosphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J Clin Oncol. 2011;29:166–73. http://www.ncbi.nlm.nih.gov/pubmed/21135276.
Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, Aura C, De Azambuja E. First results of the NeoALTTO trial (BIG 01-06/EGF 106903): a phase III, randomized, open label, neoadjuvant study of lapatinib, trastuzumab, and their combination plus paclitaxel in women with HER2-positive primary breast cancer. Presented at the 33rd annual San Antonio breast cancer symposium; 2010 (abstract S3-3)
Gianni L, Pienkowski T, Im Y-H, Roman L, Tseng L-M, Liu M-C, et al. Neoadjuvant pertuzumab (P) and trastuzumab (H): antitumor and safety analysis of a randomized phase II study (’NeoSphere’). Cancer Res 2010;70(24 Suppl):Abstract nr S3-2
Baselga J, Cortés J, Kim S-B, Im S-A, Hegg R, Im Y-H, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl. J Med. 2012;366:109–19. http://www.ncbi.nlm.nih.gov/pubmed/22149875.
Amiri-Kordestani L, Wedam S, Zhang L, Tang S, Tilley A, Ibrahim A, et al. First FDA approval of neoadjuvant therapy for breast cancer: pertuzumab for the treatment of patients with HER2-positive breast cancer. Clin Cancer Res. 2014;20:5359–64. http://www.ncbi.nlm.nih.gov/pubmed/25204553.
Saini KS, Azim HA, Metzger-Filho O, Loi S, Sotiriou C, de Azambuja E, et al. Beyond trastuzumab: new treatment options for HER2-positive breast cancer. Breast. 2011;20 Suppl 3:S20-7. http://linkinghub.elsevier.com/retrieve/pii/S0960977611702892.
Saura C, Garcia-Saenz JA, Xu B, Harb W, Moroose R, Pluard T, et al. Safety and efficacy of neratinib in combination with capecitabine in patients with metastatic human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2014;32:3626–33. http://jco.ascopubs.org/cgi/doi/10.1200/JCO.2014.56.3809.
Chow LW-C, Xu B, Gupta S, Freyman A, Zhao Y, Abbas R, et al. Combination neratinib (HKI-272) and paclitaxel therapy in patients with HER2-positive metastatic breast cancer. Br J Cancer. 2013;108:1985–93. http://www.ncbi.nlm.nih.gov/pubmed/23632474.
Martin M, Bonneterre J, Geyer CE, Ito Y, Ro J, Lang I, et al. A phase two randomised trial of neratinib monotherapy versus lapatinib plus capecitabine combination therapy in patients with HER2+ advanced breast cancer. Eur J Cancer. 2013;49:3763–72. http://linkinghub.elsevier.com/retrieve/pii/S0959804913007181.
Cortés J, Dieras V, Ro J, Barriere J, Bachelot T, Hurvitz S, et al. Afatinib alone or afatinib plus vinorelbine versus investigator’s choice of treatment for HER2-positive breast cancer with progressive brain metastases after trastuzumab, lapatinib, or both (LUX-Breast 3): a randomised, open-label, multicentre, phase 2 tr. Lancet Oncol. 2015;16:1700–10. http://dx.doi.org/10.1016/j.ccr.2012.02.022.
Weil RJ, Palmieri DC, Bronder JL, Stark AM, Steeg PS. Breast cancer metastasis to the central nervous system. Am J Pathol. 2005;167:913–20. http://www.ncbi.nlm.nih.gov/pubmed/16192626.
Montagna E, Cancello G, D’Agostino D, Lauria R, Forestieri V, Esposito A, et al. Central nervous system metastases in a cohort of metastatic breast cancer patients treated with trastuzumab. Cancer Chemother Pharmacol. 2009;63:275–80. http://www.ncbi.nlm.nih.gov/pubmed/18379783.
Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28:2698–704. http://jco.ascopubs.org/cgi/doi/10.1200/JCO.2009.26.2071.
Isakoff SJ, Baselga J. Trastuzumab-DM1: building a chemotherapy-free road in the treatment of human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2011;29:351–4. http://jco.ascopubs.org/cgi/doi/10.1200/JCO.2010.31.6679.
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91. http://www.ncbi.nlm.nih.gov/pubmed/23020162.
Miller K, Cortes J, Hurvitz SA, Krop IE, Tripathy D, Verma S, et al. HERMIONE: a randomized Phase 2 trial of MM-302 plus trastuzumab versus chemotherapy of physician’s choice plus trastuzumab in patients with previously treated, anthracycline-naïve, HER2-positive, locally advanced/metastatic breast cancer. BMC Cancer. 2016;16:352. http://bmccancer.biomedcentral.com/articles/10.1186/s12885-016-2385-z.
André F, O’Regan R, Ozguroglu M, Toi M, Xu B, Jerusalem G, et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014;15:580–91. http://linkinghub.elsevier.com/retrieve/pii/S147020451470138X.
Coley WB. The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc R Soc Med. 1910;3:1–48. http://www.ncbi.nlm.nih.gov/pubmed/19974799.
Raedler LA. Keytruda (Pembrolizumab): First PD-1 inhibitor approved for previously treated unresectable or metastatic melanoma. Am Health Drug Benefits. 2015;8:96–100. http://www.ncbi.nlm.nih.gov/pubmed/26629272.
Sul J, Blumenthal GM, Jiang X, He K, Keegan P, Pazdur R. FDA Approval summary: pembrolizumab for the treatment of patients with metastatic non-small cell lung cancer whose tumors express programmed death-ligand 1. Oncologist. 2016;21:643–50. http://www.ncbi.nlm.nih.gov/pubmed/27026676.
Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer. Proc Natl Acad Sci. 2012;109:2796–801. http://www.ncbi.nlm.nih.gov/pubmed/21825174.
Coventry BJ, Weightman MJ, Bradley J, Skinner JM. Immune profiling in human breast cancer using high-sensitivity detection and analysis techniques. JRSM Open. 2015;6:205427041560390. http://www.ncbi.nlm.nih.gov/pubmed/26464809.
Loi S, Sirtaine N, Piette F, Salgado R, Viale G, Van Eenoo F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase iii randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol. 2013;31:860–7. http://www.ncbi.nlm.nih.gov/pubmed/23341518.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34. http://www.ncbi.nlm.nih.gov/pubmed/11015443.
Ali HR, Glont S-E, Blows FM, Provenzano E, Dawson S-J, Liu B, et al. PD-L1 protein expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes. Ann Oncol. 2015;26:1488–93. http://www.ncbi.nlm.nih.gov/pubmed/25897014.
Wimberly H, Brown JR, Schalper K, Haack H, Silver MR, Nixon C, et al. PD-L1 Expression correlates with tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy in breast cancer. Cancer Immunol Res. 2015;3:326–32. http://www.ncbi.nlm.nih.gov/pubmed/25527356.
Sabatier R, Finetti P, Mamessier E, Adelaide J, Chaffanet M, Ali HR, et al. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget.2015;6:5449–64. http://www.ncbi.nlm.nih.gov/pubmed/25669979.
Mittendorf EA, Philips A V., Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 Expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2:361–70. http://www.ncbi.nlm.nih.gov/pubmed/24764583.
Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. [Internet]. 2016 [cited 2017 Feb 13];13:674–90. http://www.ncbi.nlm.nih.gov/pubmed/27184417.
Issa-Nummer Y, Darb-Esfahani S, Loibl S, Kunz G, Nekljudova V, Schrader I, et al. Prospective validation of immunological infiltrate for prediction of response to neoadjuvant chemotherapy in HER2-negative breast cancer—a substudy of the neoadjuvant GeparQuinto trial. PLoS One. 2013;8:e79775. http://www.ncbi.nlm.nih.gov/pubmed/24312450.
García-Teijido P, Cabal ML, Fernández IP, Pérez YF. Tumor-Infiltrating lymphocytes in triple negative breast cancer: the future of immune targeting. Clin Med Insights Oncol. Libertas Academica; 2016;10:31–9. http://www.ncbi.nlm.nih.gov/pubmed/27081325.
West NR, Milne K, Truong PT, Macpherson N, Nelson BH, Watson PH. Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Res. 2011;13:R126. http://www.ncbi.nlm.nih.gov/pubmed/22151962.
Desmedt C, Di Leo A, de Azambuja E, Larsimont D, Haibe-Kains B, Selleslags J, et al. Multifactorial approach to predicting resistance to anthracyclines. J Clin Oncol. 2011;29:1578–86. http://www.ncbi.nlm.nih.gov/pubmed/21422418.
Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. http://www.ncbi.nlm.nih.gov/pubmed/18097448.
Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11:215–33. http://www.ncbi.nlm.nih.gov/pubmed/22301798.
Loi S, Sirtaine N, Piette F, Salgado R, Viale G, Van Eenoo F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol. 2013;31:860–7. http://www.ncbi.nlm.nih.gov/pubmed/23341518.
Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase iii randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014;32:2959–66. http://www.ncbi.nlm.nih.gov/pubmed/25071121.
Loi S, Michiels S, Salgado R, Sirtaine N, Jose V, Fumagalli D, et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann Oncol. 2014;25:1544–50. http://www.ncbi.nlm.nih.gov/pubmed/24608200.
Nagalla S, Chou JW, Willingham MC, Ruiz J, Vaughn JP, Dubey P, et al. Interactions between immunity, proliferation and molecular subtype in breast cancer prognosis. Genome Biol. 2013;14:R34. http://genomebiology.biomedcentral.com/articles/10.1186/gb-2013-14-4-r34.
Rody A, Karn T, Liedtke C, Pusztai L, Ruckhaeberle E, Hanker L, et al. A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res. 2011;13:R97. http://www.ncbi.nlm.nih.gov/pubmed/21978456.
Sabatier R, Finetti P, Cervera N, Lambaudie E, Esterni B, Mamessier E, et al. A gene expression signature identifies two prognostic subgroups of basal breast cancer. Breast Cancer Res Treat. 2011;126:407–20. http://www.ncbi.nlm.nih.gov/pubmed/20490655.
Staaf J, Ringner M, Vallon-Christersson J, Jonsson G, Bendahl PO, Holm K, et al. Identification of Subtypes in Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer Reveals a Gene Signature Prognostic of Outcome. J Clin Oncol. 2010;28:1813–20. http://www.ncbi.nlm.nih.gov/pubmed/20231686.
Rody A, Holtrich U, Pusztai L, Liedtke C, Gaetje R, Ruckhaeberle E, et al. T-cell metagene predicts a favorable prognosis in estrogen receptor-negative and HER2-positive breast cancers. Breast Cancer Res. 2009;11:R15. http://www.ncbi.nlm.nih.gov/pubmed/19272155.
Teschendorff AE, Miremadi A, Pinder SE, Ellis IO, Caldas C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 2007;8:R157. http://www.ncbi.nlm.nih.gov/pubmed/17683518.
Callari M, Cappelletti V, D’Aiuto F, Musella V, Lembo A, Petel F, et al. Subtype-specific metagene-based prediction of outcome after neoadjuvant and adjuvant treatment in breast cancer. Clin. Cancer Res. 2016;22:337–45. http://www.ncbi.nlm.nih.gov/pubmed/26423797.
Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4+ follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. American Society for Clinical Investigation; 2013;123:2873–92. http://www.ncbi.nlm.nih.gov/pubmed/23778140.
Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, et al. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. 2008;14:5158–65. http://www.ncbi.nlm.nih.gov/pubmed/18698033.
Bianchini G, Qi Y, Alvarez RH, Iwamoto T, Coutant C, Ibrahim NK, et al. Molecular anatomy of breast cancer stroma and its prognostic value in estrogen receptor-positive and -negative cancers. J Clin Oncol. 2010;28:4316–23. http://jco.ascopubs.org/cgi/doi/10.1200/JCO.2009.27.2419.
Schmidt M, Bohm D, von Torne C, Steiner E, Puhl A, Pilch H, et al. The Humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68:5405–13. http://cancerres.aacrjournals.org/cgi/doi/10.1158/0008-5472.CAN-07-5206.
Pusztai L, Karn T, Safonov A, Abu-Khalaf MM, Bianchini G. New strategies in breast cancer: immunotherapy. Clin Cancer Res. 2016;22:2105–10. http://www.ncbi.nlm.nih.gov/pubmed/26867935.
Nanda R, Chow LQM, Dees EC, Berger R, Gupta S, Geva R, et al. Pembrolizumab in patients with advanced triple-negative breast cancer: phase Ib KEYNOTE-012 Study. J Clin Oncol. 2016;34:2460–7. http://www.ncbi.nlm.nih.gov/pubmed/27138582.
Emens LA, Braiteh FS, Cassier P, et al. Inhibition of PD-L1 by MPDL3280A leads to clinical activity in patients with metastatic triple-negative breast cancer; Presented at San Antonio Breast Cancer Symposium; December 9–13, 2014; San Antonio, TX
Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. Dove Press; 2016;9:5023–39. http://www.ncbi.nlm.nih.gov/pubmed/27574444.
Grigg C, Rizvi NA. PD-L1 biomarker testing for non-small cell lung cancer: truth or fiction? J Immunother Cancer. BioMed Central; 2016;4:48. http://www.ncbi.nlm.nih.gov/pubmed/27532023.
Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, et al. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. European Molecular Biology Organization; 2008;27:1368–77. http://www.ncbi.nlm.nih.gov/pubmed/18388863.
Ibrahim YH, García-García C, Serra V, He L, Torres-Lockhart K, Prat A, et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012;2:1036–47. http://www.ncbi.nlm.nih.gov/pubmed/22915752.
Tate CR, Rhodes L V, Segar HC, Driver JL, Pounder FN, Burow ME, et al. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res. 2012;14:R79. http://www.ncbi.nlm.nih.gov/pubmed/22613095.
Schech A, Kazi A, Yu S, Shah P, Sabnis G. Histone Deacetylase Inhibitor Entinostat inhibits tumor-initiating cells in triple-negative breast cancer cells. Mol Cancer Ther. 2015;14:1848–57. http://www.ncbi.nlm.nih.gov/pubmed/26037781.
Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15:4649–64. http://www.ncbi.nlm.nih.gov/pubmed/19567590.
Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci. 2009;106:8368–73. http://www.ncbi.nlm.nih.gov/pubmed/19416831.
Zagouri F, Sergentanis TN, Chrysikos D, Papadimitriou CA, Dimopoulos M-A, Psaltopoulou T. Hsp90 inhibitors in breast cancer: a systematic review. The Breast. 2013;22:569–78. http://www.ncbi.nlm.nih.gov/pubmed/23870456.
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. American Society for Clinical Investigation; 2011;121:2750–67. http://www.ncbi.nlm.nih.gov/pubmed/21633166.
Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437:199–213. http://www.ncbi.nlm.nih.gov/pubmed/21711248.
Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–29. http://www.ncbi.nlm.nih.gov/pubmed/20094046.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Informed consent
Informed consent for publication was obtained from all authors of this review.
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s12032-017-0989-z.
Rights and permissions
About this article

Cite this article
Corona, S.P., Sobhani, N., Ianza, A. et al. Advances in systemic therapy for metastatic breast cancer: future perspectives. Med Oncol 34, 119 (2017). https://doi.org/10.1007/s12032-017-0975-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-017-0975-5