
Abstract
Colorectal cancer (CRC) patients with peritoneal carcinomatosis (PC) have a poor prognosis. Over the last decade, increasing evidence has suggested that the Collagen Triple Helix Repeat Containing 1 (CTHRC1) gene is involved in cancer progression and invasion. In this study, we investigated the expression of CTHRC1 in CRC and its potential as a prognostic factor for CRC patients with PC. Microarray analysis of four fresh paired samples showed that the expression of CTHRC1 in peritoneal metastases was higher than that in the corresponding primary tumor. These results were validated using semi-quantitative reverse transcription and polymerase chain reaction. Finally, immunohistochemical analysis showed that CTHRC1 was increased in the peritoneal metastasis group (n = 30) and the primary cancer with peritoneal metastasis group (n = 57) compared to the primary cancer without peritoneal metastasis group (n = 54), both P < 0.001. Cox proportional hazards model analysis showed that high CTHRC1 expression was associated with poor survival (HR = 2.754, P < 0.001, 95 % CI 1.731–4.383). Overall, the results of our study suggest that increased expression of CTHRC1 is associated with PC in CRC patients and could predict poor outcome in CRC patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Peritoneal carcinomatosis (PC) is a common mode of metastasis from colorectal cancer (CRC). PC is found in approximately 7 % of patients at the time of their primary surgery. It is also found in approximately 4–19 % of CRC patients during follow-up after curative surgery [1]. CRC patients have poor prognosis, with a mean survival between 5 and 9 months [2]. Thus, it is vital to identify biologic predictors for early diagnosis and prognosis.
Gene-expression signatures of differently expressed genes might be one way to identify the patients who are most likely to develop metastatic cancer and would benefit from early diagnosis and therapy. Using a genome-wide cDNA microarray, genes that are associated with the liver metastasis of colon cancers were identified [3, 4], but few studies have aimed to uncover the genes related to PC in CRC.
Human CTHRC1 was first found in a screen for differentially expressed sequences in balloon-injured versus normal rat arteries. Increased expression of CTHRC1 was also reported to promote cell migration and inhibited collagen I synthesis in rat fibroblasts [5]. Aberrant expression of CTHRC1 is widely present in solid human tumors and seems to be associated with cancer tissue invasion and metastasis [6, 7]. Recently, some studies that focused on digestive tract tumors found that CTHRC1 is associated with Barrett’s esophagus and esophageal adenocarcinoma [8]. It has also been reported that up-regulated CTHRC1 in gastric carcinogenesis contributed to tumor cell invasion and metastasis [9].
In this study, we used an Oligo microarray containing 26,000 genes to search for differentially expressed genes between peritoneal metastases and corresponding colorectal primary tumors. Ultimately, we selected CTHRC1 for further validation. Also, using immunohistochemical (IHC) analysis, we aimed to investigate CTHRC1 expression in primary CRC with peritoneal metastases and the corresponding peritoneal metastases compared to primary CRC without peritoneal metastases. Furthermore, we wanted to find out whether overexpression of CTHRC1 is associated with poor prognosis in CRC patients.
Materials and methods
Patient samples
For microarray analysis and SqRT-PCR, tumor samples from 4 patients with sporadic colorectal adenocarcinoma were used. All patients underwent surgical resection in the Nanfang Hospital of Southern Medical University (Guangzhou, China) between April 2010 and December 2010. Clinicopathological data from these four patients are summarized in Table 1. Each resected cancer specimen was evaluated for its tumor cell content in hematoxylin and eosin stained sections. Only specimens containing more than 75 % tumor cells were used for analysis. Samples were macro dissected by pathologists, soaked in RNA later (Ambion USA) and then stored at −80 °C until further use. For immunohistochemistry, tissue samples (n = 141) from 111 CRC patients who underwent surgical resection in the Nanfang Hospital of Southern Medical University (Guangzhou, China) were used. Paraffin-embedded tissue samples from primary CRC (n = 57) and the corresponding peritoneal metastases (n = 30) were obtained from 57 of these CRC patients. All these CRC patients underwent surgical resection between April 2003 and June 2009, suffered from PC and were followed up once every 6 months after surgery. The remaining primary CRC tissue samples (n = 54) were obtained from patients who underwent surgical resection between April 2003 and June 2006 and were free from PC after 5-year follow-up. According to the International Union Against Cancer (UICC) stage classification, 13.5 % of the patients had stage I disease, 22.5 % had stage II, 12.6 % had stage III and 51.4 % had stage IV. Five patients were excluded because of incomplete follow-up data, while the rest 106 patients were included in Cox regression analysis. The study was approved by the Ethics Committee at the Nanfang Hospital of Southern Medical University and informed consent was obtained from each patient.
RNA isolation and microarray procedures
Total RNA was extracted using TRIZOL (Invitrogen, USA) according to the manufacturer’s instructions. Total RNA was then purified using a QIAGEN RNeasy mini kit (Ambion, USA). Total RNA (2 μg) was reverse transcribed into cDNA using T7-Oligo (dT) primers. The purified cDNA was used for in vitro RNA syntheses with T7 RNA polymerase, and the amino-allyl labeled aa-UTP was incorporated into synthesized aRNA production. A total of 5 μg of aRNA was used for the labeling reaction by linking the mono-reaction Cy3 dye (GE Amersham, USA) to aa-UTP in a coupling reaction. The Cy3 labeled aRNA samples were hybridized with the Whole Human Genome Oligo Microarray in a one-channel format. The microarray slide was then scanned with the LuxScan 3.0 scanner (Capital Bio Biotech, Beijing, China). The data acquisition from the scanned microarray slide was performed with GenePix pro 6.0 software (Axon, USA). The relative expression change ratio was calculated and used for further data analysis.
Bioinformatics analysis
The microarray data were analyzed using the R Statistical Software package downloaded from www.r-project.org. The statistical significance of the microarray data was determined by a non-parametric test (Empirical Bayes). A value of P < 0.05 and a fold change of greater or less than 2 were considered statistically significant. Significantly regulated genes were subsequently used to provide functional annotations and ontologies. Genes corresponding to the gene ontology (GO) terms for cell adhesion, extracellular matrix and cell migration were considered for further analysis.
Semi-quantitative reverse transcription and polymerase chain reaction (SqRT-PCR)
To confirm the cDNA microarray results, SqRT-PCRs for CTHRC1 were performed on 4 patients. Reverse transcription was performed on 5 μg of total RNA for 120 min at 42 °C using an oligo (dT) primer and Invitrogen SuperScript II Reverse Transcriptase, SS II (Invitrogen Life Technology Inc., American). PCR was performed with a Roche FastStart Universal SYBR Green Master kit (Roche Diagnostic Corporation, Switzerland) according to the manufacturer’s instructions under the following conditions: 95 °C for 10 min followed by 40 cycles of 94 °C for 10 s and 50 °C for 15 s and 72 °C for 15 s. The primer sequences for CTHRC1 were 5′-CAATAATGATGCGAGAAACT-3′(forward) and 5′-ACCAAGGAAGCCCTGAAA- 3′ (reverse).
Immunohistochemistry (IHC)
2-μm-thick sections were stained by immunohistochemistry using an anti-CTHRC1 antibody (rabbit polyclonal to CTHRC1, 1:1,000 dilution; Abcam, Ltd., USA). Immunostained slides were characterized quantitatively by digital image analysis using ImagePro-Plus 6.0 (Media Cybernetics, Silver Spring, MD). Images were captured through an Olympus BX-51 microscope fitted with a MicroImage video camera (Olympus DP72). The staining was defined via optical density and the same capturing conditions were equally applied to all images. The immunohistochemical parameters assessed in the detected area included integrated optical density (IOD) and area of interest (AOI). We used the AOD ratio (average optical density, AOD = IOD/AOI) to represent the protein expression level. A series of 5 random images were taken from each slide for each immunostained parameter to obtain a mean value for statistical comparison. All the images were reviewed by three different observers, including a pathologist.
Statistical analysis
CTHRC1 expression was represented by AOD and reported as the mean ± standard deviation. Multiple comparisons between three groups were performed by one-way ANOVA with the LSD method. The patients were further classified into two groups according to the mean value of AOD. The survival probability analysis was calculated in both groups using the Kaplan–Meier method. Significant differences in survival probability were assessed by the logrank test. The impact of various factors on survival was assessed using multivariate analysis and a Cox proportional hazards model. A P value <0.05 was considered to be statistically significant. All statistical analyses were performed using SPSS.13.0.
Results
Gene detection by microarray analysis and their functional category analysis
Using the methods described above, we identified 57 genes that were differentially expressed between primary CRC tissue and the corresponding peritoneal metastases. Comparing with the primary CRC tissue, the gene expression of CTHRC1 was up-regulated 7.5-fold in corresponding peritoneal metastases (P = 0.0055). Twenty-six genes showed increased expression and 31 genes showed decreased expression in the peritoneal metastases. To investigate the biological functions involved in the differentially expressed genes, we performed Gene Ontology category analysis. The genes were mainly associated with the extracellular region and the proteinaceous extracellular matrix (Table 2).
Validation of CTHRC1 expression by SqRT-PCR
CTHRC1 was found to be up-regulated in peritoneal metastases in our microarray analysis. The results from SqRT-PCR showed changes in gene expression consistent with the microarray data (Fig. 1).

Results of SqRT-PCR. a The melt curve of CTHRC1, b the amplification plot c the fold change trend of SqRT-PCR was similar with the microarray analysis finding
Assessment of CTHRC1 protein expression in colorectal cancer tissue samples
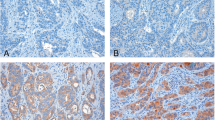
We performed immunohistochemical analysis in selected tissue samples from 57 primary colorectal cancers with peritoneal carcinoma, 54 primary colorectal cancers without peritoneal carcinoma and 30 peritoneal carcinomas. CTHRC1 was predominantly positive in the cell cytoplasm of cancer cells (Fig. 2). The mean AOD of the primary tumors with PC group, the primary tumors without PC group and the PC group were 0.25 ± 0.04, 0.19 ± 0.03 and 0.26 ± 0.04, respectively. The expression of CTHRC1 was different between the three groups (P < 0.001, Fig. 3). Furthermore, the expression of CTHRC1 was higher in primary CRC with peritoneal metastases than in primary CRC without peritoneal metastases (P < 0.001).

CTHRC1 immunohistochemical expression. CTHRC1 was predominantly positive in the cytoplasm of cancer cells. a Primary tumors without PC (200×), b primary tumors without PC (400×), c primary tumors with PC (200×), d Primary tumors with PC (400×), e PC (200×), f PC (400×)

Differential expression of CTHRC1 in three groups. Asterisk CTHRC1 expression level of primary tumor with metastasis group and peritoneal metastasis group were higher than that of primary tumor without metastasis group by one-way ANOVA and post hoc test (P < 0.001)
CTHRC1 is a predictor of survival in CRC patients
All of the enrolled patients were divided into two groups according to the mean AOD (range 0.103–0.396, mean value = 0.224). As expected, Kaplan–Meier curves revealed that patients with higher expression of CTHRC1 had a significantly poor survival than those with low CTHRC1 expression (31.36 ± 5.63 vs. 57.84 ± 3.69 months, P < 0.001) (Fig. 4). The Cox regression analysis showed that the CTHRC1 expression level was an independent negative prognostic factor along with sex, tumor invasion depth, tumor size and lymph node metastasis, with a hazard ratio (HR) of 2.754 between groups with high and low CTHRC1 expression (P < 0.001, 95 % CI 1.731–4.383) (Table 3).

Kaplan–Meier analysis of Cumulative Overall Survival of CRC patients with differential CTHRC1 expression. The prognosis of patients belonging to the low CTHRC1 expression level group was better than that of patients belonging to the high CTHRC1 expression level group (P < 0.001)
Discussion
Accumulating data suggest that not only genetic changes that are intrinsic to cancer cells, but also alterations within the tumor microenvironment at the site of metastasis can promote tumor cell invasion, angiogenesis and other important molecular and cellular processes. Genomic profiling studies suggest that differences in gene expression determine whether colorectal cancer will spread toward the peritoneal surfaces, toward the liver, or both [10]. Although several studies have identified genes, including CTGF [11], REG1A [12] and KRAS mutation [13], that play a role in PC in CRC, the molecular nature of site-specific metastasis is still largely unknown. The existence of these signatures suggests that understanding the functions of the genes that are associated with PC in CRC will not only contribute to understanding the underlying mechanisms of CRC progression, but will also bring new insights into how site-specific metastases develop.
Our microarray analysis results revealed that a series of genes were significantly different between primary and metastatic carcinomas. Using Empirical Bayes and fold change, we identified 57 differentially expressed genes, which were involved in extracellular region part (GO: 0044421), extracellular region (GO: 0005576), proteinaceous extracellular matrix (GO: 0005578) and extracellular matrix (GO: 0031012). These GO terms are associated with an excess risk of PC in CRC. Therefore, we selected genes from these GO terms for further study. Based on previously published research reports, we hypothesized that the high expression of CTHRC1 would be important for the invasion and migration of cancer cells.
Kharaishvili et al. [14] verified that CTHRC1 and periostin play important roles in breast cancer progression and the combined evaluation of CTHRC1, and periostin could serve as a potential marker for bone metastasis of breast cancer. Tang et al. [7] found aberrant CTHRC1 expression in cancers of the gastrointestinal tract and suggested that CTHRC1 protein expression is correlated with melanoma invasion and metastasis and promotes melanoma cell migration. Wang et al. [15] reported similar results and found high expression of CTHRC1 in dermatofibrosarcoma protuberans, a locally aggressive spindle cell neoplasm that frequently recurs and metastasizes. Our results are consistent with these findings and support our hypothesis that CTHRC1 not only correlates with the development of CRC but also promotes peritoneal metastasis during the colorectal carcinogenesis process. Briefly, the development of PC consists of several well-defined steps, including detachment of cells from the primary tumor, peritoneal transport, mesothelial adhesion, invasion of submesothelial layers and accessing systemic circulation [16]. During these steps, several growth factors, cytokines, proteases and signal pathways are present in this complicated microenvironment. The TGF-β and Wnt pathways are two important participants in this microenvironment, and the relationship between CTHRC1 and these two pathways may be the mechanism by which CTHRC1 promotes the formation of peritoneal metastasis in CRC patients. Collagen deposition is regulated by the signaling cascade via TGF-β activation of Smad2/3 complexes [17]. In addition, several experiments demonstrated that CTHRC1 regulates extracellular collagen deposition by inhibiting Smad2/3 phosphorylation [18]. However, sequence analysis of the CTHRC1 promoter region in melanoma cells revealed a binding site for SMAD, which is responsive to TGF-β protein regulation [7]. Interestingly, phosphorylated Smad2/3 increases the expression of CTHRC1, while up-regulated CTHRC1 prohibits the deposition of extracellular collagen that is regulated by Smad2/3 phosphorylation. Furthermore, the ability of CTHRC1 to down-regulate phosphorylated Smad2/3 may contribute to CRC invasion and metastasis because Smad2 [19] and Smad3 [20] are potent tumor suppressors in the colonic epithelium.
The best-characterized Wnt pathway is the canonical pathway, which is involved in determining cell fate and the regulation of growth, including the formation of the body axis, patterning of the neuroectoderm and amplification of neural progenitors [21]. The Wnt/planar cell polarity (PCP) pathway, which is the non-canonical Wnt pathway, controls tissue polarity and cell movement through the activation of RHOA, c-Jun N-terminal kinase (JNK) and nemo-like kinase (NLK) signaling cascades [22]. These Wnt pathways are not parallel but do interact with each other. Canonical Wnt-induced Wnt11 activates non-canonical Wnt signaling cascades to induce cellular movement and also activates the Ca2+-MAP3K7-NLK signaling cascade to inhibit canonical Wnt signaling [23]. In addition, Wnt signaling output is not intrinsically related to the Wnt protein itself but rather due to a combination of factors, including receptor availability [24]. CTHRC1 interacts with multiple extracellular components of Wnt signaling, which include both canonical and non-canonical Wnt proteins, Fzd proteins and the Wnt/PCP co-receptor Ror2. These components form a Cthrc1–Wnt–Fzd/Ror2 complex to selectively activate the Wnt/PCP pathway and suppress the canonical Wnt pathway [25]. Whether CTHRC1 promotes the movement of cancer cells by reinforcing the Wnt/pcp pathway and simultaneously inhibiting cancer cell proliferation by suppressing the canonical Wnt pathway deserves more attention in the future.
In the present study, we investigated whether increased expression of CTHRC1 was found in primary cancer and peritoneal metastases in CRC. Together with the clinical pathology features of 106 CRC patients, we used immunohistochemistry to evaluate the expression of CTHRC1 at the population level. The immunohistochemistry results demonstrated that CTHRC1 protein expression was higher in peritoneal metastases and primary CRC tumors with peritoneal metastases than in the primary CRC tumors without peritoneal metastases. Palma et al. [26] suggested that specific antibodies against CTHRC1 and Nuclear factor (erythroid-derived 2)-like 3 (NFE2L3) may be useful for the analysis of these two genes in clinical samples. The same has also been suggested for the screening of therapeutic compounds in CRC. Taken together, these data suggest that CTHRC1 might play an important role in the progression of PC and can act as a potential predictor for PC in CRC. Furthermore, our study identified a correlation between increased CTHRC1 expression in CRC and poor clinical prognosis. Therefore, it is possible that overexpression of CTHRC1 in patients results in poor outcome. These data suggest that the overexpression of CTHRC1 may be an independent prognostic factor in CRC patients. We also found that clinicopathological characteristics such as sex, tumor invasion depth, tumor size and lymph node metastasis were prognostic biological markers for CRC patients who suffered from PC.
Although several limitations may weaken the generalization of our findings, including a small sample size and the nature of a retrospective study, the novelty of this study should be emphasized. In our study, the consistent observation of an association at the molecular and population levels provides support to the credibility of the observed effect. However, detailed studies that focus on the molecular nature of CTHRC1 should be performed to uncover the mechanism underlying the role of CTHRC1 in CRC invasion and migration.
In summary, our study found that CTHRC1 is a peritoneal metastasis-related gene in colorectal carcinogenesis. Our findings demonstrate that CTHRC1 could serve as a potential marker for predicting PC in CRC. Furthermore, our results suggested that CTHRC1 can predict the survival outcome of CRC patients who suffer from PC. However, further functional experiments are needed to clarify the role of CTHRC1 in CRC progression and metastasis.
References
Koppe MJ, Boerman OC, Oyen WJ, Bleichrodt RP. Peritoneal carcinomatosis of colorectal origin: incidence and current treatment strategies. Ann Surg. 2006;243(2):212–22. doi:10.1097/01.sla.0000197702.46394.16.
Gomez-Portilla A, Cendoya I, Lopez de Tejada I, Olabarria I, Magrach L, Martinez de Lecea C, et al. Principles of the treatment of peritoneal carcinomatosis due to colorectal cancer. Current review and update. Cir Esp. 2005;77(1):6–17.
Ki DH, Jeung HC, Park CH, Kang SH, Lee GY, Lee WS, et al. Whole genome analysis for liver metastasis gene signatures in colorectal cancer. Int J Cancer J Int du Cancer. 2007;121(9):2005–12. doi:10.1002/ijc.22975.
Li M, Lin YM, Hasegawa S, Shimokawa T, Murata K, Kameyama M, et al. Genes associated with liver metastasis of colon cancer, identified by genome-wide cDNA microarray. Int J Oncol. 2004;24(2):305–12.
Pyagay P, Heroult M, Wang Q, Lehnert W, Belden J, Liaw L, et al. Collagen triple helix repeat containing 1, a novel secreted protein in injured and diseased arteries, inhibits collagen expression and promotes cell migration. Circ Res. 2005;96(2):261–8. doi:10.1161/01.RES.0000154262.07264.12.
Ip W, Wellman-Labadie O, Tang L, Su M, Yu R, Dutz J, et al. Collagen triple helix repeat containing 1 promotes melanoma cell adhesion and survival. J Cutan Med Surg. 2011;15(2):103–10.
Tang L, Dai DL, Su M, Martinka M, Li G, Zhou Y. Aberrant expression of collagen triple helix repeat containing 1 in human solid cancers. Clin Cancer Res Off J Am Assoc Cancer Res. 2006;12(12):3716–22. doi:10.1158/1078-0432.CCR-06-0030.
Orloff M, Peterson C, He X, Ganapathi S, Heald B, Yang YR, et al. Germline mutations in MSR1, ASCC1, and CTHRC1 in patients with Barrett esophagus and esophageal adenocarcinoma. JAMA. 2011;306(4):410–9. doi:10.1001/jama.2011.1029.
Wang P, Wang YC, Chen XY, Shen ZY, Cao H, Zhang YJ, et al. CTHRC1 is upregulated by promoter demethylation and transforming growth factor-beta1 and may be associated with metastasis in human gastric cancer. Cancer Sci. 2012;. doi:10.1111/j.1349-7006.2012.02292.x.
Varghese S, Burness M, Xu H, Beresnev T, Pingpank J, Alexander HR. Site-specific gene expression profiles and novel molecular prognostic factors in patients with lower gastrointestinal adenocarcinoma diffusely metastatic to liver or peritoneum. Ann Surg Oncol. 2007;14(12):3460–71. doi:10.1245/s10434-007-9557-7.
Lin BR, Chang CC, Chen RJ, Jeng YM, Liang JT, Lee PH, et al. Connective tissue growth factor acts as a therapeutic agent and predictor for peritoneal carcinomatosis of colorectal cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2011;17(10):3077–88. doi:10.1158/1078-0432.ccr-09-3256.
Astrosini C, Roeefzaad C, Dai YY, Dieckgraefe BK, Jons T, Kemmner W. REG1A expression is a prognostic marker in colorectal cancer and associated with peritoneal carcinomatosis. Int J Cancer J Int Cancer. 2008;123(2):409–13. doi:10.1002/ijc.23466.
Gillern SM, Chua TC, Stojadinovic A, Esquivel J. KRAS status in patients with colorectal cancer peritoneal carcinomatosis and its impact on outcome. Am J Clin Oncol. 2010;33(5):456–60. doi:10.1097/COC.0b013e3181b4b160.
Kharaishvili G, Cizkova M, Bouchalova K, Mgebrishvili G, Kolar Z, Bouchal J. Collagen triple helix repeat containing 1 protein, periostin and versican in primary and metastatic breast cancer: an immunohistochemical study. J Clin Pathol. 2011;64(11):977–82. doi:10.1136/jclinpath-2011-200106.
Wang L, Xiang YN, Zhang YH, Tu YT, Chen HX. Collagen triple helix repeat containing-1 in the differential diagnosis of dermatofibrosarcoma protuberans and dermatofibroma. Br J Dermatol. 2011;164(1):135–40. doi:10.1111/j.1365-2133.2010.10050.x.
Ceelen WP, Bracke ME. Peritoneal minimal residual disease in colorectal cancer: mechanisms, prevention, and treatment. Lancet Oncol. 2009;10(1):72–9. doi:10.1016/S1470-2045(08)70335-8.
Ghosh AK. Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp Biol Med (Maywood). 2002;227(5):301–14.
LeClair RJ, Durmus T, Wang Q, Pyagay P, Terzic A, Lindner V. Cthrc1 is a novel inhibitor of transforming growth factor-beta signaling and neointimal lesion formation. Circ Res. 2007;100(6):826–33. doi:10.1161/01.res.0000260806.99307.72.
Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, et al. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. 1996;86(4):543–52.
Arai T, Akiyama Y, Okabe S, Ando M, Endo M, Yuasa Y. Genomic structure of the human Smad3 gene and its infrequent alterations in colorectal cancers. Cancer Lett. 1998;122(1–2):157–63.
Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi:10.1146/annurev.cellbio.20.010403.113126.
Katoh M. WNT/PCP signaling pathway and human cancer (review). Oncol Rep. 2005;14(6):1583–8.
Katoh M. Integrative genomic analyses of WNT11: transcriptional mechanisms based on canonical WNT signals and GATA transcription factors signaling. Int J Mol Med. 2009;24(2):247–51.
Mikels AJ, Nusse R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 2006;4(4):e115. doi:10.1371/journal.pbio.0040115.
Yamamoto S, Nishimura O, Misaki K, Nishita M, Minami Y, Yonemura S, et al. Cthrc1 selectively activates the planar cell polarity pathway of Wnt signaling by stabilizing the Wnt-receptor complex. Dev Cell. 2008;15(1):23–36. doi:10.1016/j.devcel.2008.05.007.
Palma M, Lopez L, Garcia M, de Roja N, Ruiz T, Garcia J, et al. Detection of collagen triple helix repeat containing-1 and nuclear factor (erythroid-derived 2)-like 3 in colorectal cancer. BMC Clin Pathol. 2012;12:2. doi:10.1186/1472-6890-12-2.
Acknowledgments
We thank Professor Yongjian Deng for his support in collecting the paraffin-embedded tissue samples and Dr. Xinhua Zhou for her critical review of the images of the immunostained slides.
Conflict of interest
The authors have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tan, F., Liu, F., Liu, H. et al. CTHRC1 is associated with peritoneal carcinomatosis in colorectal cancer: a new predictor for prognosis. Med Oncol 30, 473 (2013). https://doi.org/10.1007/s12032-013-0473-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-013-0473-3