
Abstract
MicroRNA-34 family has anti-proliferative and apoptotic roles. Recent studies have shown that p53 upregulates miR-34 family leading to direct repression of several key oncogenes. Inactivation of miR-34a has been reported in multiple types of malignancies including breast cancer. The critical role of miR-34a in p53-mediated cell cycle arrest and apoptosis invokes studies focusing on the specific role of miR-34a dysregulation in carcinogenesis. While presence of p53 mutations has frequently been described in breast cancer, still most of the breast tumors do not show any variation in the p53 coding sequence or protein expression. Therefore, it is important to clarify possible involvement of other mediators of p53 pathway in breast cancer. In this study, expression of mature miR-34a in breast tumors with wild-type p53 was investigated in order to find any correlation between dysregulation of miR-34a expression and breast cancer. In about 40 % of the wild-type p53 samples, miR-34a was significantly downregulated. Neither hypermethylation of the miR-34a promoter nor genetic variations of the p53-binding site were detected in tumor samples with downregulated miR-34a. This study has provided evidence that miR-34a expression can be affected in a significant proportion of breast tumors independent of p53. Furthermore, downregulation of miR-34a was significantly associated with metastasis, while there was a significant correlation between upregulation of miR-34a and non-metastatic condition indicating a protective role for miR-34a against more invasive disease. Knowledge of miR-34a status may provide additional useful information regarding the nature of breast tumors, especially when p53 testing does not show any aberration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is a multi-stage process during which normal cell acquires a complex of several alterations in cell physiology including self-sufficiency, resistance to growth-inhibitory signals and apoptosis, unlimited replicative capacity, continuous angiogenesis and metastasis [1]. MicroRNAs include a class of non-coding small RNAs, consisting of about 22 nucleotides in length [2]. They are principally involved in post-transcriptional regulation of gene expression and affect all main aspects of cellular physiology including developmental transitions, stress response, metabolism, proliferation and apoptosis [3]. These mechanisms are frequently altered during malignant transformation. Epigenetic changes including dysregulation of miRNAs have been increasingly described as an important player in cancer development during recent years [4–8]. Members of miR-34 family, including miR-34a, b and c, were reported as direct targets of p53 protein by several investigators in 2007 [9–15]. These studies showed that upregulation of miR-34 family by p53 leads to cell cycle arrest and apoptosis through its inhibitory effect on some key regulators of cell cycle progression, like E2F3, BCL-2, c-Myc, CDK4, etc. [15]. However, miR-34a has been identified as the major form of miR-34 family induced after p53 activation [10, 12–15]. P53 gene mutations have been reported in almost all types of cancer, and existence of these mutations has usually been regarded as a poor prognostic indicator [16]. While dysregulation of several miRNAs like let-7, miR-10b, miR-17-5p, miR-21, miR-27a, miR-125a, miR-125b, miR-205 and miR-206 have been strongly linked with development of breast cancer [17], there is no established model between p53 and miR-34a in breast tumorigenesis. Also, silent information regulator 1 (SIRT1) has been identified as another important component of p53-miR-34a pathway. Yamakuchi et al. [18] studied miR-34a targets and found that SIRT1 is regulated by miR-34a. SIRT1 protein expression has been shown to be frequently increased in several types of cancer including prostate cancer [19], acute myeloid leukemia [20] and colon cancer [21]. SIRT1 has been known to play a role in tumorigenesis, principally by deacetylation of p53 which subsequently decreases the ability of p53 to promote cell cycle arrest and apoptosis [18].
Inactivation or downregulation of miR-34a could be one mechanism by which tumor cells can escape apoptosis and survive despite an intact p53. Therefore, deregulation of miR-34a expression in breast tumors with wild-type p53 has been suggested as a possible explanation for tumor development and a subject for further investigation [17, 22]. CpG islands exist in the promoter region of most genes [23]. Transcriptional silencing by promoter hypermethylation has been strongly linked to cancer development through inactivation of tumor suppressor genes [24]. This mechanism has also been suggested for downregulation of tumor suppressor miRNAs in cancer [23]. Because of the existence of a CpG island in miR-34a promoter located on chromosome 1p36, hypermethylation of miR-34a promoter may be responsible for downregulation of miR-34a expression in breast tumors with wild-type p53.
In this study, we aimed to understand whether miR-34a expression level is affected in human breast cancer with wild-type p53, and whether miR-34a expression can independently provide useful information in regards to outcome prediction in breast cancer. Some previous studies have provided evidence that SIRT1 is regulated by miR-34a at post-transcriptional level [18, 25]. Therefore, we also aimed to study the expression level of SIRT1 mRNA in malignant and normal breast tissues and to understand whether it has any specific correlation with p53, miR-34a and clinicopathologic features of the patients.
Materials and methods
Sample collection
A total of 46 unselected breast tumor samples and their normal adjacent tissue (NAT) were obtained from Iran National Tumor Bank (INTB). Breast cancer (BC) patients included in this study did not receive any treatment before sampling and had provided written consent. All samples were diagnosed as infiltrating ductal carcinoma. Clinicopathologic characteristics of the patients including, age, tumor size, lymph node involvement, metastasis status, tumor grade, estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) status were collected and included in the final analysis.
Immunohistochemistry
Immunohistochemical (IHC) staining of the paraffin sections was performed using mouse monoclonal anti-human p53 antibody (clone DO-7, DakoCytomation). Visualization was performed by LSAB™+/HRP kit and DAB+ Chromogen (DakoCytomation). For negative controls, mouse IgG2b (DakoCytomation) with the same concentration as the primary antibody was used.
DNA and RNA isolation
Before DNA and RNA isolation, frozen tissue samples were disrupted by a MagNA Lyser homogenizer (Roche Applied Science, Mannheim, Germany). Genomic DNA was isolated from each tumor sample and its NAT using DNeasy Blood & Tissue Kit (Qiagen GmbH, Hilden, Germany) according to manufacturer’s instructions. RNA was isolated by High Pure miRNA Isolation Kit (Roche) according to the protocol for isolation of total RNA including small RNA.
P53 mutation detection by HRM and DNA sequencing
DNA samples were used for mutation detection in p53 exons 5–8 by high-resolution melt (HRM) technique on a Rotor-Gene™ 6000 (Corbett Research, Qiagen, Germany) real-time analyzer. Primers for p53 exons 5, 6, 7 and 8 were designed by Vector NTI Advance® 11.5.1 (Invitrogen™, Life Technologies, Grand Island, USA) and validated by the BLAST sequence analysis tool (NCBI, Bethesda, USA) (Table 1). HRM reactions were prepared by SensiMix™ HRM (Quantace Ltd., London, UK) in 10 μl volumes containing 5 μl SensiMix™ HRM, 0.4 μl EvaGreen™ dye (Biotium Inc.), forward and reverse primers at 200 nM final concentration, water and 100 ng of genomic DNA. HRM reactions with 4 replicates for each sample were performed with a ramp of 75–95 and 0.1 °C temperature rising. Two DNA samples of healthy individuals with confirmed wild-type p53 sequence were included in each HRM experiment as wild-type controls. Samples which could not be confirmed as wild-type or mutated by HRM were sequenced by appropriate primers covering either exons 5–6 or 7–8 fragments (Table 1). PCR products were visualized on a 2 % agarose gel to confirm presence of the desired product and were sequenced by an Applied Biosystems 3730xl DNA Sequencer (Macrogen Inc., Seoul, Korea). Sequencing data were analyzed by Sequence Scanner software v1.0 (Applied Biosystems, Foster City, CA, USA) and Mutation Surveyor v3.30 (SoftGenetics LLC, State College, PA, USA) for mutational analysis.
MiR-34a quantitative real-time PCR
First strand cDNA synthesis was performed using miRCURY LNA™ Universal RT microRNA PCR system (Exiqon, Vedbæk, Denmark). Equal amounts of RNA samples (20 ng/reaction) were reverse transcribed in triplicates according to manufacturer’s instructions. For qPCR experiments, miRCURY LNA™ SYBR® Green Master (Exiqon) was used with miRCURY LNA™ PCR primer sets (Exiqon) for hsa-miR-34a as target and hsa-miR-132 as the normalizer gene. Reactions were run in 10 μl volumes on a Rotor-Gene™ 6000 real-time analyzer for 45 cycles.
Sequence analysis of p53-binding site
P53-binding site (p53BS) which is located 39 bp upstream of the first non-coding exon of miR-34a on chromosome 1 [13, 14] was analyzed for sequence changes in 13 tumor samples which were identified to have wild-type p53 and downregulated miR-34a expression. A 268-bp fragment containing the p53BS was amplified using a primer set as indicated in Table 1. PCR products were visualized by agarose gel electrophoresis and were sequenced by an Applied Biosystems 3730xl DNA Sequencer (Macrogen). Sequencing data were analyzed by Sequence Scanner v1.0 (Applied Biosystems) and Mutation Surveyor v3.30 (SoftGenetics LLC).
Bisulfite treatment of genomic DNA
Bisulfite conversion of the DNA samples was carried out by EpiTect® Bisulfite Kit (Qiagen). Generally, about 1 μg of each DNA sample was modified according to manufacturer’s protocol, and the final product was eluted in 20 μl of the elution buffer.
Methylation-specific PCR (MSP)
CpG methylation of the miR-34a promoter in the breast tumor DNA samples was assessed using primers mentioned in Table 1 according to Lodygin et al. [23] and Vogt et al. [26]. For methylated and unmethylated controls, reactions with 10 ng of human EpiTect® Control DNA (Qiagen) were also included in this experiment. Methylated and unmethylated PCR products were 122 and 126 bp, respectively which were visualized on a 3 % agarose gel with ethidium bromide staining. Intensity of the bands was analyzed by ImageJ 1.45s image analysis software (National Institute of Health, USA).
P53 and SIRT1 quantitative real-time PCR
Quantitative real-time PCR was performed using specific primers for p53, SIRT1 (Table 1) and RealQ PCR Master (Ampliqon, Denmark) on a Rotor-Gene™ 6000 real-time analyzer. β-Actin was selected as the housekeeping gene.
Data analysis
For all qPCR experiments, comparative quantitation between tumor samples and their respective NAT was performed by REST 2009 (Relative Expression Software Tool, Qiagen) based on Pair Wise Fixed Reallocation Randomization Test® [27]. GraphPad Prism 5 (GraphPad Software Inc., La Jolla, USA) was used for all other statistical tests and production of charts.
Results
Clinicopathologic features and IHC
All patients were female with mean age of 48.8 (range 28–74 years, SD: 12.9). More than half of the study population were at premenopausal stage (n = 25, 54.3 %) (Table 2). Tumor size was between 1 and 13 cm (4 ± 0.36; mean ± SEM) including T1 (n = 10), T2 (n = 22), T3 (n = 12), and T4 (n = 2). Lymph node involvement was positive in 37 samples (85 %). There were 17 patients with metastatic disease. Tumor grades included grade I (n = 17), II (n = 22) and III (n = 7). 39, 28 and 17 samples were ER+, PR+ and HER2+ respectively. Only 2 samples were triple-negative (4 %). Overexpression of p53 protein was detected by immunohistochemistry in 12 breast tumor samples (26.1 %, n = 46).
P53 mutational analysis
In the HRM experiments, only when all four replicates of any sample showed evidence of variation based on a temperature shift of 0.2 °C or more, the sample was regarded as mutated (Fig. 1). Nucleotide variations in p53 exons 5–8 were detected in 13 breast tumors (28.3 %, n = 46) using HRM, while 33 other samples (71.7 %) did not show any confirmed variation in p53 exons 5–8 (Table 3). There was no genetic variation in the NATs.

HRM analysis on p53 exon 6. Left panel normalized graph curves showing different genotypes based on curve shifting. Right panel difference graphs helping visual interpretation of the difference in fluorescence of a sample to the control DNA at each temperature transition
MiR-34a expression level in breast tumors with wild-type and mutated p53
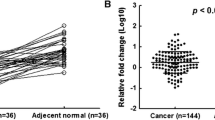
We selected miR-132 as the normalizer for qPCR experiments because other types of RNA, like ribosomal RNAs are sub-optimal normalizers for miRNA expression data due to different sensitivity to degradation. Therefore, miRNAs are preferred over mRNA or ribosomal RNA for normalization of miRNA data. The choice of miRNAs for normalization depends on the tissue of interest, and this needs to be investigated for every tissue separately. We selected and tested miR-132 and miR-374 which were already assessed in BC samples [28]. They quantified 249 mature miRNAs in BC samples for this purpose and identified miR-132 and miR-374 as the best normalizers based on data analysis by geNorm [29] and NormFinder [30] softwares. We selected miR-132 as the single normalizer in our qPCR experiments. Specificity of real-time PCR reaction for miR-34a and miR-132 was confirmed by melt-curve analysis (Fig. 2). A total of 17 tumor samples showed significant downregulation of mature miR-34a compared with their NATs (40 % of the WT-p53 group and 37 % of all samples) (Fig. 3; Table 3). In this group, the mean relative expression of miR-34a in tumor tissues to NATs was about 0.2 (standard error or SEM = 0.04, 95 % confidence interval (CI), 0.116–0.282) (Fig. 4). Among the 17 tumor samples with downregulated miR-34a, 14 samples did not show any significant change in p53 mRNA level compared with their respective NATs, while in the remaining three, p53 mRNA was significantly downregulated. In 17 tumor samples, miR-34a was significantly upregulated compared to their respective NATs with a mean of 3.97 (SEM = 0.66, 95 % CI, 2.59–5.39). Study of the relationship between clinicopathologic features of the BC patients (Table 2) showed statistically significant correlation between miR-34a dysregulation and metastatic disease (Fisher’s exact test, P = 0.0324). Statistical test of the difference between proportions indicated that downregulation of miR-34a was significantly associated with metastatic disease (58.8 %, P < 0.05), while miR-34a upregulation was more prevalent in metastasis-free patients (48.3 %, P < 0.05).

Melt-curve analyses of mature hsa-miR-34a, hsa-miR-132 showing the specificity of the qPCR reactions. Charts produced by Rotor-Gene 6000 Series Software, version 1.7.87 (Corbett Research, Qiagen)

Pie charts showing the distribution of different miR-34a expression levels among a all tumor samples and b tumor samples with wild-type p53 (WT p53)

Expression of mature miR-34a in tumor samples shown as fold change compared to NATs. a miR-34a expression in 17 BC samples which had wild-type p53 and downregulated miR-34a, b miR-34a upregulation detected in 17 BC samples mostly with mutated p53 gene
Sequence analysis of the p53BS
DNA sequencing and mutational analysis of the p53BS did not show any variation in breast tumor samples with downregulated miR-34a.
MSP analysis of the miR-34a promoter
Tumor samples with downregulated miR-34a and wild-type p53 exons 5–8 were analyzed for methylation status of promoter region of miR-34a located on chromosome 1p36. Among the 13 samples analyzed, only one sample showed weak methylation of miR-34a promoter and 12 samples were negative for methylation of the miR-34a promoter (Fig. 5).

MSP analysis of the miR-34a promoter in 13 breast tumor samples and control DNA. a MSP fragments visualized on a 3 % agarose gel showing methylated (M) and unmethylated (U) products for each sample. Control EpiTect human control DNA (Qiagen). b Intensity of the bands measured by image analysis.*Weakly methylated compared to methylated control DNA
Dysregulation of SIRT1 mRNA in breast tumors
Comparison of tumor samples with their respective NATs for SIRT1 mRNA expression using qPCR showed significant upregulation in 23 BC samples with a mean expression ratio of 2.38 (SEM = 0.197, 95 % CI, 1.97–2.79) and downregulation in 14 BC samples (mean = 0.33, SEM = 0.065, 95 % CI, 0.19–0.47) (Fig. 6). Fisher’s exact test did not show any significant correlation between dysregulation of SIRT1 mRNA and any clinicopathologic feature. Frequency of SIRT1 upregulation was similar between BC tumors with up- and down-regulated miR-34a (53 % for each group). In wild-type and mutated p53 groups, SIRT1 was upregulated in 30.4 and 50 % of cases, respectively. Statistical analysis with Fisher’s exact test did not show a significant correlation between upregulation of SIRT1 mRNA and either miR-34a expression or p53 status.

SIRT1 mRNA dysregulation in 37 tumor samples and their respective NATs. a Overexpression in 23 tumor samples and b downregulation in 14 tumor samples
Discussion
Involvement of p53 mutations in almost all types of cancer, including breast cancer, and the close cooperation of p53, miR-34a and SIRT1 with their apoptotic or anti-apoptotic features have created a network with different possibilities regarding promotion or inhibition of cancer. Chang et al. [31] showed that Myc plays a critical role in tumorigenesis by controlling the expression of some miRNAs including miR-34a. Application of miRNAs which are suppressed by Myc has been suggested as a therapeutic option in cancer [32]. A methylation study of miR-34a promoter in several BC cell lines identified 25 % of cases as methylated [23]. In another research including 10 breast tumor tissues, miR-34a and miR-b/c promoter was methylated in 60 and 90 % of cases, respectively [26]. Foekens et al. [28] described a positive association of miR-34b with tumor size and steroid hormone receptor levels. He et al. [12] showed that there is a precise correlation between the expression level of miR-34 family and p53 status in wild-type and p53-deficient mouse embryonic fibroblasts. We utilized Locked Nucleic Acid (LNA™) technology which has been known as superior method for detection of short RNA or DNA sequences. LNA™-enhanced primers provide several advantages for detection of miRNAs. These include high-affinity biding to complementary RNA and high stability for in vitro applications like PCR and real-time PCR, while it also improves specificity for the target [33]. Using this method, we found downregulation of miR-34a in both wild-type and mutated p53 breast tumors with a frequency of 39.4 and 30.8 %, respectively. However, 13 out of 17 tumor samples (76.5 %) showing downregulation of miR-34a belonged to the wild-type p53 group. This finding shows that reduced levels of miR-34a in breast tumors were not possibly the consequence of mutations in the p53 exons 5–8 encoding its DNA-binding domain. Meanwhile, we found BC cases with either unchanged or upregulated miR-34a expression indicating that p53 mutations do not always lead to miR-34a downregulation in breast tumors. In a previous study [34], the reduced expression of miR-34a in neuroblastoma was not associated with either mutations of p53 exons 2–11 or mutations of miR-34a p53-binding site. Lodygin et al., reported frequent CpG methylation of the miR-34a in 25 % of breast cancer cell lines they studied [23], and later, Vogt et al. [26] found methylation of miR-34a promoter in 6 out of the 10 (60 %) breast tumor samples they studied. We examined the p53-binding site of the miR-34a promoter for possible genetic variations, and then, we studied methylation status of the miR-34a promoter. Neither genetic variations nor hypermethylation of miR-34a promoter were detected in the samples with wild-type p53 and downregulated miR-34a. Our results do not support a strong role for miR-34a promoter hypermethylation in breast cancer. These are indicating that miR-34a might be regulated by other factors which work independent of p53 and require further investigation. Enerly and colleagues used miRNA microarray hybridization and showed that miR-34a is not differentially expressed based on the p53 mutational status in primary breast tumors [35]. This was against previous reports about the regulatory effect of p53 on miR-34a, and they concluded that this might indicate a lack of feedback loop between miR-34a and p53. We found that in a significant proportion of breast tumors (39.4 %) with wild-type p53 exons 5–8, mature miR-34a was downregulated compared to the normal breast tissue. Knowledge of miR-34a expression in such cases can be helpful, because attempts toward restoration of p53 pathway in cancer patients without information on miR-34a expression level may not produce the expected results. We also found that among several clinicopathologic features of the BC patients, only metastasis status had a significant correlation with dysregulation of miR-34a. Downregulation of miR-34a was significantly related to metastatic disease, while its upregulation was more related to non-metastatic cases. Li and colleagues studied the effect of ectopic expression of miR-34a in hepatocellular carcinoma and concluded that miR-34a has a suppressive role in tumor migration and invasion through modulation of the c-Met signaling pathway [36]. In another study on BC patients, it was shown that high expression of miR-34a is associated with a lower risk of metastasis and recurrence in breast cancer [37]. In our study, we found that among 17 patients which showed upregulation of miR-34a, only 3 patients had metastasis either early or later in their course of follow-up, while in the patients with downregulated miR-34a (n = 17), there were 10 cases with metastatic disease. Correlation between miR-34a downregulation and increased risk of metastasis has also been reported in prostate cancer [38, 39]. Based on these reports and our results, it seems that lower expression of miR-34a may have significant implications in regards to tumor progression and recurrence in BC patients. However, upregulation of miR-34a may provide a protective effect against metastasis which requires further investigation by functional studies.
SIRT1 has been described to be upregulated mainly at protein level in breast tumors [25]. Overexpression of SIRT1 has been reported in several other types of cancer including acute myeloid leukemia, prostate and colon cancers [19–21]. These studies have provided evidence supporting a role for SIRT1 as tumor promoter. However, there has been significant controversy around the role of SIRT1 as a tumor promoter or suppressor [40, 41]. In regards to breast cancer, Wang et al. [42] found that expression of SIRT1 depends on mutational status of BRCA1, and also BRCA1 deficiency causes reduced levels of SIRT1 which may lead to malignant transformation. We found dysregulation of SIRT1 mRNA in about 80 % of the BC samples. This indicates that SIRT1 is affected at transcriptional level in breast tumors. Sung et al. [25] did not find any significant change at transcriptional level based on a semi-quantitative RT-PCR approach. We detected overexpression of SIRT1 mRNA in 50 % and its downregulation in 30 % of the tumor samples. However, there was no significant correlation between SIRT1 mRNA dysregulation and any of the clinicopathologic features. Yamakuchi et al. [18] found that SIRT1 is a direct target of miR-34a. Based on their findings, miR-34a regulates SIRT1 at translational level and does not affect SIRT1 at transcriptional level. In contrast to that, Fujita et al. [43] studied the effect of ectopic expression of miR-34a in a prostate cancer cell line PC3 and found that miR-34a-induced SIRT1 inhibition occurred at the transcriptional but not post-transcriptional level. We found frequent dysregulation of SIRT1 mRNA in BC tumors (80.4 %, n = 46), but there was no significant correlation with either miR-34a expression or p53 status. So, there are possibly other factors, independent of either miR-34a or p53 which may cause SIRT1 mRNA dysregulation during tumorigenesis.
Conclusion
In this study, we found that miR-34a is frequently downregulated in wild-type p53 breast tumors. Downregulation of miR-34a in wild-type p53 samples was not the consequence of genetic variations or hypermethylation of the miR-34a promoter. Metastatic breast cancer was significantly associated with lower expression of miR-34a. This finding suggests miR-34a expression as a useful biomarker for metastasis. Increased levels of miR-34a in breast tumors of the metastasis-free patients might indicate a protective role for miR-34a against metastasis and recurrence. This needs to be functionally evaluated in future. Based on our results knowledge of miR-34a expression in breast tumors may provide additional useful data when combined with other known biomarkers.
References
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70.
Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107(7):823–6.
Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;113(6):673–6.
Cho WC. OncomiRs: the discovery and progress of microRNAs in cancers. Mol Cancer. 2007;6:60.
Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM. MicroRNA expression and function in cancer. Trends Mol Med. 2006;12(12):580–7.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54.
Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302(1):1–12.
Wang B, Zhang Q. The expression and clinical significance of circulating microRNA-21 in serum of five solid tumors. J Cancer Res Clin Oncol. 2012;138(10):1659–66.
Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, MacDougald OA, Cho KR, Fearon ER. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17(15):1298–307.
Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, Arking DE, Beer MA, Maitra A, Mendell JT. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26(5):745–52.
Corney DC, Flesken-Nikitin A, Godwin AK, Wang W, Nikitin AY. MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 2007;67(18):8433–8.
He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447(7148):1130–4.
Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26(5):731–43.
Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6(13):1586–93.
Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA. 2007;104(39):15472–7.
Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253(5015):49–53.
Ng EK, Wong CL, Ma ES, Kwong A. MicroRNAs as new players for diagnosis, prognosis, and therapeutic targets in breast cancer. J Oncol. 2009;2009:305420.
Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008;105(36):13421–6.
Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, Elgavish A, Nagy TR. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67(14):6612–8.
Bradbury CA, Khanim FL, Hayden R, Bunce CM, White DA, Drayson MT, Craddock C, Turner BM. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19(10):1751–9.
Stunkel W, Peh BK, Tan YC, Nayagam VM, Wang X, Salto-Tellez M, Ni B, Entzeroth M, Wood J. Function of the SIRT1 protein deacetylase in cancer. Biotechnol J. 2007;2(11):1360–8.
Iorio MV, Casalini P, Tagliabue E, Menard S, Croce CM. MicroRNA profiling as a tool to understand prognosis, therapy response and resistance in breast cancer. Eur J Cancer. 2008;44(18):2753–9.
Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Korner H, Knyazev P, Diebold J, Hermeking H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7(16):2591–600.
Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17(2):193–9.
Sung JY, Kim R, Kim JE, Lee J. Balance between SIRT1 and DBC1 expression is lost in breast cancer. Cancer Sci. 2010;101(7):1738–44.
Vogt M, Munding J, Gruner M, Liffers ST, Verdoodt B, Hauk J, Steinstraesser L, Tannapfel A, Hermeking H. Frequent concomitant inactivation of miR-34a and miR-34b/c by CpG methylation in colorectal, pancreatic, mammary, ovarian, urothelial, and renal cell carcinomas and soft tissue sarcomas. Virchows Arch. 2011;458(3):313–22.
Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30(9):e36.
Foekens JA, Sieuwerts AM, Smid M, Look MP, de Weerd V, Boersma AW, Klijn JG, Wiemer EA, Martens JW. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci USA. 2008;105(35):13021–6.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034.
Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64(15):5245–50.
Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40(1):43–50.
Frenzel A, Loven J, Henriksson MA. Targeting MYC-regulated miRNAs to combat cancer. Genes Cancer. 2010;1(6):660–7.
Jensen SG, Lamy P, Rasmussen MH, Ostenfeld MS, Dyrskjot L, Orntoft TF, Andersen CL. Evaluation of two commercial global miRNA expression profiling platforms for detection of less abundant miRNAs. BMC Genomics. 2011;12:435.
Feinberg-Gorenshtein G, Avigad S, Jeison M, Halevy-Berco G, Mardoukh J, Luria D, Ash S, Steinberg R, Weizman A, Yaniv I. Reduced levels of miR-34a in neuroblastoma are not caused by mutations in the TP53 binding site. Genes Chromosom Cancer. 2009;48(7):539–43.
Enerly E, Steinfeld I, Kleivi K, Leivonen SK, Aure MR, Russnes HG, Ronneberg JA, Johnsen H, Navon R, Rodland E, Makela R, Naume B, Perala M, Kallioniemi O, Kristensen VN, Yakhini Z, Borresen-Dale AL. miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS One. 2011;6(2):e16915.
Li N, Fu H, Tie Y, Hu Z, Kong W, Wu Y, Zheng X. miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett. 2009;275(1):44–53.
Peurala H, Greco D, Heikkinen T, Kaur S, Bartkova J, Jamshidi M, Aittomaki K, Heikkila P, Bartek J, Blomqvist C, Butzow R, Nevanlinna H. MiR-34a expression has an effect for lower risk of metastasis and associates with expression patterns predicting clinical outcome in breast cancer. PLoS One. 2011;6(11):e26122.
Watahiki A, Wang Y, Morris J, Dennis K, O’Dwyer HM, Gleave M, Gout PW. MicroRNAs associated with metastatic prostate cancer. PLoS One. 2011;6(9):e24950.
Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, Wiggins JF, Bader AG, Fagin R, Brown D, Tang DG. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17(2):211–5.
Luo J, Altieri DC. SIRTing through breast cancer is just a survivin’ game. Mol Cell. 2008;32(2):159–60.
Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci. 2009;5(2):147–52.
Wang RH, Zheng Y, Kim HS, Xu X, Cao L, Luhasen T, Lee MH, Xiao C, Vassilopoulos A, Chen W, Gardner K, Man YG, Hung MC, Finkel T, Deng CX. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell. 2008;32(1):11–20.
Fujita Y, Kojima K, Hamada N, Ohhashi R, Akao Y, Nozawa Y, Deguchi T, Ito M. Effects of miR-34a on cell growth and chemoresistance in prostate cancer PC3 cells. Biochem Biophys Res Commun. 2008;377(1):114–9.
Acknowledgments
We would like to thank Dr. Reza Mirfakhraie, Dr. Mohammad Zaefizadeh and Ms. Tayebeh Majidizadeh for their technical advice. This study was supported by a Research Grant (No. 370) from the National Institute of Genetic Engineering and Biotechnology.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Javeri, A., Ghaffarpour, M., Taha, M.F. et al. Downregulation of miR-34a in breast tumors is not associated with either p53 mutations or promoter hypermethylation while it correlates with metastasis. Med Oncol 30, 413 (2013). https://doi.org/10.1007/s12032-012-0413-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-012-0413-7