
Abstract
Sorafenib is a multi-target oral anticancer drug used as first-line treatment for patients with advanced human hepatocellular carcinoma (HCC). But the exact mechanism of sorafenib involved in HCC treatment is not clear yet. In this study, a comparative proteomic approach was performed to identify novel sorafenib-related proteins in HCC. Proteomes of HepG2 cells treated with sorafenib and the control (without sorafenib) were obtained by two-dimensional differential gel electrophoresis. Comprehensive analysis of proteins was focused on total protein spots to filtrate the different protein spots between the two groups. The differentially expressed proteins were identified by peptide mass fingerprinting with high-performance liquid chromatography–tandem mass spectrometry. Then, Western blot and immunohistochemistry were used to verify the expression of some candidate proteins. Results indicated that 19 protein spots were differentially expressed with significant changes, including 6 up-regulated proteins and 13 down-regulated proteins. It was confirmed by Western blot that expressions of Annexin A1 and cyclophilin A were down-regulated in sorafenib-treated HCC cell lines. Immunohistochemical study revealed their oncogenic role in HCC tissues. These observations might be novel findings leading to bring new insights into the exact mechanism of sorafenib and identify possible therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human hepatocellular carcinoma (HCC) is the fifth major cause of cancer death worldwide, of which 50% were reported in China, and surgical resection is one of the only curative therapies [1, 2]. However, it has limitations for patients with dissemination in early stage. Moreover, the effects of chemotherapy are very limited and the objective response rate of single-drug treatment is only about 10%. The advent of targeted agents, inhibiting pivotal molecules involved in the regulation of signal transduction pathways central to tumorigenesis and progression, has provided new options for the treatment of patients with advanced HCC [3]. Sorafenib, one of the molecular targeted drugs, is a multi-target oral anticancer drug used as a first-line treatment for patients with advanced HCC [4]. Studies demonstrated a significantly longer overall survival (OS) time and median time to progression for patients in the sorafenib arm and showed evidence of the potential for combining sorafenib with other anticancer agents. But the exact mechanism of sorafenib involved in HCC treatment is not yet clear.
The development of theory and technology in proteome has provided new ideas and research fields for cancer research. Proteome can be used not only for elucidating the mechanism of carcinogenesis, but also for seeking the biomarkers diagnosis and therapy of cancer [5]. High-throughput proteome profiling and bioinformatics tools allow for examining global changes in protein expression of HCC in response to sorafenib. Identification of differentially expressed proteins may lead to a better understanding of the molecular events involved in sorafenib anti-tumor mechanism, identify possible novel therapeutic targets, and bring new insights into HCC carcinogenesis.
In the present study, we detected the effect of sorafenib on human hepatocellular carcinoma HepG2 cell lines and performed comprehensive proteome analysis of HepG2 cells treated with sorafenib and the control (without sorafenib treatment), and 19 different expressed proteins were identified. Expression of Annexin A1 and cyclophilin A was also verified by both Western blot and immunohistochemistry.
Materials and methods
Cell lines and cell culture
The human hepatocellular carcinoma cell line HepG2, Hep3B, and Bel-7402 were purchased from the American Tissue Culture Collection. The cell line was grown in RPMI-1640 culture media (Hyclone, USA), supplemented with 10% (v/v) fetal bovine serum (Hyclone, USA), 100 U/ml of penicillin (Sigma, USA), and 0.1 mg/mL of streptomycin (Sigma, USA). All cells were grown in identical culture conditions in a 37°C, 5% CO2 incubator; culture media was changed thrice weekly.
Cell cytotoxicity assay
The tetrazolium salt MTT (3-[4,5,-dimethylthiazol-2]-2,5-diphenyltetrazolium bromide, Sigma, USA), as previously described, was used to test the capacity of growth inhibition of sorafenib in HepG2 cells [6]. Sorafenib-containing culture medium at various concentrations was added into 96-well cell culture plates and incubated for 24 h or 48 h. After reaction with MTT solution dissolved in phosphate-buffered saline (PBS, 1 mg/mL) for 4 h, dimethylsulfoxide (DMSO) was added to solubilize the reduced tetrazolium salts. Measurement of the absorbance at 570 nm in a microtitre plate reader was performed. The experiments were repeated in triplicate.
Analysis of apoptosis
Cells were cultured for 24 h in the presence of sorafenib according the result from the cell cytotoxicity assay. Apoptosis in the HepG2 cell line was quantified by staining with annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI). The samples were analyzed using flow cytometry (FACSCalibur, BD Biosciences, San Jose, CA).
Cell cycle analysis
Cells were cultured for 24 h in the presence of sorafenib according the result from cell cytotoxicity assay. After harvesting by trypsinization and washing with PBS, cells were fixed in ice-cold 70% ethanol, washed, and resuspended in 1 mL PBS, treated with 10 μL RNase, and stained with 150 μL propidium iodide (PI) for 30 min at room temperature. The stained cells were analyzed by flow cytometry (FACSCalibur, BD Biosciences, San Jose, CA), and DNA content was quantified using Coulter System software (Beckman Coulter, Inc., Fullerton, CA).
Protein sample preparation
Proteins were extracted from intact cell pellets (6 × 105 cells) treated with sorafenib at 20 μmol/L for 24 h, using a buffer consisting of 7 M urea, 2 M thiourea, 4% CHAPS, 2% NP-40, 2% pharmalyte, 5 mM PMSF, 1% Triton X-100, 100 mM DTT, and 0.5 Mm EDTA and centrifuged for 1 h at 4 000 rpm. The supernatant was precipitated with acetone and dried. Protein concentrations were determined using the Bradford assay. All samples were stored at −80°C prior to electrophoresis.
Two-dimensional gel electrophoresis (2-DE)
First, dimension isoelectric focusing (IEF) was performed at 20°C for 30,000 VhT using IPGphor™ Isoelectric Focusing Unit (Amersham Pharmacia Biotech AB, Uppsala, Sweden). The second dimension gel electrophoresis was performed using 12.5% gradient SDS polyacrylamide gels with buffer running solutions containing 1.5 M Tris and 10% SDS. Gels were stained with 0.2% (w/v) silver nitrate [7]. ScanMaker8700 (Amersham Pharmacia Biotech AB, Uppsala, Sweden) was used for scanning of gels, and Image analysis software (Image Master 2D Platinum Software 5.0, Geneva Bioinformatics, Geneva, Switzerland) was used to construct average gels from three independent experiments and a comparison between those average gels was performed. Protein spots that showed more than a twofold difference in the normalized spot volume between sorafenib-treated cells and the control (or which were “undetectable” in one of the cell lines) were chosen for further analysis.
High-performance liquid chromatography–tandem mass spectrometry (HPLC–MS)
After digestion of interested differently expressed spots, a HPLC system (Surveyor, ThermoFinnigan, San Jose, CA) was applied. Peptides were eluted by using a gradient from buffer A (0.1% formic acid) to buffer B (90% vol/vol acetonitrile, 0.1% formic acid). The HPLC column eluent was eluted directly into the electrospray ionization source of a LCQ-Deca ion trap mass spectrometer (ThermoFinnigan, San Jose, CA). Automated peak recognition, dynamic exclusion, and daughter ion scanning of the top two most intense ions were performed using the Xcalibur software [8]. Spectra were scanned over the range 400–2,000 mass units.
Database searching and data interpretation
MS/MS data were analyzed using SEQUEST, a computer program that allows the correlation of experimental data with theoretical spectra generated from known protein sequences [9]. All matched peptides were confirmed by visual examination of the spectra, and all spectra were searched against the latest version of the public non-redundant protein database of the NCBI or SWISS-PROT.
Western blot analysis
In order to validate the obtained data, the expression of two selected proteins was determined by Western blot analysis in three HCC cell lines. In brief, 20 μg of total protein extracts from sorafenib-treated HCC cells lines were separated on 12% polyacrylamide gel and the proteins were then transferred electrophoretically onto polyvinylidene difluoride membranes. After blocking for 1 h with blocking buffer (1× PBS, 0.5% Tween-20 with 5% nonfat dry milk), the membranes were incubated overnight, at 4°C, with antibodies against Annexin A1 (rabbit polyclonal IgG, 1:500; AbCam Biotechnology, Abcam China, Hong Kong) or cyclophilin A (mouse monoclonal IgG, 1:200; AbCam Biotechnology). The primary antibodies were detected by use of horseradish peroxidase (HRP)-conjugated secondary antibodies (goat anti-rabbit or mouse IgG; Santa Cruz Biotechnology). Visualization of the immunoreactive proteins was accomplished by the use of ECL Plus reagents (Amersham Biosciences) and exposed to X-ray film. β-actin was used as a loading control.
Immunohistochemical analysis
Since patients with sorafenib treatment always gave up surgery, it was very difficult to collect the samples of tumor tissues. We therefore detected the expression of Annexin A1 and cyclophilin A in HCC tissues compared to normal hepatic tissues (native liver tissue adjacent to hepatic hemangioma) by immunohistochemistry (IHC). A total of 40 samples, including 20 HCC tissues and 20 native liver tissues adjacent to hepatic hemangioma, were obtained from the First Affiliated Hospital of Xi’an Jiaotong University. The study protocol was approved by the ethics committee and the human research review committee of Xi’an Jiaotong University, and written informed consent was obtained from all subjects. Tissue specimens were fixed in neutral-buffered formalin (10% v/v formalin in water; pH 7.4) and embedded in paraffin wax. Serial sections of 4-μm thickness were cut and mounted on charged glass slides. Conditions for Annexin A1 and cyclophilin A were optimized and evaluated by two independent pathologists. The rabbit polyclonal antibody against Annexin A1 and cyclophilin A was used at dilutions of 1:1,000 or 1:500, respectively. The streptavidin-peroxidase technique (Golden Bridge International: SP-9000) was used as described [10]. An irrelevant rabbit antiserum served as a negative control. Sections were counterstained with Mayer’s hematoxylin. The staining results were evaluated as described previously [11]. For the purpose of statistical analysis, the median of this series (30% of malignant cells showing a stronger intensity than normal hepatic cells) was used as a cutoff value to distinguish tumors with a low (<30) or high (≥30%) level of Annexin A1 or cyclophilin A expression.
Statistical analysis
The student’s t test was used to compare the behavioral responses and Western blot results. χ 2 analysis was done to evaluate the significance of differences for immunohistochemical analysis between the two groups. A Fisher’s exact text was used to analyze the relationship between expression levels of Annexin A1 and cyclophilin A and various clinicopathologic characteristics. The difference was considered statistically significant if the P value was less than 0.05.
Results
Growth inhibition of HepG2 cells by sorafenib
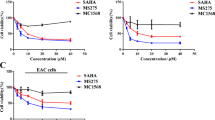
The HepG2 cell lines were cultured in the presence of various concentrations of sorafenib for 24 and 48 h, and the cell viabilities were measured by the MTT assay. As shown in Fig. 1a, the viabilities of HepG2 were inhibited by sorafenib in dose- and time-dependent manners. The IC50 value of 24 h was 15.35 μmol/L.

Sorafenib inhibits growth of HepG2 cells. a HepG2 cells were treated with various concentrations of sorafenib and incubated for 24 h or 48 h. Cell growth was analyzed by the MTT assay. The percent viabilities relative to control cells are shown. HepG2 were inhibited by sorafenib in dose- and time-dependent manners. b Induction of apoptosis was analyzed by annexin V/PI analysis. HepG2 cells were incubated with the indicated concentrations of sorafenib for 24 h. Results of annexin V/PI analysis were quantified. c Representative dot blot figures for cells apoptosis analyzed by flow cytometry. d Quantification of the results of cell cycle induced by sorafenib. A marked increase in S-phase cells and a decrease in G1 cells are observed. ▲ P < 0.05, compared to control group (sorafenib = 0 μmol/L)
Apoptosis induced by sorafenib
To further quantify the cell death, annexin V/PI analysis was performed. Sorafenib induced annexin V single-positive and annexin V/PI double-positive cells in a dose-dependent manner (Fig. 1b).
Cell cycle arrest by sorafenib
Since cell survival was inhibited by sorafenib in HepG2 cells, we examined the cell cycles of sorafenib-treated cells. As shown in Fig. 1c, sorafenib treatment resulted in dose-dependent increase in cells at the S-phase and decreases at the G1 phase as described before [12].
2-DE and the analysis of gel images
To isolate proteins efficiently, the 2-DE was performed with a 12.5% separation gel in the second dimension. The resulting images were analyzed using PDQUEST software. A typical 2-DE proteome spot pattern of treated and control tissue is shown in Fig. 2a. Under the same experimental conditions, six gels, three for control and three for sorafenib-treated were analyzed. Using gel matching and analysis, the proteomic profile of treated HCC cells and controls were shown to be very similar. In the 2-DE maps of the sorafenib-treated cells and the control, 19 protein spots were found to be differentially expressed with changes in the stain density of twofold or more, including 6 up-regulated proteins and 13 down-regulated proteins (P < 0.05). Two representative different spots were magnified and are shown in Fig. 2b and c.

Representative example of differential protein expression profiles. a Proteins from sorafenib-treated HepG2 cells (left) and the control cells (right, without sorafenib treatment). b Representative example of highly expressed proteins with sorafenib treatment. c Representative example of lowly expressed proteins with sorafenib treatment. Protein spot numbers were from Table 1
HPLC–MS analysis and protein identification
Nineteen differentially expressed spots were identified by HPLC–MS analysis. All identified different proteins are shown in Table 1. An example (No. 1 as shown in Table 1) of the tandem mass spectrum of one of the nineteen peptides found, along with the SEQUEST output data for that spectrum, was shown in Fig. 3. The identified proteins were next categorized into possible functions according to the classification systems [13]. Detected proteins were mainly involved in energy metabolism, protein synthesis, and signal transduction (Fig. 4).

An example tandem mass spectrum (MS/MS) of a peptide identified from sorafenib-treated HepG2 cells. The area highlighted in Fig. 2a was analyzed by HPLC–MS. This spectrum represents one example (No. 1 as shown in Table 1) of a peptide identified by HPLC–MS and SEQUEST database searching of the resulting spectra as being from HepG2 cells (see also Table 1). The SEQUEST output data for this spectrum is also indicated

Comparison of protein identification results. Detected proteins were mainly involved in energy metabolism, protein synthesis, and signal transduction
Confirmation of down-regulated expression of Annexin A1 and cyclophilin A by Western blot analysis
Western blot analysis of protein extracts from sorafenib-treated HCC cells lines was used to confirm different expression of Annexin A1 and cyclophilin A, as shown in Fig. 5. In all sorafenib-treated cases, Annexin A1 expression was lower with the treatment dose of sorafenib. Expression of cyclophilin A was also much lower than the control.

Western blot analysis of Annexin A1 and cyclophilin A in sorafenib-treated human hepatocellular carcinoma cell lines. Western blot analysis revealed markedly down-regulated Annexin A1 and cyclophilin A expression in sorafenib-treated human hepatocellular carcinoma cell lines HepG2, Hep3B, and Bel-7402. Cells were treated with different dose of sorafenib, and β-actin was used as a loading control
Confirmation of up-regulated expression of Annexin A1 and cyclophilin A by IHC
Expression of Annexin A1 and cyclophilin A in 20 tumor and non-tumor tissues was also confirmed by IHC. Positive staining of Annexin A1 and cyclophilin A located in the cytoplasm was detected by IHC, showing that they were expressed mildly in normal tissues but strongly in cancer tissues (Fig. 6). For all 20 HCC samples, Annexin A1-positive staining was observed in 80% of tumor tissues (16/20) compared with only 35% of non-tumor tissues (7/20), while positive staining of cyclophilin A was observed in 75% of tumor tissues (15/20) compared with only 30% of non-tumor tissues (6/20). Differences between intensity of staining of Annexin A1 and cyclophilin A in tumor and non-tumor tissues were significant (P < 0.05). High expression levels of both proteins were significantly associated with tumor grade (Table 2).

Immunohistochemical study of Annexin A1 and cyclophilin A in HCC. Typical immunohistological features with high levels of Annexin A (up) and cyclophilin A (down) expression in HCC (a, c) and normal tissues (b, d). Annexin A1 and cyclophilin A were expressed more strongly in cancer than that in normal tissues. The slides were viewed by microscopy (magnification 400)
Discussion
This study identifies 19 proteins differentially expressed in sorafenib-treated human hepatocellular carcinoma HepG2 cell lines compared with control cells (Table 1). We have performed the first proteomic study of human hepatocellular carcinoma HepG2 cell lines treated with sorafenib to determine the possible common mechanisms. Although several of these proteins have been previously implicated in various types of cancers, they have never been linked with sorafenib, thus providing an interesting insight into the mechanisms of sorafenib in HCC.
In our study, some identified down-regulated proteins function as potential tumor-associated proteins. Elevated expression of Cofilin-1 and Prx1 has been observed in several human cancers [14–17]. And, there are more reports for Annexin A1 and cyclophilin A. It was found that Annexin A1 was up-expressed in HCC, pancreatic cancer, and breast cancer and played an important role in the malignant transformation process either through modulation of cytosolic phospholipase A2 activity or EGFR function [18–21]. Recent studies that used analytic methods such as MALDI-TOF, protein profiling, and bioinformatics demonstrated that many cancers including small cell lung cancer, pancreatic cancer, breast cancer, colorectal cancer, squamous cell carcinoma, and melanoma show over-expression of cyclophilin A [22–27]. Although there are several articles about the oncogenic role of Annexin A1 and cyclophilin A, few reports revealed the expression and function of these two proteins in HCC. The limited results showed Annexin A1 might play a critical role in the process of HCC metastasis and combination of cisplatin with cyclophilin A inhibitors increased apoptotic cell death in HCC cells [28, 29]. So, we chose Annexin A1 and cyclophilin A for further experiments. Our results demonstrated that over-expression of Annexin A1 and cyclophilin A in HCC compared to normal hepatic tissues, and sorafenib down-expressed Annexin A1 and cyclophilin A in HCC cell lines in a dose-dependent manner. High expression levels of both proteins were significantly associated with tumor grade, showing an oncogenic role in HCC. These results suggest that sorafenib may inhibit the growth of HepG2 cells partly by down-regulating the expression of certain tumor-associated proteins, especially Annexin A1 and cyclophilin A.
We also found some proteins related to chemoresistance. Ribonucleotide reductase M1 (RRM1) is a key enzyme involved in DNA synthesis, catalyzing the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides. Studies indicated that higher levels of RRM1 were associated with chemoresistance to gemcitabine-based therapies [30–34]. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is a multifunctional protein that acts at the intersection of energy metabolism and stress response in tumor cells. Phadke et al. [35] recently reported that GAPDH depletion induced cell cycle arrest without inducing apoptosis and defined GAPDH as an important determinant of cellular sensitivity to antimetabolite chemotherapy. Our result showed that RRM1 and GAPDH were down-regulated in sorafenib-treated HepG2 cells, laying the foundation for the combination of sorafenib with other anticancer drugs. For up-regulated proteins, Rab27a is a protein function in the exocytic transport of lysosome-related organelles. Mutations of the Rab27a gene were discovered to cause pigmentary dilution and immunodeficiency in human Griscelli syndrome [36]. We hypothesize that Rab27a may be involved in the dermatologic adverse events, and more research is need to verify the accurate role of Rab27a in sorafenib treatment.
We can also see that 8 of the 19 proteins found altered upon sorafenib treatment are enzymes of the monosaccharide metabolism, most of which are involved in glycolysis. The relationship between glycolysis and cancer has been fully reviewed [37]. Meanwhile, recent report revealed that inhibition of lactic dehydrogenase was a way to increase the anti-proliferative effect of sorafenib, which is consistent with our experiment that sorafenib can down-regulate expression of two types of l-lactate dehydrogenase [38]. We can conclude that sorafenib had an effect on the glycolysis, but whether it was the reason or result of its anticancer ability needs further study.
In conclusion, our in vitro study identified a group of proteins that are differentially expressed between sorafenib-treated HepG2 cell lines and the control. There were further questions that arose which require more in-depth study. Firstly, are the protein expression changes identified central to the mechanism of adverse events, such as rash and desquamation? Further research on clinical patients with toxic side effects will provide further information. Secondly, are any of the elements of this proteomic profile predictive of clinical responses to sorafenib in patients with HCC? On-going research to correlate in vivo protein expression changes and clinical response will address this issue. Third, are the proteins detected possible novel therapeutic targets? Functional assays that relate differential expression of these novel proteins to the generation of cell cytotoxicity or drug resistant will improve our understanding of the roles of these proteins in HCC.
References
Bosch FX, Ribes J, Díaz M, Cléries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127(5 suppl 1):S5216.
Lai EC, Lau WY. The continuing challenge of hepatic cancer in Asia. Surgeon. 2005;3(3):210–5.
Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4(5):361–70.
Dal Lago L, D’Hondt V, Awada A. Selected combination therapy with sorafenib: a review of clinical data and perspectives in advanced solid tumors. Oncologist. 2008;13(8):845–58.
Wong SC, Chan CM, Ma BB, Lam MY, Choi GC, et al. Advanced proteomic technologies for cancer biomarker discovery. Expert Rev Proteomics. 2009;6(2):123–34.
Wosikowski K, Regis JT, Robey RW, Alvarez M, Buters JT, et al. Normal p53 status and function despite the development of drug resistance in human breast cancer cells. Cell Growth Differ. 1995;6(11):1395–403.
Mortz E, Krogh TN, Vorum H, Görg A. Improved silver staining protocols for high sensitivity protein identification using matrix-assisted laser desorption/ionization-time of flight analysis. Proteomics. 2001;1(11):1359–63.
Andon NL, Hollingworth S, Koller A, Greenland AJ, Yates JR III, et al. Proteomic characterization of wheat amyloplasts using identification of proteins by tandem mass spectrometry. Proteomics. 2002;2(9):1156–68.
Yates JR III, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67(8):1426–36.
Chen HJ, Xiao JR, Yuan W. Loss of p16INK4, alone and with overexpression of osteopontin, correlates with survival of patients with spinal metastasis from hepatocellular carcinoma. Med Oncol. 2010;27(3):1005–9.
Wang J, Zhang M, Zhang L, Cai H, Zhou S, et al. Correlation of Nrf2, HO-1, and MRP3 in gallbladder cancer and their relationships to clinicopathologic features and survival. J Surg Res. 2010;164(1):e99–105.
Liu L, Cao Y, Chen C, Zhang X, McNabola A, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66(24):11851–8.
Yang Y, Liu X, Fang X, Wang X. Proteomic analysis of mitochondria in Raji cells following exposure to radiation: implications for radiotherapy response. Protein Pept Lett. 2009;16(11):1350–9.
Wang W, Goswami S, Lapidus K, Wells AL, Wyckoff JB, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64(23):8585–94.
Yanagawa T, Iwasa S, Ishii T, Tabuchi K, Yusa H, et al. Peroxiredoxin I expression in oral cancer: a potential new tumor marker. Cancer Lett. 2000;156(1):27–35.
Noh DY, Ahn SJ, Lee RA, Kim SW, Park IA, et al. Overexpression of peroxiredoxin in human breast cancer. Anticancer Res. 2001;21(3B):2085–90.
Cha MK, Suh KH, Kim IH. Overexpression of peroxiredoxin I and thioredoxin 1 in human breast carcinoma. J Exp Clin Cancer Res. 2009;28:93.
Masaki T, Tokuda M, Ohnishi M, Watanabe S, Fujimura T, et al. Enhanced expression of the protein kinase substrate annexin in human hepatocellular carcinoma. Hepatology. 1996;24(1):72–81.
Bai XF, Ni XG, Zhao P, Liu SM, Wang HX, et al. Overexpression of annexin 1 in pancreatic cancer and its clinical significance. World J Gastroenterol. 2004;10(10):1466–70.
Ahn SH, Sawada H, Ro JY, Nicolson GL. Differential expression of annexin I in human mammary ductal epithelial cells in normal and benign and malignant breast tissues. Clin Exp Metastasis. 1997;15(2):151–6.
de Coupade C, Gillet R, Bennoun M, Briand P, Russo-Marie F, et al. Annexin 1 expression and phosphorylation are upregulated during liver regeneration and transformation in antithrombin III SV40 T large antigen transgenic mice. Hepatology. 2000;31(2):371–80.
Campa MJ, Wang MZ, Howard B, Fitzgerald MC, Patz EF Jr. Protein expression profiling identifies macrophage migration inhibitory factor and cyclophilin A as potential molecular targets in non-small cell lung. Cancer Res. 2003;63(7):1652–6.
Li M, Zhai Q, Bharadwaj U, Wang H, Li F, et al. Cyclophilin A is overexpressed in human pancreatic cancer cells and stimulates cell proliferation through CD147. Cancer. 2006;106(10):2284–94.
Zheng J, Koblinski JE, Dutson LV, Feeney YB, Clevenger CV. Prolyl isomerase cyclophilin A regulation of Janus-activated kinase 2 and the progression of human breast cancer. Cancer Res. 2008;68(19):7769–78.
Wong CS, Wong VW, Chan CM, Ma BB, Hui EP, et al. Identification of 5-fluorouracil response proteins in colorectal carcinoma cell line SW480 by two-dimensional electrophoresis and MALDI-TOF mass spectrometry. Oncol Rep. 2008;20(1):89–98.
Huang CF, Sun ZJ, Zhao YF, Chen XM, Jia J, et al. Increased expression of peroxiredoxin 6 and cyclophilin A in squamous cell carcinoma of the tongue. Oral Dis. 2011;17(3):328–34.
Al-Ghoul M, Brück TB, Lauer-Fields JL, Asirvatham VS, Zapata C, et al. Comparative proteomic analysis of matched primary and metastatic melanoma cell lines. J Proteome Res. 2008;7(9):4107–18.
Lee J. Novel combinational treatment of cisplatin with cyclophilin A inhibitors in human hepatocellular carcinomas. Arch Pharm Res. 2010;33(9):1401–9.
Dai Z, Liu YK, Cui JF, Shen HL, Chen J, et al. Identification and analysis of altered alpha1, 6-fucosylated glycoproteins associated with hepatocellular carcinoma metastasis. Proteomics. 2006;6(21):5857–67.
Goan YG, Zhou B, Hu E, Mi S, Yen Y. Overexpression of ribonucleotide reductase as a mechanism of resistance to 2, 2-difluorodeoxycytidine in the human KB cancer cell line. Cancer Res. 1999;59(17):4204–7.
Cao MY, Lee Y, Feng NP, Xiong K, Jin H, et al. Adenovirus-mediated ribonucleotide reductase R1 gene therapy of human colon adenocarcinoma. Clin Cancer Res. 2003;9(12):4553–61.
Davidson JD, Ma L, Flagella M, Geeganage S, Gelbert LM, et al. An increase in the expression of ribonucleotide reductase large subunit 1 is associated with gemcitabine resistance in non-small cell lung cancer cell lines. Cancer Res. 2004;64(11):3761–6.
Rosell R, Danenberg KD, Alberola V, Bepler G, Sanchez JJ, et al. Ribonucleotide reductase messenger RNA expression and survival in gemcitabine/cisplatin-treated advanced non-small cell lung cancer patients. Clin Cancer Res. 2004;10(4):1318–25.
Phadke MS, Krynetskaia NF, Mishra AK, Krynetskiy E. Glyceraldehyde 3-phosphate dehydrogenase depletion induces cell cycle arrest and resistance to antimetabolites in human carcinoma cell lines. J Pharmacol Exp Ther. 2009;331(1):77–86.
Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25(2):173–6.
Wang JS, Wang FB, Zhang QG, Shen ZZ, Shao ZM. Enhanced expression of Rab27A gene by breast cancer cells promoting invasiveness and the metastasis potential by secretion of insulin-like growth factor-II. Mol Cancer Res. 2008;6(3):372–82.
Gatenby RA, Gillies RJ. Glycolysis in cancer: a potential target for therapy. Int J Biochem Cell Biol. 2007;39(7–8):1358–66.
Fiume L, Vettraino M, Manerba M, Di Stefano G. Inhibition of lactic dehydrogenase as a way to increase the anti-proliferative effect of multi-targeted kinase inhibitors. Pharmacol Res. 2011;63(4):328–34.
Acknowledgments
Supported by National Natural Science Foundation of China (30872483).
Author information
Authors and Affiliations
Corresponding author
Additional information
Aili Suo and Mingxin Zhang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Suo, A., Zhang, M., Yao, Y. et al. Proteome analysis of the effects of sorafenib on human hepatocellular carcinoma cell line HepG2. Med Oncol 29, 1827–1836 (2012). https://doi.org/10.1007/s12032-011-0013-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12032-011-0013-y