
Abstract
Cisplatin (DDP)-based adjuvant chemotherapy is widely used for the treatment of esophageal cancer. However, DDP resistance has become more common and thus new approaches are required to be explored. Cisplatin was used to induce autophagy in the human esophageal cancer cell line, EC9706 cells, and the effect of autophagy on the survival of EC9706 cells was investigated using an autophagy inhibitor 3-MA. Cell viability was measured by CCK8 assay. Apoptosis and cell cycle were detected by flow cytometry. Monodansylcadaverine (MDC) was used to detect autophagy. Western blotting assay was used to investigate the molecular changes that occurred in the course of treatment. DDP inhibited cell proliferation, induced cell death and cell cycle arrest at S phage. Moreover, autophagy was activated through class III PI3K pathway. The expression of autophagy-related Beclin1 and LC3-I was up-regulated and part of LC3-I was converted into LC3-II. However, after the combination treatment of 3-MA and DDP, the cell inhibitory rate increased; the apoptosis rate and the numbers of cells in S phase also increased. Furthermore, the accumulation of autophagic vacuoles was decreased; the expression of Beclin1 and LC3 was significantly down-regulated and the release of cytochrome c was decreased. DDP-induced apoptosis in EC9706 cells can be enhanced by the inhibitor of autophagy, 3-MA. Autophagy might play a role as a self-protective mechanism in DDP-treated esophageal cancer cells, and its inhibition could be a novel strategy for the adjuvant chemotherapy of esophageal cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Esophageal squamous cell carcinoma (ESCC) is one of the most prevalent malignant tumors in developing countries, especially in China [1]. Several studies have shown survival benefits of platinum-based chemotherapy followed by radical esophagectomy. Good and bad effects of chemotherapy depend on the level of tumor sensitivity to chemotherapeutic drugs. However, it is reported that cells of ESCC have developed resistance to chemotherapeutic drugs, thus resulting in a decrease in the 5-year survival rate for ESCC [2]. Some evidence indicates that cisplatin not only can kill cells through the induction of apoptosis but also induce cell death by nonapoptotic forms of cell death [3]. The significance of nonapoptotic cell death in chemotherapy and the mechanisms by which it is induced remain unclear now. Provided that cancer cells cannot survive in the face of apoptosis, it would be viable if therapeutic agents could kill cancer cells of anti-apoptosis by other means.
Autophagy is a cellular self-catabolic process where cellular components are engulfed and trafficked to lysosomes for proteolytic degradation [4]. The main function of autophagy is to maintain quality control for proteins and organelles in order to enhance survival under conditions of scarcity or starvation. The autophagic process has received increased attention after the recognition that it represents a novel form of cell death, autophagic cell death (type II programmed cell death), distinct from apoptosis and necrosis [5]. Autophagy has been proved as a self-defense mechanism activated in response to stressful stimuli, such as exposure to chemo-radiotherapy [6, 7]. However, autophagy may also result in cell death, if it proceeds to completion. Nevertheless, more and more studies have indicated the importance of autophagy in the cancer development and progression, and the role and relationship of autophagy and apoptosis in cancer cells are still unclear and indicate more complex than ever thought.
Previous study suggested that the protein level of Beclin1 (the autophagy-related gene) was higher in ESCC tissue than that in normal tissue [8]. In present study, we investigated the role of autophagy in the apoptotic cell death induced by DDP on EC9706 cells, detected molecular changes that occurred with the addition of 3-MA and examined the changes occurring during the apoptosis process.
Materials and methods
Cell line and cell culture
A poorly differentiated ESCC cell line EC9706 was obtained from the State Key Laboratory of Molecular Oncology, Chinese Academy of Medical Sciences (Beijing, China). The cells were cultured in RPMI-1640 medium (Gibco, Rockville, USA) containing 10% fetal bovine serum (FBS) (Hyclone Laboratories, Logan, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in the presence of 95% air, 5% CO2 with medium changes every 2 days. Cells in the mid-log phase were used in the experiments. The cells were divided into three groups: control, DDP, and DDP plus 3-MA groups. After 24 h, the culture medium were replaced with fresh medium containing none (control group), DDP (DDP group), and the combination (DDP plus 3-MA group). EC9706 cells were treated with 1.5 mg DDP (IC50 = 1.44 ± 0.15 mg) and/or 5 mM 3-MA for 24 h.
Chemicals and reagents
Cisplatin (DDP) was purchased from Shandong Qilu Pharmaceutical Product Factory, Jinan, China. 3-MA (Sigma) was used as an inhibitor of autophagy. 3-MA (100 mg) was dissolved in phosphate-buffered saline (PBS) to make a 100 mM stock solution and kept at room temperature. The future dilution was made in RPMI-1640 medium to obtain the required concentration. Monodansylcadaverine (MDC, Sigma) was used to detect autophagy.
Cell cytotoxicity assay by cell counting kit-8
Cells were seeded at 5 × 103 per well onto flat-bottomed 96-well culture plates. On the day of measuring the growth rate of EC9706 cells, 100 μL of spent medium was replaced with an equal volume of fresh medium containing 10% CCK8 (WST-8, Dojindo Laboratories, Tokyo, Japan). Cells were incubated at 37°C for 1 h, and the absorbance of the solution was finally determined at 450 nm using microplate spectrophotometer. Cell inhibitory rate was calculated according to the following formula: Cell inhibitory rate (%) = [1−A450 (sample)/A450 (control)] × 100.
Detection of apoptosis and cell cycle by flow cytometry
Apoptotic cells were detected by flow cytometry. Cells harvested were washed twice with cold PBS and resuspended in PBS. After adding 5 μL of AnnexinV-FITC solution and 5 μL of PI solution according to the instructions of AnnexinV-FITC Apoptosis Detection Kit (KeyGen Biotechnology, Nanjing, China), cells were incubated for 15 min at room temperature in the dark. Then, the cells were analyzed on a flow cytometry system (FACScan, Becton–Dickinson, San Jose CA, USA). Data were analyzed using Cell Quest software (Bection Dickinson). Cell cycle analysis was performed with the ethanol-fixed cells resuspended in 1 mL PBS with 40 μg PI and 100 μg of RNase A for 30 min at 37°C. Samples were then subjected to analysis of their DNA contents by flow cytometer system.
Analysis of autophagy
A fluorescent compound, MDC, has been proposed as a special tracer for autophagic vacuoles [9]. The autophagic vacuoles were labeled with MDC by incubating cell growth on cover-slips with 0.05 mmol/l MDC in PBS at 37°C for 1 h. After incubation, cells were washed three times with PBS and immediately analyzed by fluorescence microscopy using an inverted microscope (Nikon Eclipse TE 300, Germany) equipped with a filter system (excitation wavelength 380 nm, emission filter 525 nm). Cell number was counted to normalize the measurement, and the percentage of MDC incorporation in cells was calculated.
Western blot analysis
Cells were harvested from cultured dishes and were lysed for 20 min in cold lysis buffer. Cell extracts were collected and centrifuged at 12,000 rpm for 5 min. Total proteins (20 μg) from whole cell lysates were boiled for 5 min in 1× SDS buffer, resolved by 12% SDS–PAGE, and then electrotransferred to nitrocellulose membranes (Amersham, Uppsala, Sweden) by a semi-dry transferor. After the electrophoretic transfer, the membrane was blocked overnight at 4°C and after blocking, membranes were incubated for 1 h with primary antibodies at recommended concentration. The antibodies for Beclin1, LC3, and β-actin were purchased from Santa Cruz Technologies. Also, anti-class III PI3K and anti-cytochrome c were purchased from Abcam Technologies and Cell Signaling Technologies, respectively. Then, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (anti-rabbit or anti-mouse IgG) (Santa Cruz). The blots were developed using enhanced chemiluminescence (Amersham Biosciences, Piscataway NJ, USA) and developed on Kodak X-omat LS film (Eastman Kodak Company, New Haven CT, USA). Densitometry was performed with Kodak ID image analyses software (Eastman Kodak Company).
Measurement of cytochrome c release
Mitochondria and cytosol fractions were prepared by different centrifugation. Adherent and floating cells were collected and suspended in cold buffer (250 mM sucrose, 1 mM EDTA, 50 mM Tris–HCl pH 7.5 supplemented with 1 mM PMSF, 1 μg/mL leupeptin, 1 μg/mL pepstatin A, 5 μg/mL aprotinin, and 5 mM DTT). Then, cells were mechanically lysed using a Dounce cell homogenizer (Sigma) and centrifuged at 1000 g for 10 min at 4°C to remove nuclei and unbroken cells. Then, the fraction enriched with mitochondria (heavy membrane fraction) was pelleted by centrifugation at 10,000 g for 20 min at 4°C. Finally, the supernatant was centrifuged further at 100,000 g for 1 h at 4°C to remove the light membrane portion, so as to get the cytosol fraction.
Statistical analysis
All data represent at least three independent experiments and are expressed as means ± SD. For statistical analysis, Student’s t-test was used as appropriate. An association was considered significant when the exact significant level of the test was P < 0.05.
Results
Cell death in ESCC cells induced by DDP treatment
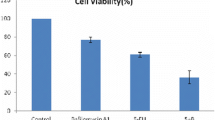
After EC9706 cells were treated with DDP at the indicated concentrations for 48 h, cell inhibitory rate was examined by CCK8 assay. As shown in Fig. 1a, EC9706 cell growth was effectively inhibited by DDP in a dose- and time-dependent manner, and the IC50 for 48 h of DDP treatment in EC9706 cells was 1.44 ± 0.15 mg. Based on this result, we used 1.5 mg of DDP for 48 h in EC9706 cells for further experiments.

a EC9706 cells were incubated with different concentrations of DDP for 24 and 48 h. Cell inhibitory rate was measured with CCK8 assay. b 3-MA enhances DDP-induced EC9706 cell death. Control (no treatment), DDP (treated with 1.5 mg of DDP), DDP plus 3-MA (treated with both 5 mM of 3-MA and 1.5 mg of DDP) for 24 h. a P < 0.05 compared with control group
DDP-induced EC9706 cell death is enhanced by 3-MA treatment
Next, the changes of EC9706 cell death were assessed using DDP with or without 3-MA.Cells were divided into three groups: control (no treatment), DDP (treated with 1.5 mg DDP), and combination (treated with both of 1.5 mg DDP and 5 mM 3-MA). In the DDP and 3-MA group, the inhibitory rate of EC9706 cell increased faster than in the DDP group. After treatment for 24 h, the combination drove 57.3% of the cells to death, a 26.1% increase compared with the rate of cell death in the DDP group (Fig. 1b). This result suggests that 3-MA can enhance DDP-induced cell death in ESCC cells.
3-MA is one of the most widely used inhibitor of autophagy, and many studies have indicated that autophagy can play an important role in the response of cancer cells to chemotherapeutics. Then, we detected changes of autophagy activity by observing the fluorescence of MDC, which has been known as a specific marker for autophagic vacuoles [10]. In addition, the DDP treatment also induced an autophagic response in ESCC cells (Fig. 2). The number of autophagic vacuoles stained by MDC in the DDP group was much higher than in the control. With the addition of 3-MA, the number of vacuoles was lower than in the DDP group. The quantification result of MDC incorporated into cells indicated that there was a 1.56-fold increase in the DDP group compared with the control and that 3-MA decreased by 1.08-fold (Fig. 2).

3-MA inhibits the increased autophagy induced by DDP. The autophagic vacuoles were observed with MDC. In the DDP group, the autophagic vacuoles were increased in EC9706 cells compared with control group, which was counteracted by 3-MA. a P < 0.05 compared with control group
Apoptosis has been reported to be associated with the effect of chemotherapeutic drugs in ESCC cells. We used AnnexinV-FITC and propidium iodide (PI) staining assay to observe the apoptotic cell death and cell cycle progression in DDP-treated ESCC cells. As illustrated in Fig. 3, compared with the DDP group, the apoptosis rate of the EC9706 cells that were co-treated with 3-MA significantly increased from 19.76 to 29.12% (P < 0.05) and the percentage of cells in S phase increased from 36.80 to 44.50% (P < 0.05), which was accompanied by a decrease in cells at G0/G1 phase (P < 0.05).

3-MA increases DDP-induced apoptotic cell death and cell cycle arrest at S phase. Photographs (a, b, c) are the flow cytometry results of the control group, DDP group, and DDP+3-MA group, respectively. Compared with the DDP group, the apoptosis rate of the EC9706 cells that were co-treated with 3-MA significantly increased from 19.76 to 29.12% (P < 0.05), and the percentage of cells in S phase increased from 36.80 to 44.50% (P < 0.05), which was accompanied by a decrease in cells at G1 phase (P < 0.05)
To obtain clues as to the mechanism by which DDP induces autophagy, western blot analysis was performed. The cytochrome c and the mitochondria play a central role in inducing the cell to begin the process of the programmed cell death, apoptosis [11]. Thus, we examined the release of cytochrome c from mitochondria into cytosol. The fractions of mitochondria and cytosol were isolated by different centrifugation. We found that DDP treatment alone caused translocation of cytochrome c from mitochondria to the cytosol and 3-MA caused an increase of cytochrome c release in ESCC cells (Fig. 4). Beclin-1 is a Bcl-2-interacting protein that promotes autophagy, associated with inhibition of cellular proliferation and tumorigenesis [12]. As shown in Fig. 5, DDP significantly up-regulated the expression of Beclin-1. However, after the addition of 3-MA, the increase in protein Beclin1 was inhibited. Microtubule-associated protein light chain 3(LC3) is another commonly used molecular marker to assess autophagy. LC3 occurs in two forms in cells; cytoplasmic LC3-I and lipidated LC3-II, which is associated with autophagosomal membranes and forms cytoplasmic speckles [13]. So LC3-II is localized to preautophagosomes and autophagosomes, making this protein an autophagosomal marker [14]. Thus, LC3 antibody was able to recognize both the upper LC3-I band and the lower LC3-II. After a 24-h incubation with DDP, the LC3-I protein level was significantly increased, which indicated that DDP up-regulated the expression of LC3-I and part of LC3-I was converted into LC3-II. However, after the addition of 3-MA, compared with the DDP group, no significant alteration in the expression of LC3-I was found, but the conversion of LC3-I to LC3-II was significantly decreased.

Effect of 3-MA on the cytochrome c release by DDP treatment. The mitochondria (M) and the cytosol (C) fractions were isolated and subjected to immunoblotting for the detection of cytochrome c. In the right graphs, C/(C + M) was calculated and presented as mean ± SEM from three independent experiments to represent the extent of the release of cytochrome c from mitochondria (M) to cytosol (C)

The expression of autophagy-related proteins. Beclin1 and PI3K III were up-regulated in the DDP group, and 3-MA counteracted the up-regulation of Beclin1 and PI3K III. LC3-I and LC3-II were also up-regulated in the DDP group. After co-treatment with 3-MA, LC3-I decreased and the conversion of LC3-I to LC3-II was also reduced. a P < 0.05 compared with control group
In the autophagic pathways, there are two main molecular regulation mechanisms, phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/mTOR (the mammalian target of rapamycin) and class III PI3K signal transduction pathways. In the process of autophagy, the complex of Beclin-1/class III PI3K-dependent autophagy has an important function in mediating the localization of other autophagy-related proteins to the preautophagosomal membrane [15]. We then detected the expression of PI3K III proteins during our experiment. As illustrated in Fig. 5, there was a significant up-expression of PI3K III protein after DDP treatment. However, after the addition of 3-MA, the increase in protein PI3K III was inhibited. These results suggest that DDP may induce autophagy through the activation of class III PI3K pathway in the cells. The data above indicate that autophagy is also involved in the cell response to DDP treatment and an inhibitor of autophagy could enhance the apoptotic cell death induced by DDP. From a clinical application, autophagy inhibitor may sensitize ESCC cells to DDP-based chemotherapy.
Discussions
Cisplatin (DDP) is a clinically highly relevant anticancer drug used for the treatment of esophageal cancer. The major mechanism underlying its antitumor activity has been ascribed to the induction of DNA damage by platinum compounds [16]. In our experiment, we found that DDP could induce EC9706 cells to death (Fig. 1a), and this cell death included apoptotic cell death (Fig. 3). At present, drugs inducing apoptosis remain the main chemotherapeutic agents in medical oncology [17]. Although cisplatin-based chemotherapy has shown promising effects with various types of carcinomas including ESCC, acquired resistance to chemotherapy has been one of the most important clinical problems [18, 19]. Therefore, it is still an urgent need to explore new therapeutic strategies.
3-MA is a classical specific inhibitor of autophagy. It stops autophagy at the sequestration step in mammalian cells through inhibiting class III PI3K without markedly affecting protein synthesis or ATP levels [20, 21]. Some evidence has shown that 3-MA can enhance the effect of chemotherapeutic drugs by triggering apoptosis in several cancer cells. In human prostate cancer cells, 3-MA exacerbated the release of cytochrome c to cytosol and then increased PC-3 cell apoptosis induced by sulforaphane [22]. In human colon cancer cells, sulforaphane-induced WiDr cell death was also increased by 3-MA treatment [23]. In our experiment, we observed that in EC9706 cells, the cell death induced by the combination of DDP and 3-MA was obviously higher than DDP alone (Fig. 1b). Thus, this result suggests that 3-MA enhances DDP-induced EC9706 cell death and that inhibition of autophagy improves chemotherapeutic effect of DDP in ESCC cells. Autophagy is a cellular pathway involved in protein and organelle degradation. Autophagy principally serves an adaptive role to protect organisms against diverse pathologies, including infections, cancer, neurodegeneration, aging, and heart disease [4]. Recently, several reports have indicated that autophagy is readily induced in response to stressful stimuli, such as metabolic stress [24, 25] and exposure to anticancer drugs [26–28]. So it is believed that autophagy has an important role in tumorigenesis and progression.
Despite present controversies on the exact role of autophagy in the process of tumor generation and progression, either by promoting cell survival or contrarily by inducing cell death, it is certainly an intricate target for cancer therapy [29, 30]. As expected, treatment of EC9706 cells in the DDP group resulted in increased formation of autophagic vacuoles, as well as up-regulation of Beclin1 protein, induction of LC3-I, and recruitment of processed LC3-II to autophagosomes, which are the typical features of autophagy induction, compared with the control group (Figs. 2, 5). Indeed, autophagy induced by anticancer drugs in cancer cells was often observed [31]. Autophagy might serve as a protective response through degrading damaged proteins or organelles by anticancer treatments; and its inhibition can enhance the effect of anticancer treatment (Fig. 1b). Furthermore, we found that, in ESCC cells, 3-MA inhibited the increase of autophagy induced by DDP (Figs. 2, 5) and improved DDP-induced apoptosis by increasing the release of cytochrome c and the number of cells in S phase (Figs. 3, 4). Bafilomycin A1, a vacuolar type H+-ATPase inhibitor, which suppresses autophagy by preventing acidification of lysosomes, was also used to inhibit cancer cell growth and increase apoptotic cell death in various cancer cells induced by irradiation or chemotherapy [7, 32]. Therefore, we suppose that autophagy inhibitors might sensitize ESCC cells to DDP chemotherapy by improving the rate of apoptotic cell death induced by DDP or by converting the autophagic process to an apoptotic process.
In the formation of autophagosomes, there are two main signal transduction pathways, PI3K/Akt/mTOR and class III PI3K pathway. The Beclin 1/PI3K-III complex is involved in the formation of autophagosomes and initiation of autophagy [15]. Meanwhile, 3-MA, the specific inhibitor of autophagy, is known to target the class III PI3K through competition for adenosine triphosphate (ATP) binding in the active site of its kinase domain and inhibit the autophagic vesicle formation [33]. We found that 3-MA could inhibit the increase of PI3K III protein induced by DDP treatment (Fig. 5). Thus, it is possible that DDP-induced autophagy was activated through the class III PI3K pathway. These findings are supported by similar reports that inhibition of autophagy induced by overexpression of mda-7/interleukin-24 can strongly augment the antileukemia activity in vitro and in vivo [34].
On the other hand, autophagy induction in response to anticancer drugs not only represents a survival mechanism by counteracting the effects of treatment on cancer cells but also can potentially result in tumor cell death, which is also called autophagic cell death—type II programmed cell death(PCD), when autophagy exceeds the safe threshold [35]. Otherwise, it has been demonstrated that rapamycin can exert its antitumor effect on malignant glioma cells by inducing autophagy through inhibiting mTOR-signaling pathways [36, 37]. The data above suggest that autophagy may have different effects in different ways in different cancer cells lines at different stages of tumorigenesis and progression. It also appears to have different responses to anticancer drug-induced stress.
In conclusion, we show that DDP-induced apoptosis in EC9706 cells is enhanced by the inhibitor of autophagy, 3-MA, through activation of class III PI3K pathway, release of cytochrome c and S cell cycle arrest. Moreover, we also find that cell death induced by DDP is increased when autophagy is inhibited. These results provide evidence that inhibition of autophagy may be an effective way to improve the chemotherapy of anticancer agents. These findings suggest that autophagy inhibitor should be investigated as a potential novel chemotherapeutic agent for the adjuvant treatment of human ESCC.
References
Pisani P, Parkin DM, Bray F, Ferlay J. Estimates of the worldwide mortality from 25 cancers in 1990. Int J Cancer. 1999;83:18–29.
Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–52.
Sun Y, Peng ZL. Programmed cell death and cancer. Postgrad Med J. 2009;85:134–40.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42.
Kroemer G, et al. Classification of cell death: recommendations of the nomenclature committee on cell death. Cell Death Differ. 2005;12:1463–7.
Li J, et al. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann Surg Oncol. 2009;16:761–71.
Lomonaco SL, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer. 2009;125:717–22.
Chen Y, Lu Y, Lu C, Zhang L. Beclin-1 expression is a predictor of clinical outcome in patients with esophageal squamous cell carcinoma and correlated to hypoxia-inducible factor (HIF)-1alpha expression. Pathol Oncol Res. 2009. doi: 10.1007/s12253-008-9143-8.
Munafo DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–29.
Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66:3–14.
Goodsell DS. The molecular perspective: cytochrome c and apoptosis. Stem Cells. 2004;22:428–9.
Liang XH, et al. Induction of autophagy and inhibition of tumorgenesis by Beclin1. Nature. 1999;402:672–6.
Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8.
Galluzzi L, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryote. Cell Death Differ. 2009;16:1093–107.
Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–5.
Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33:9–23.
Lefranc F, Kiss R. Autophagy, the Trojan horse to combat glioblastomas. Neurosurg Focus. 2006;20:E7. doi:10.3171/foc.2006.20.4.4.
Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79.
Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478:23–43.
Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA. 1982;79:1889–92.
Lum JJ, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48.
Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome c and apoptosis in human prostate cancer cells. Cancer Res. 2006;66:5828–35.
Nishikawa T, et al. Inhibition of autophagy potentiates sulforaphane—induced apoptosis in human colon cancer cells. Ann Surg Oncol. 2009 [Epub ahead of print]. doi: 10.1245/s10434-009-0696-x.
Degenhardt K, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64.
Karantza-Wadsworth V, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–35.
Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007;14:548–58.
Pan J, et al. Autophagy induced by farnesyltransferase inhibitors in cancer cells. Cancer Biol Ther. 2008;7:1679–84.
Park MA, et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol Ther. 2008;7:1648–62.
de Bruin EC, Medema JP. Apoptosis and non-apoptotic death in cancer development and treatment response. Cancer Treat Rev. 2008;34:737–49.
Maiuri MC, et al. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009;16:87–93.
Ding WX, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–10.
Wu YC, et al. Inhibition of macroautophagy by bafilomycin A1 lowers proliferation and induces apoptosis in colon cancer cells. Biochem Biophys Res Commun. 2009;382:451–6.
Gozuacik D, Kimch A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906.
Yang C, et al. Inhibition of autophagy induced by overexpression of mda-7/interleukin-24 strongly augments the antileukemia activity in vitro and in vivo. Cancer Gene Ther. 2009 [Epub ahead of print]. doi: 10.1038/cgt.2009.57.
Chen N, Karantza-Wadsworth V. Role and regulation of autophagy in cancer. Biochim Biophys Acta. 2009;1793:1516–23.
Iwamaru A, et al. Silencing mammalian target of rapamycin signaling by small interfering RNA enhances rapamycin-induced autophagy in malignant glioma cells. Oncogene. 2007;26:1840–51.
Takeuchi H, et al. Synergistic augmentation of rapamycin-induced auto-phagy in malignant glioma cells by phosphatidylinositol 3-kinase protein kinase B inhibitors. Cancer Res. 2005;65:3336–46.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, D., Yang, Y., Liu, Q. et al. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med Oncol 28, 105–111 (2011). https://doi.org/10.1007/s12032-009-9397-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12032-009-9397-3