
Abstract
No biomarker has been available to detect early lung cancer so far. The aim of this study is to screen biomarker patterns for early diagnosis of non-small cell lung cancer (NSCLC) using laser capture microdissection (LCM) and surface-enhanced laser desorption ionization time-of-flight mass spectrometry (SELDI-TOF-MS). The 3 groups of the interested cells from 13 NSCLC tissues, 11 normal lung tissues (out of the 13 NSCLC patients), and 6 benign lung diseased tissues (BLD) were successfully separated by LCM, respectively, and the homogeneities of each type of the cell populations in the three groups were estimated to be over 95%. One-hundred- and twenty-three M/Z peaks were found in the NSCLCs and normal lungs, and between the two groups the relative intensity of 98 M/Z peaks was significantly different (P < 0.05) using SELDI-TOF-MS. The diagnostic pattern constructed using support vector machine (SVM) including three proteins, M/Z 4282, 3201, and 4252 Da, respectively, showed maximum Youden Index (YI). The pattern was validated by leave-one-out cross validation (LOOCV) and the results showed that the sensitivity was 100.0%, specificity 90.9%, and positive predictive value (PPV) 92.9%. In the NSCLCs and BLDs 188 M/Z peaks were determined and 54 showed statistically difference (P < 0.05). The sensitivity, specificity, and PPV of the diagnostic pattern consisting of two proteins, M/Z 3204 and 3701 Da, were all 100.0%. So, by using LCM we have successfully purified the interested cells and solved the problem of heterogeneity of lung cancer tissue. SELDI protein chip coupled with SVM could effectively screen the differentially expressional protein profiles and eventually establish biomarker patterns with high sensitivity and specificity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lung cancer is one of the most malignant tumors with the highest incidence and mortality rate in the world [1]. Pathogenesis of lung cancer remains unclear and the current five-year overall survival rate for patients is only 10–15% mainly because of lack of available methods for early diagnosis [2]. In recent years, proteomics has become one of the most effective methods to study the pathogenesis, screen the early diagnostic markers and the therapeutic molecular targets of tumor. However, the proteomic study of lung cancer tissue, especially the comparative proteomic study between lung cancers and benign lung diseases (BLD), is rare due to the obvious heterogeneity of lung cancer tissue [3, 4]. Based on clinical and histopathological features, lung cancer comprises small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) which accounts for about 80%. To find helpful evidence for early diagnosis of NSCLC, we compared the differentially expressional protein profiles between NSCLCs and normal lungs and between NSCLCs and BLDs by surface-enhanced laser desorption ionization time-of-flight mass spectrometry (SELDI-TOF-MS). The biomarker patterns were finally established by a support vector machine (SVM) after separating and purifying the parenchyma cells from the tissues of NSCLCs, normal lungs, and BLDs by laser capture microdissection (LCM).
Materials and methods
Materials
All patients suffering from lung cancer or BLDs included in this study attended the Second Affiliated Hospital of Medical School of Xi’an Jiaotong University from May 2004 to October 2006. Informed consent was obtained from every subject before this study and the privacy of each subject was guaranteed according to ethical and legal practice. The average age of the 13 NSCLC patients [7 squamous cell cancers (SCC) and 6 adenocarcinomas (AD)] was 59.4 years old (45–69 years), including 9 males and 4 females. All patients did not undergo chemotherapy or radiotherapy before operation. Complete histopathological information for each case was available. The P-TNM stages (UICC, 2007) were that 2 cases were at stage Ia, 5 stage Ib, 3 stage IIa, 3 stage IIb. Distributions of tumoral differentiation were that 4 cases were well, 7 moderate, and 2 poor. The average age of 6 BLDs control was 56.3 years old (45–65 years), and 4 patients were males and 2 females. The diagnosis of these BLDs was pathologically confirmed as follows: 1 case was inflammatory pseudotumor, 1 mediastinal cyst, 1 bronchiogenic cyst, 1 tuberculoma, 1 hamartoma, and 1 benign pleural endothelioma. Lung tissues of the eleven normal cases were cut at least 5 cm away from the edge of the tumors. The patients averaged 59.9 years (from 45 to 69), and 8 were males and 3 females. Seven of them were matched to SCC and four to AD.
Preparation of tissue specimens
Tissues from lung tumors, BLDs, and normal lung tissues were immediately cut and washed and stored at −80°C after operation. One section was used to confirm diagnosis pathologically and identify cells of interest by routine hematoxylin and eosin (H&E). Then a serial of frozen section at 8 μm was stained by slightly improved H&E for LCM. Briefly, the procedures of slightly improved H&E were: staining with hematoxylin for 1 s; washing with distilled water for 10 s; staining with eosin for 10 s; dehydrating with 85%, 90%, 100% ethanol for 10 s, respectively; clearing with xylene I and xylene II for 2 min, respectively.
LCM
Cells of interest from normal bronchial and alveolar epithelia, tumoral and BLD tissues were microdissected from the frozen tissue sections using the PixCell. Laser Capture Microdissection Microscope (ARCTURUS Inc, American) basically as described by Bonner et al. [5]. The condition of LCM were laser beam diameter, 7.5 μm; duration time, 15.5 ms; and energy, 80 mW.
Cell sample preparation and protein chip array analysis
All LCM samples were thawed on ice for 30–60 min. Ten microliters of U9 buffer (9 M urea, 2% CHAPS, 1% DTT) was added to 5 μl of each LCM sample in a 96-well cell culture plate, which was then agitated on a platform shaker at 4°C for 30 min. Next, 185 μl of sodium acetate (100 mM, pH4) was added to the U9 and was further agitated on a platform shaker at 4°C for 2 min. IMAC3 chips were activated by adding 200 μl of sodium acetate and agitated for 5 min twice. Diluted samples (100 μl) were applied to each spot of the bioprocessor (Ciphergen Biosystems, Ciphergen Biosystem Inc, American) which contained the Protein Chip arrays. The bioprocessor was then sealed and agitated on a platform shaker at 4°C for 60 min. The chips were then washed three times with 200 μl of sodium acetate and another two times with deionized water. Finally, the chips were removed from the bioprocessor and air-dried at room temperature. Before SELDI-TOF-MS analysis, 1 μl of a saturated solution of SPA (Sigma Inc American) in 0.5 l/l CAN, and 5 ml/l TFA was applied onto each chip twice and the chips were again air-dried.
The chips were detected by the PBS-II plus mass spectrometer reader (PBS II+ SELDI-TOF-MS Ciphergen Biosystem Inc, American). Data were obtained by averaging 144 laser shots with an intensity of 185, a detector sensitivity of 7, a high mass of 100000 Da, and an optimized range of 2000–20000 Da. Mass accuracy was calibrated to <0.1% with the All-in-one peptide molecular mass standard (Ciphergen Biosystems).
Bioinformatics and statistics analysis
The total experimental data including through data preprocessing and model construction were arrived at the Zhejiang University Cancer institute’s Protein Chip Data Analysis System (ZUCIPDAS, www.zlzx.net) designed by Yu Jiekai. Firstly, the original data were denoised using the undecimated discrete wavelet transform (UDWT) method. Secondly, the spectra were subjected to baseline correction by aligning with a monotone local minimum curve and mass calibration (adjusting the intensity scale according to three labeled peaks that appeared in all the selected spectra). The proteomic peaks were detected and quantified by an algorithm which takes into account the maximal height of every denoised, baseline-corrected, and calibrated mass spectrum. Thirdly, the peaks were filtered to maintain an S/N of more than three. The S/N of a peak was the ratio of the height of the peak above the baseline to the wavelet-defined noise. Finally, to match peaks across spectra, we pooled the detected peaks whose relative difference in their mass sizes was not more than 0.3%. The minimal percentage of each peak, appearing in all the spectra, was specified to ten. The matched peaks across the spectra were defined as a peak cluster. If there was not a peak in a spectrum within a given cluster, the maximal height within the cluster would be assigned to its peak value. Normalization was performed only with the identified peak clusters.
The preprocessed data were used to establish models. In this experiment, we used a non-linear SVM classifier with a radial-based function kernel, and with a parameter Gamma of 0.6, and with a cost of the constraint violation of 19 to discriminate the different groups. The diagnostic model was evaluated and validated by LOOCV. The principle of validation is that in the approach one sample was taken out each time as the test set and the remaining samples were kept as the training set, and then the test was repeated until each sample has been taken once as a test sample.
Each peak in the experimental data was estimated by the P value of Wilcoxon test. The top ten peaks with the smallest P value were selected for further analysis. Combinations with the highest accuracy in distinguishing different groups of data were selected as potential biomarkers. The SVM model with the highest YI was selected as the model for diagnosing NSCLC.
Results
LCM
Fresh tissue specimens cut from lung tumors and BLDs and normal lungs were prepared (Fig. 1a). A section from NSCLC was stained with routine H&E (Fig. 1b). A Cryostat section 8 μm thickness was stained with slightly improved H&E for LCM (Fig. 1c, d). Cells of interest were collected on an LCM cap (Fig. 1f), while the interstitial tissues remained on section after LCM (Fig. 1e). Approximately 4000 laser hits to each LCM cap resulted in 12000 cells captured in NSCLCs, 3500 laser hits in 7000 cells in BLDs, and 3000 laser hits in 6000 cells in normal lung tissues. Each type of cell populations on LCM cap was estimated to be over 95% homogeneous as determined by microscopic visualization.

LCM process of No. 2 NSCLC tissue specimen: (a) Fresh sample after operation; (b) 10 μm thick section fixed in formalin and paraffin embedded and stained with routine H&E (20 × 10 folds); (c) 8 μm thick frozen section stained with improved H&E (20 × 10 folds) (d) LCM after improved H&E (20 × 10 folds); (e) Interstitial tissue remained on section after LCM (20 × 10 folds); (f) Interested cells collected on LCM cap (20 × 10 folds)
Analysis of the differential pattern
A total of 123 qualified M/Z peaks in an optimized range of 2000–20000 Da were profiled by an IMAC3 protein chip in NSCLCs and normal lungs, and the relative intensity of the peaks between NSCLCs and normal lungs was analyzed by the P value of Wilcoxon test. The results showed the relative intensity of 98 peaks had statistical difference (P < 0.05). Among the 98 peaks, 43 were over-expressed in NSCLCs while being down-expressed in normal lungs. As opposed to the above, the remaining 55 were over-expressed in normal lungs while being down-expressed in NSCLCs. The top ten peaks (Table 1) with the smallest P value were randomly combined and then fed into SVM. The accuracy of each combination in distinguishing NSCLCs from normal lungs was analyzed, and the combination with the highest YI containing three M/Z peaks of 4282, 3201, and 4252 Da, respectively, was chosen as the diagnostic pattern. The three M/Z peaks were all over-expressed in normal lungs while being down-expressed in NSCLCs. When the diagnostic pattern was evaluated by LOOCV, the specificity, sensitivity, and positive predictive value (PPV) were 90.9%, 100.0%, and 92.9%, respectively (Fig. 2 and Table 2).

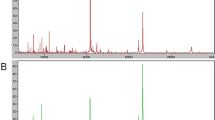
Representative spectra (a) and gel views (b) of the 4253 Da M/Z peaks in NSCLCs and normal lungs
All 188 qualified M/Z peaks were found in NSCLCs and BLDs and the relative intensity of 54 peaks among them showed statistical difference (P < 0.05), 16 were over-expressed in NSCLCs while being down-expressed in BLDs and the remaining 38 over-expressed in BLDs while being down-expressed in NSCLCs. The diagnostic pattern with the highest YI comprised two M/Z peaks of 3204 and 3701 Da (Fig. 3 and Table 3). When the diagnostic pattern was validated by LOOCV, the specificity, sensitivity, and PPV were all 100%.

Representative spectra (a) and gel views (b) of the 3204 Da M/Z peaks in NSCLCs and benign lung diseases
Discussion
At present, an urgent need in lung cancer study is to seek early diagnostic biomarkers. Traditional examinations, such as chest X-ray, sputum cytology, etc. cannot discover lung cancer immediately because there are no obvious symptoms and signs at its early stage [6]. Clinical studies show that existing lung cancer markers have shortcomings of low sensitivity and specificity [7]. SELDI-TOF-MS, a new and powerful proteomic technology, has advantages of high sensitivity throughout and can detect many biomarkers quickly and simultaneously. Indeed, the application of this technology has made a great progress in tumoral study [8–10]. However, the complex pathology and marked heterogeneity of lung cancer tissue have severely hampered proteomic study [11].
LCM, a revolutionary technology in tumoral study that emerged in late 1990 s, can exactly separate single cell or the same kind of cell populations from complex tissues and effectively solve the problem of tissue heterogeneity in experiments [12]. To our knowledge, so far only one published paper by Zhukov et al. [13] studied lung cancer tissue proteomics by applying LCM and SELDI technology. Regretfully, only 4 patients with lung cancer (1 SCC, 1 AD, 1 bronchioloalveolar carcinoma, and 1 mixed) were included in the work.
The SVM classification technique, firstly described by Vapnik, is a new mechanical learning method based on statistical theory. The SVM can solve problems such as the generalization of the medium and small samples in pattern recognition, pattern selection, over-fitting, etc. [14, 15]. The SVM is specifically used for the finite samples to get the optimal solution with available information other than the optimal solution with the sample number tending to be infinite in theory; the overall optimal point can be obtained to solve the local extremum problem which is unsolvable in the artificial neural network method [16]. LOOCV is utilized to determine the accuracy of the classifier. Independent test sets are used to further evaluate the accuracy of our proposed models. All these steps ensure that the selection of biomarkers is not influenced by systematic biases.
In this study, we slightly improved the H&E stain method for LCM. Clearly, stained cells appearing with complete structure and parenchyma and interstitial cells were easily recognized (Fig. 1c). The interested cells were successfully separated by LCM, and each type of cell populations in the three groups was evaluated to be over 95% homogeneous, which coincided with literature [13, 17].
A total of 123 qualified peaks were detected and the relative intensity of 98 peaks had statistical difference (P < 0.05) between NSCLCs and normal lung tissues using SELDI-TOF-MS in this study. The combination with the highest YI including three peaks with 4282, 3201, and 4252 Da was chosen as the diagnostic model. When the diagnostic model was examined by LOOCV, the specificity, sensitivity, and PPV were 90.9%, 100.0%, and 92.9%, respectively. Similarly, among 188 qualified peaks determined between NSCLCs and BLDs, the relative intensity of 54 peaks showed significant difference (P < 0.05). The diagnostic pattern consisting of two proteins, h M/Z 3204 and 3701 Da, yielded 100% in specificity, sensitivity, and PPV, respectively. The above results indicated that the SELDI patterns can effectively discriminate the NSCLCs from normal controls and BLDs, respectively, and have superb diagnostic power as compared with carcinoembryonic antigen (CEA), cytokeratin-19 fragments (Cyfra21-1), and so on [11].
In conclusion, LCM coupled with SELDI-TOF-MS applied in lung cancer research can effectively solve the problem of tissue heterogeneity and screen tumor biomarkers more accurately. It is possible that these candidate biomarkers will be applied in clinical practice in the future. However, the sample size in this study is still limited and more samples are being collected for further study.
References
Yang DC, Yang SY. The current status and progress of lung cancer diagnosis. Chin J Tuberc Respir Dis 2004;27:18–9.
Granville CA, Dennis PA. An overview of lung cancer genomics and proteomics. Am J Respir Cell Mol Biol 2005;32:169–76.
Zhong L et al. Identification of circulating antibodies to tumor-associated proteins for combined use as markers of non-small cell lung cancer. Proteomics 2004;4:1216–25.
Alfonso P et al. Proteomic analysis of lung biopsies: differential protein expression profile between peritumoral and tumoral tissue. Proteomics 2004;4:442–7.
Bonner RF et al. Laser capture micro-dissection: molecular analysis of tissue. Science 1997;278:1481–3.
Swensen SJ et al. Lung cancer screening with CT: mayo clinic experience. Radiology 2003;226:756–61.
Liao P et al. Detection of serum protein biomarkers by surface-enhanced laser desorption and ionization time of flight mass spectrometry (SELDI-TOF-MS) in patients with lung adenocarcinoma. Bull Chin Cancer 2006;15(4):258–61.
Service RF. Genetics and medicine. Recruiting genes, proteins for a revolution in diagnostic. Science 2003;300:236–9.
Cazares LH et al. Normal, benign, preneoplastic and malignant prostate cells have distinct protein expression profiles resolved by surface enhanced laser desorption/ionization mass spectrometry. Clin Cancer Res 2002;8:2541–52.
Seibert V, Ebert MP, Buschmann T. Advance in clinical cancer proteomics: SELDI-TOF-mass spectrometry and biomarker discovery. Brief Funct Genomic Proteomic 2005;4:16–26.
Yang SY et al. Application of serum SELDI proteomic patterns in diagnosis of lung cancer. BMC Cancer 2005;5:83.
Emmert-Buck MR et al. Laser capture microdissection. Science 1996;274:998–1001.
Zhukov TA, Johanson RA, Cantor AB, Clark RA, Tockman MS. Discovery of distinct protein profiles specific for lung rumors and pre-malignant lung lesions by SELDI mass spectrometry. Lung Cancer 2003;40:267–79.
Byvatov E, Schneider G. Support vector machine applications in bioinformatics. Appl Bioinformatics 2003;2:67–77.
Jorissen RN, Gilson MK. Virtual screening of molecular databases using a support vector machine. Chem Info Model 2005;45:549–61.
Wang JX et al. Application of serum protein fingerprint in diagnosis of papillary thyroid carcinoma. Proteomics 2006;6:5344–9.
Baker H et al. Proteome-wide analysis of head and neck squamous cell carcinomas using laser-capture microdissection and tandem mass spectrometry. Oral Oncol 2005;41:183–99.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 30570795) and Program for New Century Excellent Talents in University (No. NCET06-0845) and the Key Program in Science and Technology of Shaanxi Province Shaanxi [No. 2007K09-01(3)].
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yang, S., Nan, Y., Tian, Y. et al. Study of distinct protein profiles for early diagnosis of NSCLC using LCM and SELDI-TOF-MS. Med Oncol 25, 380–386 (2008). https://doi.org/10.1007/s12032-008-9050-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12032-008-9050-6