
Abstract
Spinal and bulbar muscular atrophy (SBMA) is a neuromuscular disease caused by expansions of a polyglutamine (polyQ) tract in the androgen receptor (AR) gene. SBMA is associated with the progressive loss of lower motor neurons, together with muscle weakness and atrophy. PolyQ-AR is converted to a toxic species upon binding to its natural ligands, testosterone, and dihydrotestosterone (DHT). Our previous patch-clamp studies on a motor neuron-derived cell model of SBMA showed alterations in voltage-gated ion currents. Here, we identified and characterized chloride currents most likely belonging to the chloride channel-2 (ClC-2) subfamily, which showed significantly increased amplitudes in the SBMA cells. The treatment with the pituitary adenylyl cyclase-activating polypeptide (PACAP), a neuropeptide with a proven protective effect in a mouse model of SBMA, recovered chloride channel current alterations in SBMA cells. These observations suggest that the CIC-2 currents are affected in SBMA, an alteration that may contribute and potentially determine the pathophysiology of the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chloride (Cl−) plays an important role in regulating the excitability of neurons and muscles through changes in the membrane potential (Arosio and Ratto 2014). In the nervous system, impaired Cl− transport is involved in, e.g., Alzheimer’s disease (De Koninck 2007), epilepsy (Huberfeld et al. 2007), neuropathic pain (Price et al. 2009), and in the regulation of synaptic inhibition in mature brain (Mahadevan and Woodin 2016).
Cl− homeostasis is regulated by several proteins; among them, the largest family of proteins involved in the Cl− flow across cell membranes is represented by the ClC chloride channels and transporters (Jentsch and Pusch 2018). ClC proteins are important for several physiological processes: in addition to the regulation of excitability by stabilizing the membrane potential, they serve mainly in the regulation of cell volume and the transepithelial transport (Jentsch et al. 2002; Accardi and Picollo 2010; Poroca et al. 2017; Jentsch and Pusch 2018).
ClC proteins are expressed in both intracellular and plasma membranes. Nevertheless, based on the ion transport properties, the 9 members of ClC family can be separated into two main groups. In mammals, ClC-3 to ClC-7 are 2Cl−/H+-exchangers located in endolysosomal membranes, while ClC-1, ClC-2, ClC-Ka, and ClC-Kb are voltage-gated Cl− channels strictly expressed in the plasma membrane (Jentsch et al. 2002; Accardi and Picollo 2010; Poroca et al. 2017; Jentsch and Pusch 2018). ClC-K isoforms are differentially expressed in epithelial cells of the nephron, the inner ear, and salivary glands. In contrast, ClC-2 channels are widely and almost ubiquitously expressed in epithelial and nonepithelial cells (Thiemann et al. 1992) whereas ClC-1 is highly specific for skeletal muscle (Aromataris and Rychkov 2006).
Molecular, cellular, and pharmacological studies indicated that ClC-2 channels perform multiple functional roles, including regulation of cell volume, neuronal excitability, and cell proliferation in physiological conditions (Bi et al. 2014; Jentsch and Pusch 2018). Disruption of ClC-2 function leads to azoospermia and retinal degeneration, as observed in ClC-2 knock-out mice (Bösl et al. 2001). Likewise, altered expression and function of ClC-2 channels have been associated with human diseases although their physiological role is still not completely uncovered (Accardi and Picollo 2010; Bi et al. 2014; Jentsch and Pusch 2018).
Experimental evidence supports the idea that altered function of ClC-2 channels contributes to metabolic/endocrinological diseases, e.g., primary aldosteronism (Fernandes-Rosa et al. 2018) and brain diseases (Bi et al. 2014) such as leukoencephalopathy with ataxia (Guo et al. 2019); besides, the megalencephalic leukoencephalopathy with subcortical cysts (MLC) is caused by a recessive mutation of either MLC1 or GLIALCAM genes, where GlialCAM is an auxiliary subunit of ClC-2 channel (Jeworutzki et al. 2012).
Polyglutamine-expansions (PolyQ) in proteins have been identified as the primary cause of neurodegenerative disorders such as Huntington’s disease, dentatorubral-pallidoluysian atrophy, six spinocerebellar ataxias, and spinal and bulbar muscular atrophy (SBMA) (Lieberman et al. 2019). SBMA, also known as Kennedy’s disease, is an adult-onset, genetically inherited (X-linked) neuromuscular disorder (Kennedy et al. 1968). The causative mutation in SBMA is a glutamine-encoding trinucleotide CAG repeat expansion in the first exon of the androgen receptor (AR) gene (La Spada et al. 1991). In normal conditions, the polyQ-AR tract ranges from 9 to 36 residues. However, a repeat number higher than 38 is considered pathogenic (La Spada et al. 1991). Under pathological conditions, the stimulation of the AR by the natural agonists testosterone or dihydrotestosterone (DHT) is required for the induction of polyQ-expanded AR-mediated toxicity (Parodi and Pennuto 2011). After androgen binding, the activated AR translocates to the nucleus, where it interacts with specific sequences on the DNA, ultimately resulting in either the up- or downregulation of the target genes involved in the control of various aspects of the cell behavior. This mechanism could produce changes in ion channel expression or activity (Asuthkar et al. 2015). The influence of testosterone on the activity of ion channels has been demonstrated in cardiac myocytes, where testosterone, progesterone, and a high-concentration of 17-estradiol acutely shortened cardiac repolarization time by regulating L-type Ca2+ current (ICaL) and slowly activating delayed rectifier K+ current (IKs) (Furukawa and Kurokawa 2008).
We previously showed that polyQ-expanded AR is phosphorylated by Akt at serine 215 and serine 792. This post-translational modification reduces mutant AR toxicity both in vitro (Palazzolo et al. 2007) and in vivo (Palazzolo et al. 2009). Conversely, phosphorylation at serine 96 is enhanced in SBMA motor neurons, and its attenuation by pituitary adenylyl cyclase-activating polypeptide (PACAP), a neuropeptide with proven neuroprotective effects in neurodegenerative diseases (Polanco and Pennuto 2018), halts degeneration in vitro and in vivo (Polanco et al. 2016; Jimenez-Garduño et al. 2017).
SBMA is characterized by fasciculation, weakness, and atrophy of bulbar and limb muscles besides a marked loss of motor neurons in the spinal cord and brainstem (Kennedy et al. 1968) which is correlated with the high expression of AR in these neuronal tissues (Polanco et al. 2016). Consequently, damage to skeletal muscle cells/motor neurons and impaired neuromuscular transmission are characteristics of SBMA (Querin et al. 2018). Interestingly, a recent work by genome-wide transcriptome analysis found that motor neurons exhibit increased expression of synapse-related gene sets involved in neuromuscular transmission, proposing the neuromuscular synapse as putative therapeutic target for SBMA (Onodera et al. 2020).
Chloride homeostasis alterations have been reported in neurodegenerative diseases (Poroca et al. 2017), and are also associated with the pathophysiology of polyglutamine disorders (Hsu et al. 2018). In addition, altered overall membrane excitability has been observed in several polyQ spinocerebellar ataxias (Lieberman et al. 2019). Specifically, modifications of the resting membrane potential of SBMA cells have been associated with depletion of chloride channels, together with other voltage-gated channels, expressed in the surface membrane (Oki et al. 2015).
According to our previous electrophysiological results demonstrating that the expression of polyQ-expanded AR causes an androgen-dependent reduction of the macroscopic membrane ionic currents in polyQ-AR motor neuron-derived (MN-1) cells (Jimenez-Garduño et al. 2017), we hypothesized that also chloride currents could be involved into ionic current alterations in SBMA.
Therefore, the aim of this study was to identify the chloride membrane conductance and its putative alterations in a cell model of SBMA. Our electrophysiological and pharmacological approaches, combined with molecular biology, identify a membrane conductance in polyQ-AR MN-1 cells that resembles the ion current kinetics of voltage-gated ClC-2 channels. We found an alteration of ClC-2 currents in DHT-stimulated cells expressing AR with an expansion of 100 glutamine residues (AR100Q) which are considered a pathogenic model of SBMA and recognized to recapitulate AR toxicity.
Additionally, to deepen the applied pathophysiology and pharmacology of ClC-2 in SBMA cells, we evaluated the treatment with the neuropeptide PACAP which, as reported above, ameliorated the disease outcome (Polanco et al. 2016; Jimenez-Garduño et al. 2017; Polanco and Pennuto 2018). In this study, the incubation with PACAP produced an attenuation of the chloride current defects in SBMA MN-1 cells, uncovering a functional mechanism through PACAP might exert his efficacy. Hence, we propose the defects/alterations of the electrophysiological correlate (i.e., the cell electrochemical machinery) as a suitable cellular marker of the disease. Furthermore, our results suggest that ClC-2 channels may represent a novel target for therapeutic intervention in SBMA.
Materials and Methods
Cells
MN-1 are hybridoma cells derived from embryonic mouse spinal cord motor neurons and neuroblastoma cells (clone 2F1.10.14.7) (Salazar-Grueso et al. 1991). MN-1 cells were stably transduced with lentiviral vectors expressing a human AR transgene with either a non-pathological 24Q tract (AR24Q) or an expanded 100Q tract (AR100Q) as well as the soluble enhanced green fluorescent protein (eGFP) (Scaramuzzino et al. 2015; Pennuto and Basso 2016). Cells were routinely cultured with DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 units/mL penicillin G, and 100 μg/mL streptomycin, and incubated in 5% CO2, 100% humidity, at 37 °C. AR24Q and AR100Q cells were seeded at 5 × 104 cells/mL on poly-L-lysine coated glass coverslips. Twenty-four hours before patch-clamp recordings, the cells were cultured in 10% Charcoal Dextran Stripped (CDS) serum with either 10 nM DHT (Sigma, Germany) or vehicle (Ethanol). These cells recapitulate features of SBMA, such as mutant AR aggregation, toxicity, and mitochondrial pathology (Ranganathan et al. 2009; Scaramuzzino et al. 2015; Polanco et al. 2016).
Electrophysiology
Patch-clamp recordings in the whole-cell configuration (Jimenez-Garduño et al. 2017; Roncador et al. 2017; Tripathy et al. 2020) were done in polyQ-AR MN-1 cells at non-sterile conditions and room temperature. The standard bath solution had the following composition: (mM) NaCl 140, KCl 2.8, HEPES 10, MgCl2 1, CaCl2 2 and glucose 10 mM, pH 7.4, and 310 mOsm. The pH of this solution was lowered to 6.9 as needed. The pipette solution had the following composition (mM): CsCl 120, TEA-Cl 20, CaCl2 1, MgCl2 2, EGTA 11, HEPES 10, GTP 0.1 and ATP 1, pH 7.3, and 295 mOsm. In the channel selectivity experiments, NaCl was replaced by NaI 140 mM or Na-gluconate 137 mM. Patch electrodes were fabricated from GB150-8p (OD 1.5 mm, ID 0.86 mm) borosilicate glass capillaries (Science Product) using a PIP6 temperature-controlled pipette puller (HEKA). Working pipettes had tip resistances of 3–6 MΩ. Cells were placed and inspected under an upright microscope Nikon FN1 (Nikon Instruments). Agar bridges were used to eliminate the voltage drift caused by the use of extracellular lower chloride solutions. Bioelectric signals were picked up using an ELC-03XS amplifier (Npi) connected to a personal computer via a breakout-box interface (INT-20X, Npi). High-precision positioning was provided by a piezo-drive micromanipulator (Sensapex). Instant patch-clamp parameters (e.g., Rpipette, Rseal, Cm, Raccess) were monitored with the signal acquisition software (WinWCP Electrophysiology Software, John Dempster, University of Stratchlyde, Glasgow, UK). Voltage commands and current response values were stored in a PC. The acquisition rate of the current signals was 5 KHz and signals were filtered at 2 KHz. For the identification of chloride channels, two different voltage protocols were applied: Protocol I, holding potential 0 mV, test pulses from + 40 to − 160 mV in 20-mV hyperpolarizing steps, followed by a constant + 60-mV short pulse (100 ms, 0 ms of delay); Protocol II, holding potential 0 mV and 20-mV voltage steps between − 155 and + 125 mV, followed by a – 125-mV long pulse (Thiemann et al. 1992; Enz et al. 1999; Fahlke 2001; Stölting et al. 2014; Jentsch and Pusch 2018). For the pharmacological identification of chloride currents, corresponding treatment was prepared as follows: Chloride channel blocker 5-nitro-2-(3 phenylpropylamino)-benzoate (NPPB) and GaTx2 (both from Tocris) were used at 500 μM (in EtOH) and 0.1 μM (in water), respectively. Stock solutions were diluted to final concentration in bath solution and perfused for 1 min at 500 μL/min flow rate. PACAP 27 (Calbiochem Merck, Life Sciences), diluted in water, was used at a concentration of 100 nM with an incubation of 24 h before the experiments.
Protein Extraction and Immunoblotting
Cultured cells were harvested and resuspended into PBS. Cells were lysed in urea buffer with the following composition: 2 M thiourea, 7 M urea, 4% (w/v) CHAPS, 1% (w/v) DTT, and protease inhibitor (ROCHE complete). Lysates were incubated 10 min at 35 °C, and equal amounts of protein (40 μg) were loaded in a 6% SDS-polyacrylamide gel. The proteins were blotted onto nitrocellulose membranes. Then, the membrane was incubated with ClC-2 antibody (1:1000, Santa Cruz, sc-377284) in PBS and 5% low-fat milk. β-actin (1:5000) was used as a housekeeping protein for expression level comparison. The membrane was developed using ECL Prime Western Blotting System (GE Healthcare) with the ChemiDoc MP Imaging System (Bio-Rad, Hercules).
Immunofluorescence Analysis
Cells fixed in 4% paraformaldehyde for 10 min at room temperature (RT) were permeabilized with 0.05% triton in PBS and blocked with 3% bovine serum albumin (BSA). The ClC-2 primary antibody (Santa Cruz, sc-377284) was incubated 1:200 overnight at 4 °C in PBS plus 1% BSA. Alexa Fluor-568 anti-mouse was used at 1:2000 and incubated for 1 h at RT. Microscopy analyses were performing using confocal (SP5, Leica) and epifluorescence (DP70, Olympus) setup. Fluorescence analysis was performed with the image processing program ImageJ (https://imagej.nih.gov/ij/).
Quantitative Real-Time PCR Analysis
Total RNA from MN1 cells was extracted using TRIzol reagent following the manufacturer instructions (Invitrogen). The mRNA reverse transcription was performed with the Thermo Scientific RevertAid First Strand cDNA Synthesis Kit using Oligo-dT primers. RT-PCR analysis was carried out using the KAPA SYBR FAST qPCR Kit (KAPA Biosystems) in the CFX Connect™ Real-Time PCR Detection System (BioRad), using HRPT1 as reference gene for quantification. The primers used for the PCR and RT-PCR were the following: ClC-2 forward (5′-GACTCACATGGCCGGTATGTA-3′), ClC-2 reverse (5′-CACGGTCTACAGCGTAAACAA-3′), HRPT1 forward (5′-TTGCTGACCTGCTGGATTAC-3′) and reverse (5′-CCCGTTGACTGATCATTACAGTAG -3′). RT-PCR reaction conditions used were as follows: one initial step of 95 °C for 3 min followed by 40 cycles of 95 °C for 2 s (denaturation) and 60 °C for 25 s (annealing and extension). The resulting Ct values were used to calculate the fold change of ClC-2 gene using the “delta delta Ct” (∆∆Ct) analysis.
Data Analysis and Statistics
Patch-clamp digitalized data were analyzed off-line. To generate I-V plots, the amplitude values measured by 100 msec (steady-state) were normalized (I/Imax) to current amplitudes at − 100 mV. Data are presented as mean ± standard error of the mean (SEM). G-V curves were calculated by fitting the slope of I-V curves to the linear relation: Y = mX + c. To obtain an estimate of gating conductance, steady-state currents as a function of voltage were adjusted by a Boltzmann distribution of the form: G = G0 + (Gmax − G0)/(1 + exp.[(V − V0.5)/k]. where G, G0, and Gmax are conductance as a function of voltage, residual conductance independent of voltage, and maximal conductance at full activation (extrapolated), respectively. V0.5 is the voltage at which 50% activation occurs, and k is the slope factor (Zúñiga et al. 2004). Maximum conductivity recorded was used as the maximum conductivity (Gmax) to normalize the conductivity of each cell. For I-V as well as G-V calculations, the AR24Q cells (exposed to DHT) were employed to data normalization. Data analysis was performed using WinWCP and OriginPro 8.1. Fluorescence intensity analysis was performed by epifluorescence-acquired images at × 20 magnification. All controls were acquired and analyzed with the same imaging parameters as experimental samples. The images were subject to background subtraction, and the signals were normalized to ClC-2 fluorescence signal in AR24Q cells. RT-PCR data analysis was performed using the “delta-delta Ct” (∆∆Ct) method; fold averages are presented ± SEM. In electrophysiological and molecular experiments, the n was calculated on the number of tested cells, and the number of independent experiments (defined as cultures performed on different days) is given in the respective figure legends.
Statistical analyses were performed using two-way ANOVA followed by Holm-Sidak test (HS) for multiple comparisons. A value of P ˂ 0.05 was considered statistically significant. All statistical analyses were performed using Prism 7 (Graphpad Software, Inc.).
Results
ClC-2 Is Functionally Expressed in polyQ-AR MN-1 Cells
To investigate the activity of the ClC channels in MN-1 cells, we recorded currents with the patch-clamp technique in the whole-cell configuration. The dissection of hyperpolarizing-activated anion currents was performed using an intracellular solution with cesium chloride and tetraethylammonium (TEA)-chloride (Clark et al.1998). We used two different protocols to functionally characterize ClC channels: Protocol-I with a final + 60 mV step to elicit the canonical tail currents of ClC-2 channels (Thiemann et al. 1992; Enz et al. 1999; Jentsch and Pusch 2018), and Protocol-II to identify currents from ClC-1, CFTR, ClC-4, and ClC-5 channels (Fahlke 2001; Stölting et al. 2014).
To model SBMA in vitro, we used the SBMA motor neuron-derived MN-1 cells that we generated expressing either a non-expanded AR with 24 glutamine residues (non-pathological AR24Q) or a polyQ-expanded AR with 100 glutamine residues (pathological AR100Q). These cells represent a valuable in vitro system to model SBMA, as they express similar levels of non-expanded AR and polyQ-AR, and they show activation of caspases and premature death, AR aggregation, and mitochondrial pathology (Palazzolo et al. 2007; Pennuto and Basso 2016; Polanco et al. 2016; Ranganathan et al. 2009). Pulse stimulation of AR24Q cells with Protocol-I showed the typical electrophysiological features of ClC-2 channels: (1) strongly inward rectifying current amplitudes, (2) time-dependent opening, and (3) tail currents (Fig. 1a). On the other hand, stimulation with Protocol-II did not produce changes in AR24Q cells (Fig. 1b). Moreover, we did not detect tail currents during the final – 125-mV stimulation. Based on the known channel localization and the selectivity of the protocols employed, these electrophysiological observations indicate that ClC-1, ClC-4, and ClC-5 could be not expressed (Bennetts and Parker 2013; Stölting et al. 2014). By analyzing the current density for the AR24Q cells in the presence of vehicle (EtOH condition), we obtained a value of 3.73 pA/pF for the maximal amplitude of the current evocated with the “Protocol II,” whereas with the “Protocol I,” we calculated a value of 74.67 pA/pF for the maximal delayed inward rectifying component which thus represents 95.24% of the total chloride current.

Isolation and characterization of chloride currents in polyQ-AR MN-1 cells. Comparison between current traces obtained by applying voltage steps protocols to evoke ClC-2 and ClC-1 chloride currents in the AR24Q cells treated with vehicle. a Typical ClC-2-type membrane currents evoked by application of the Protocol-I (top-left) in whole-cell configuration. The voltage stimulation elicited sharp hyperpolarizing chloride currents. b The application of the ClC-1-evoking Protocol-II (top-right) did not elicit chloride currents
Taken together, these results demonstrate that AR24Q cells present a strong predominance of chloride currents that resemble the kinetics of the currents described for ClC-2 channels (Thiemann et al. 1992; Stölting et al. 2014; Jentsch and Pusch 2018).
Effect of ClC Channel Blockers in polyQ-AR MN-1 Cells
Taking into account that no high-affinity blockers exist for ClC-2 (Jentsch and Pusch 2018), and that ClC-2 is to some extent sensitive to classical chloride channel blockers, to test the ClC-2 channel’s identity in MN-1 cells, we used (1) the blocker 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB) (Clark et al. 1998) and (2) the peptide toxin “Gating modifier Toxin of anion channels 2” (GaTx2) (Thompson et al. 2009), which are the most used blockers for ClC-2 pharmacological characterization (Jentsch and Pusch 2018).
We first investigated the effect of broad-spectrum Cl− channel blockers on the inwardly rectifying currents. The application of NPPB decreased the amplitude of the currents in AR24Q and AR100Q cells treated with DHT (Fig. 2a).

Chloride channel blocking by NPPB. a ClC-2 chloride currents and the effect of NPPB on MN-1 cells. Top panel shows AR24Q-DHT before (left) and after (left) NPPB (500 μM) application. Bottom panel shows AR100Q DHT before (left) and after NPPB application (right). The applied potential in the voltage protocol ranged from − 10 to − 100 mV. b Comparison of the I-V curves given by the treatment with NPPB and control in DHT-treated cell conditions. The current amplitude was measured at the end of the pulse and normalized (I/Imax) to the current at − 100 mV. c Histogram showing the NPPB effect on chloride currents at the most hyperpolarized value (− 100 mV) in the four conditions. *P < 0.05, by two-way ANOVA + HS. d Normalized conductance (G/Gmax) curves obtained at each membrane potential of the currents in panel A. Lines are fits to the Boltzmann equation. Maximum conductivity recorded for each cell was used as the Gmax to normalize the conductivity of each cell. n = 4 for each condition
The analysis of I-V curve at the higher hyperpolarizing value of − 100 mV revealed inhibition of 50.6% in AR24Q Veh cells, 46.7% in AR24Q DHT, 34.6% in AR100Q Veh, and 57.7% in AR100Q DHT (Fig. 2b, c). Interestingly, there was a statistically significant increase (P = 0.048) of the ClC-2 current amplitude in AR100Q DHT cells compared to AR24Q cells, a current increase that could contribute to some aspect of the disease. Furthermore, application of NPPB attenuated the increase of chloride current under pathological conditions (Fig. 2c). To determine the voltage dependence of chloride currents, conductance (i.e., the reciprocal of resistance)-voltage (G-V) curves were constructed from the currents evoked by the pulse protocol of stimulation. Figure 2 d depicts the G-V plot of DHT-treated AR24Q and AR100Q cells. From these fits, we obtained the membrane potential values at which the activation of chloride currents is one half of its maximum (V0.5). The V0.5 of ClC-2-like current was − 86.4 ± 1.3 mV in DHT-treated AR24Q cells and − 73.4 ± 3.8 mV in DHT-treated AR100Q, as calculated by the single Boltzmann distribution, demonstrating that DHT application produced an increment in the conductance of chloride currents as well as a drift to the depolarized values in the V0.5 of ClC-2-like conductance.
Upon GaTx2 perfusion, DHT-treated AR100Q pathological cells showed a lower inward current with a value of the reduction of 25.8% at − 100 mV (Fig. 3a bottom traces, Fig. 3b bottom, and Fig. 3c). This effect also was observed in control AR24Q and AR100Q cells, but no statistically significant differences were observed (Fig. 3c). In the case of DHT-treated AR24Q cells, GaTx2 application did not show a clear effect at the evaluated concentration (Fig. 3a, top traces, and 3b) as further confirmed by the bar plot in Fig. 3c. The steady-state G-V relationships (as above) showed an increase in the conductance of AR100Q cells exposed to DHT (Fig. 3d). Likewise, the conductance of the non-pathological cells was decreased by pharmacological treatment. These results suggest a not significant effect for GaTx2 in both AR24Q and AR100Q cells.

Effect of GaTx2 on ClC-2-like currents. a Representative current traces of the GaTx2 effect on MN-1 cells. Top panel shows AR24Q-DHT before (left) and after GaTx2 (0.1 μM) application (right). Bottom panel shows AR100Q-DHT before (left) and after GaTx2 application (right). The voltage protocol was the same as described in Fig. 2. b Comparison of the I-V curves given the treatment with GaTx2 and control in DHT-treated conditions. The current amplitude was normalized (I/Imax) using control current amplitudes at − 100 mV. The normalized currents were plotted against the test potential. c Summary data for the effects of GaTx2 application on chloride currents in the four conditions in comparison with controls. d G-V relationships at different voltages of the currents in panel a. A fit to the Boltzmann equation is shown as solid lines. n = 4 per condition
Taken together, our results demonstrate that MN-1 cells expressing polyQ-expanded AR have functional ClC-2 channels that are substantially blocked by NPPB and, to a lesser extent, by GaTx2.
Acidic pH Activates ClC-2 Channels in polyQ-AR MN-1 Cells
ClC-2 channels are sensitive to extracellular pH acidification, which results in an enhancement of ClC-2 current (Clark et al. 1998). Therefore, we exposed the cells to an extracellular standard solution adjusted to a pH of 6.9. After a 1-min period of perfusion with an acidic external solution, AR24Q and AR100Q cells showed enhanced responses (Fig. 4a and corresponding I-V curves in Fig. 4b). The amplitude of ClC-2-like currents was increased significantly afterward acidic pH exposure in DHT-treated AR24Q cells by 99.1% and Veh-treated AR100Q cells by 76.9%, but not in AR24Q cells treated with Veh (48%) or AR100Q cells treated with DHT (40.4%) (Fig. 4c).

Sensitivity of ClC-2-type currents to acidification. a Representative traces of the chloride currents evoked by pH 6.9 and 7.4 in the bath solution. Figures show DHT-treated AR24Q and AR100Q cells in physiological (left, top, and bottom) and acidic (6.9, right, top, and bottom) bath solutions. Data recorded from 4 independent cells for each condition are summarized in I-V plots. The voltage protocol was the same as described in Fig. 2. b The amplitude of the voltage-activated currents was enhanced for the acidic external solution. The current amplitude was normalized (I/Imax) using control current amplitudes at − 100 mV. c Histogram showing the acidic pH (6.9) effect on chloride currents of the four conditions in comparison with controls at the most hyperpolarized value (− 100 mV). *P < 0.05, by two-way ANOVA + HS. d Steady-state G-V plots of the currents shown in panel a. A fit to the Boltzmann function is shown as solid lines. n = 4 per condition
Moreover, we observed an increase in the conductance of ClC-2-like current from AR24Q and AR100Q cells under acidic pH without changing its voltage dependence (Fig. 4d). So, our data showed that the recorded currents are sensitive to changes in extracellular pH and are activated by moderate acidification, which also represents a functional marker of ClC-2 channel activity (Clark et al. 1998).
Ion Selectivity of ClC-2 Channels in polyQ-AR MN-1 Cells
To further characterize ClC-2 channels in MN-1 cells, we applied standard substitution protocols employing Na-Iodide (I−) or Na-gluconate (G−) solutions, which discern anion selectivity for ClC-2 channels (Thiemann et al. 1992; Enz et al. 1999). Figure 5 a shows representative traces after iodide and gluconate exposure in control conditions, Veh-treated AR24Q and AR100Q cells. Both conditions showed a reduction in current amplitude (Fig. 5b). In Veh-treated AR24Q and AR100Q cells, removal of extracellular Cl− by NaI or NaG decreased the current amplitudes with the following order Cl− ˃ I− ˃ G− that is in line with the relative anion selectivity reported for ClC-2 channels (Thiemann et al. 1992; Clark et al. 1998; Fahlke 2001). However, the selectivity of ClC-2 has been studied in different models and shows differences in its permeability depending on the species origin and on the cell type (Fahlke 2001). Furthermore, data fitting of the recordings with the extracellular permeant anions Cl−, I−, and G, confirmed the presence of decreases in conductance when Cl−, as the permeant ion, is replaced (Fig. 5c). Taken together, our results match another fingerprint of ClC-2 channels, which is a lower permeability to iodide than to chloride (Fahlke 2001).

Ion selectivity of chloride channels to iodide and gluconate substitution. a Representative current traces of the chloride currents in Veh-treated AR24Q (top) and AR100Q (bottom) cells recorded in different anionic bath solutions. Current amplitudes decreased in NaI, slightly more in Na-gluconate. Data recorded from 5 independent cells for each condition are summarized in I-V plots. The voltage protocol was the same as described in Fig. 2. b Comparison of the I-V curves corresponding to the conditions as in a. The data amplitude was normalized (I/Imax) using current amplitudes at − 100 mV in NaCl physiological bath solution. c Steady-state G-V plots of the currents shown in panel a. Lines are fitted to the Boltzmann equation. n = 4 per condition
Molecular Characterization of ClC-2 Channels in polyQ-AR MN-1 Cells
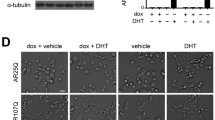
To confirm the presence of ClC-2-like currents observed by electrophysiological recording, we next sought to assess the expression of ClC-2 channels by western blot on polyQ-AR MN-1 cells treated with Veh or DHT. Bands of expected sizes (95 kDa) for ClC-2 were identified in AR24Q as well as AR100Q cells lysates (Fig. 6a): They show that AR24Q and AR100Q cells express endogenous ClC-2 channels. While the upper isoform might correspond to the product of ClC-2 association to accessory molecules such as GlialCAM (Jeworutzki et al. 2012) or channel heterodimerization (Stölting et al. 2014), the lower isoform corresponds to the molecular weight of ClC-2 previously reported (Cortez et al. 2010; Nighot and Blikslager 2012).

Molecular identification of ClC-2 channels in polyQ-AR MN-1 cells. a ClC-2 immunoblot in AR24Q and AR100Q cells. Equal amounts of protein were loaded with actin serving as a loading control. b Confocal photomicrographs of immunostaining for ClC-2 channels in 24Q (top) and 100Q (bottom) MN-1 cells. Co-expression of ClC-2 channel protein (red) and eGFP expression (green) is shown for the vehicle- and DHT-treated cells. Scale bar, 50 μm. c Fluorescence intensity analysis of normalized ClC-2 signal. *P < 0.05, by two-way ANOVA + HS. See methods for details. d Real-time PCR analysis for ClC-2 expression in the four experimental conditions. The values are expressed as the ratio between Veh and DHT treatments for the two polyQ-AR cells types, respectively. *P < 0.05, as in c
By further immunofluorescence and confocal microscopy analysis, we observed the ClC-2 protein expression in whole cells under the four conditions tested (Fig. 6b). The quantitative analysis of fluorescence intensity revealed that DHT did not modify ClC-2 presence in AR24Q cells compared to control conditions. However, the AR100Q cells exposed to DHT showed a statistically significant (P = 0.032) increase of fluorescence intensity, i.e., ClC-2 expression (Fig. 6c). Finally, we evaluated the mRNA expression of ClC-2 channels by RT-PCR in AR24Q and AR100Q cells treated with DHT. This treatment in AR100Q showed a statistically significant (P = 0.036) increase of the transcript levels of ClC-2 gene (Fig. 6d).
The quantitative analyses showing an increase of ClC-2 total protein expression, which positively correlates with the ClC-2 augmented mRNA transcript levels, demonstrate that ClC-2 channel’s upregulation is consistent with the pathological condition. The molecular data obtained suggest that the mechanisms associated with DHT toxicity in SBMA MN-1 cells drive to modifications of ClC-2 channels at transcriptional and protein levels.
PACAP Ameliorates Alterations in ClC-2 Currents in polyQ-AR MN-1 Cells
In order to elucidate whether the increase of the current flow of ClC-2 observed in AR100Q DHT cells correlates to the SBMA cellular phenotype, we measured ClC-2 currents in DHT-activated AR100Q cells treated with PACAP-27, a neuropeptide with protective effects in SBMA cell and mouse models (Polanco and Pennuto 2018; Jimenez-Garduño et al. 2017). PACAP activates the adenylyl cyclase/protein kinase A (PKA) pathway, which downregulates the phosphorylation of polyQ-AR at Ser96, thereby reducing its toxicity (Polanco et al. 2016). PACAP ameliorated the altered macroscopic ionic current in MN-1 cells modeling SBMA (Jimenez-Garduño et al. 2017) and improved the phenotype in in vitro and in vivo SBMA models (Reglodi et al. 2011; Polanco et al. 2016; Rinaldi et al. 2016). We aimed to test whether the ClC-2 function could be recovered upon PACAP treatment. PACAP significantly (P = 0.015) decreased ClC-2 currents in AR100Q DHT after 24-h (Fig. 7a, b). The reduction in the current amplitude at the maximum hyperpolarizing value was 40.6% in AR24Q Veh, 62.1% in AR24Q DHT, 39.1% in AR100Q Veh, and 58% in AR100Q DHT (Fig. 7c).

Effect of the PACAP treatment on ClC-2-type currents. a Representative traces of the chloride currents of DHT-treated AR24Q (left, top) and AR100Q (left, bottom) cells and incubated with PACAP for 24 h. The voltage protocol was the same as described in Fig. 2. b Comparison of the I-V curves corresponding to the conditions shown in panel a given by the chloride currents of the DHT-treated cells (black and blue lines in top and bottom plots, respectively) and PACAP-treated cells (red line). Currents were normalized (I/Imax) using the current amplitudes at − 100 mV. c Histogram showing the PACAP effect on chloride currents for the four conditions in comparison with controls. The voltage-activated currents were substantially reduced by PACAP incubation (100 nM). *P < 0.05, by two-way ANOVA + HS. d G-V plots calculated from the currents shown in a. The data fitted to the Boltzmann function is shown as solid lines. n = 4 per condition
The V0.5 of ClC-2-like current was − 86.4 ± 1.3 mV in DHT-treated AR24Q cells and − 72.3 ± 2.5 in DHT-treated AR100Q cells. However, PACAP induced a decrease in the normalized conductance both in non-pathological and pathological conditions (Fig. 7d). These results further confirm the hypothesis that ClC-2 functional alteration in an SBMA cellular model is directly associated with the altered cellular phenotype, since amelioration of the polyQ-AR toxicity restores ClC-2 function.
Nevertheless, the association between ClC-2 and the SBMA pathological phenotype needs to be further investigated to elucidate the specific role of ClC-2 during the onset of SBMA.
Discussion
Our study presents for the first time a specific voltage-gated ion channel as a possible future candidate in the treatment of SBMA.
Modifications of neuronal excitability by means of alterations in the membrane properties—the structure-function relationship of cell ion channels—play a crucial role in the pathogenesis, development, and pharmacological efficacy of neurodegenerative disorders (Kumar et al. 2016). Evidence has been reported for polyglutamine diseases (Chopra and Shakkottai 2014), such as Huntington’s disease (Akopian et al. 2016), spinocerebellar ataxia types 1, 2, 3, and 6 (SCA1, SCA2, SCA3, SCA6) (Matsuyama et al. 1999; Jeub et al. 2006; Dell'Orco et al. 2017; Bushart et al. 2018; Chopra et al. 2018), and SBMA (Sculptoreanu et al. 2000; Jimenez-Garduño et al. 2017).
In a previous study (Jimenez-Garduño et al. 2017), we investigated macroscopic currents and dissected single cationic currents in an in vitro model of SBMA. We proposed electrophysiological alterations of SBMA cells as a suitable reference point for further studies and possible treatments of the disease. On this line, the investigation of chloride channels involved in the neuronal excitability in SBMA model cells led us to identify and characterize ionic currents and conductance whose behavior most resembled that of the typical ClC-2 (Fahlke 2001; Stölting et al. 2013, 2014).
We found that the majority of the standard protocols for the electrophysiological characterizations of the ClC-2 currents were positive and effective (hyperpolarizing voltage stimulations, sensitivity of blockers, sensitivity to pH variations, iodide/gluconate selectivity). In all the measurements, we always checked that mutant and control cells have a similar cell volume, as calculated by cell capacitance (data not shown) since cell volume variations are a recognized and important triggering factor for ClC activity (Jentsch et al. 2002). ClC-2 channel is a hyperpolarization-activated chloride channel whose gating produces a Cl− outflow that shows strong inwardly rectifying currents (conversely depolarization produces Cl− influx with small outwards currents). Exclusive opening at voltages negative to the chloride reversal potential allows for ClC-2 to regulate chloride flow and neuronal excitability (Stölting et al. 2014).
We hypothesized that altered ClC-2 function drives a misregulation of neuronal excitability, which could ultimately lead to muscle atrophy. This hypothesis is supported by the role exerted by ClC-2 to facilitate post-synaptic inhibition (Smith et al. 1995). Recent findings correlate neuron hyperexcitability with intracellular chloride concentration alterations linked to inhibitory GABAergic stimulus (Huberfeld et al. 2007; Sulis Sato et al. 2017).
According to the evidence that skeletal muscles are a crucial component of SBMA pathogenesis (Palazzolo et al. 2009; Cortes and La Spada 2014; Milioto et al. 2017), other findings demonstrate that the hyperexcitability of SBMA muscle fibers is due to loss of chloride and other voltage-gated membrane channels (Yu et al. 2006). Moreover, the loss of ClC-2 also causes hyperexcitability in CA1 pyramidal cells (Rinke et al. 2010), and it reduces spike activity independently of the concentration of intracellular chloride (Ratté and Prescott 2011). On the other hand, a putative role of ClC-2 channels in the pathogenesis of epilepsy is controversial (Bi et al. 2014; Niemeyer et al. 2010). In addition, it has been shown that ClC-2 promotes neuroprotection in the hippocampus of aging mice (Cortez et al. 2010), and it contributes to the differentiation of oligodendrocyte precursors and to the regulation of myelin synthesis (Hou et al. 2018).
In addition, WB analysis gives molecular evidence of the ClC-2 protein expression in polyQ-AR MN-1 cells. Interestingly, as assessed by immunofluorescence and quantitative PCR, DHT-treated polyQ-AR cells showed an increase in ClC-2 expression compared to control cells. These results provide support to the increasing of ClC-2 like membrane conductance which might be the product of the ClC-2 gene-upregulated expression.
Our findings—the first obtained for a polyQ repeat disorder—on ClC-2 channel regulation in SBMA motor neuron-derived cells MN-1 may represent a breakthrough insight on neuronal excitability. Nevertheless, to further strengthen our findings, further experiments are necessary to unravel the pathophysiological activity of putative ClC-2 in knockout models (following Polanco et al. 2016) and SBMA disease-specific iPSCs (according to Palazzolo et al. 2007, and Narayanan et al. 2017), as well as by silencing the channel expression.
Moreover, we also provided evidence that the treatment with neuroprotective drugs, such as the neuropeptide PACAP, reverts the CIC-2 over-functionality in a cell model of SBMA.
PACAP is a neuropeptide of the vasoactive intestinal peptide (VIP)/secretin/glucagon family with pleiotropic signaling and neuroprotection properties (Tsuchikawa et al. 2012). In SBMA, the impact of the canonical signaling of polypeptides such as VIP or PACAP has been tested. As reported for knock-in SBMA mice, a PACAP analog promoted polyQ-AR degradation and ameliorated disease phenotype (Polanco et al. 2016).
However, PACAP might modulate neuronal function also through the modulation of intrinsic membrane currents which determine neuronal excitability. In support of these hypothesis, the effect of PACAP on chloride current is consistent with its role on neuronal excitability that we previously demonstrated for the macroscopic currents in the SBMA cell model (Jimenez-Garduño et al. 2017). Accordingly, a facilitating and modulatory role of PACAP at the bioelectric level has been recently emphasized by studies showing how PACAP signaling is able to induce plasticity at autonomic synapses (Jayakar et al. 2014) and to trigger both short- and long-term plasticity at nicotinic synapses (Starr and Margiotta 2017). Additionally, it has been demonstrated that PACAP protects rat motor neurons against glutamate-induced excitotoxicity in vitro (Tomimatsu and Arakawa 2008). Similarly, the VIP/PACAP regulatory machinery provides pharmacological profiles and targets towards neuroprotection which are widely distributed in the central nervous systems (Zawilska et al. 2005; Pilzer and Gozes 2006; Reglodi et al. 2011; Korkmaz et al. 2012).
Even if the mechanisms associated with the changes of increased ClC-2 expression/activity shown by our analysis are uncovered, the effectiveness of PACAP treatment in the recovering of normal function of ClC-2 suggests that activation of PKA pathways could be involved in these modifications. Indeed, it is clear the decreasing of ClC-2-like current amplitude exerted by PACAP, indicating that its regulatory function over ClC-2 channel activity may occur in both pathological and non-pathological conditions. Accordingly, we speculate that phosphorylation mechanisms could be involved also in the regulation of ClC-2 channel activity during SBMA as demonstrated for other ionic channels (Gray et al. 1998). Furthermore, taken into account that VIP activates Cl− inwardly rectifying conductances by the contribution of ClC-2 channels (Kajita et al. 2000) and shares a high degree of homology with PACAP-27, our findings open also a novel experimental scenario about VIP neuroprotective role in SBMA through the modulation of the cellular electrical activity.
In conclusion, this work highlights the power of combining electrophysiology and pharmacology study of ion channels involved in SBMA. As a whole, our characterization of a ClC ion channel and relative currents in a SBMA cell model show novel evidence for the functional role of ClC-2 in SBMA pathophysiology. Moreover, our findings indicate a pharmacological target for future therapeutic interventions and the assessment of the disease’s putative biomarkers.
References
Accardi A, Picollo A (2010) CLC channels and transporters: proteins with borderline personalities. Biochim Biophys Acta 1798:1457–1464
Akopian G, Barry J, Cepeda C, Levine MS (2016) Altered membrane properties and firing patterns of external globus pallidus neurons in the R6/2 mouse model of Huntington’s disease. J Neurosci Res 94:1400–1410
Aromataris EC, Rychkov GY (2006) ClC-1 chloride channel: matching its properties to a role in skeletal muscle. Clin Exp Pharmacol Physiol 33:1118–1123
Arosio D, Ratto GM (2014) Twenty years of fluorescence imaging of intracellular chloride. Front Cell Neurosci 8:258
Asuthkar S, Demirkhanyan L, Sun X, Elustondo PA, Krishnan V, Baskaran P, Velpula KK, Thyagarajan B, Pavlov EV, Zakharian E (2015) The TRPM8 protein is a testosterone receptor: II. Functional evidence for an ionotropic effect of testosterone on TRPM8. J Biol Chem 290:2670–2688
Bennetts B, Parker MW (2013) Molecular determinants of common gating of a ClC chloride channel. Nat Commun 4:2507
Bi MM, Hong S, Zhou HY, Wang HW, Wang LN, Zheng YJ (2014) Chloride channelopathies of ClC-2. Int J Mol Sci 15:218–249
Bösl MR, Stein V, Hübner C, Zdebik AA, Jordt SE, Mukhopadhyay AK, Davidoff MS, Holstein AF, Jentsch TJ (2001) Male germ cells and photoreceptors, both dependent on close cell–cell interactions, degenerate upon ClC-2 Cl− channel disruption. EMBO J 20:1289–1299
Bushart DD, Chopra R, Singh V, Murphy GG, Wulff H, Shakkottai VG (2018) Targeting potassium channels to treat cerebellar ataxia. Ann Clin Transl Neurol 5:297–314
Chopra R, Shakkottai VG (2014) The role for alterations in neuronal activity in the pathogenesis of polyglutamine repeat disorders. Neurotherapeutics 11:751–763
Chopra R, Bushart DD, Shakkottai VG (2018) Dendritic potassium channel dysfunction may contribute to dendrite degeneration in spinocerebellar ataxia type 1. PLoS One 13:e0198040
Clark S, Jordt SE, Jentsch TJ, Mathie A (1998) Characterization of the hyperpolarization-activated chloride current in dissociated rat sympathetic neurons. J Physiol 506:665–678
Cortes CJ, La Spada AR (2014) Motor neuron degeneration in spinal and bulbar muscular atrophy is a skeletal muscle-driven process: relevance to therapy development and implications for related motor neuron diseases. Rare Dis 2:e962402
Cortez MA, Li C, Whitehead SN, Dhani U, D’antonio C, Huan LJ, Bennett AL et al (2010) Disruption of ClC-2 expression is associated with progressive neurodegeneration in aging mice. Neuroscience 167:154–162
De Koninck Y (2007) Altered chloride homeostasis in neurological disorders: a new target. Curr Opin Pharmacol 7:93–99
Dell'Orco JM, Pulst SM, Shakkottai VG (2017) Potassium channel dysfunction underlies Purkinje neuron spiking abnormalities in spinocerebellar ataxia type 2. Hum Mol Genet 26:3935–3945
Enz R, Ross BJ, Cutting GR (1999) Expression of the voltage-gated chloride channel ClC-2 in rod bipolar cells of the rat retina. J Neurosci 19:9841–9847
Fahlke C (2001) Ion permeation and selectivity in ClC-type chloride channels. Am J Physiol Ren Physiol 280:F748–F757
Fernandes-Rosa FL, Daniil G, Orozco IJ, Göppner C, El Zein R, Jain V et al (2018) A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet 50:355–361
Furukawa T, Kurokawa J (2008) Non-genomic regulation of cardiac ion channels by sex hormones. Cardiovasc Hematol Dis Drug Targets 8(4):245–251
Gray PC, Scott JD, Catterall WA (1998) Regulation of ion channels by cAMP-dependent protein kinase and A-kinase anchoring proteins. Curr Opin Neurobiol 8:330–334
Guo Z, Lu T, Peng L, Cheng H, Peng F, Li J, Lu Z, Chen S, Qiu W (2019) CLCN2-related leukoencephalopathy: a case report and review of the literature. BMC Neurol 19:156
Hou X, Zhang R, Wang J, Li Y, Li F, Zhang Y, Zheng X, Shen Y, Wang Y, Zhou L (2018) CLC-2 is a positive modulator of oligodendrocyte precursor cell differentiation and myelination. Mol Med Rep 17:4515–4523
Hsu YT, Chang YG, Chern Y (2018) Insights into GABAAergic system alteration in Huntington’s disease. Open Biol 8:180165
Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C (2007) Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci 27:9866–9873
Jayakar SS, Pugh PC, Dale Z, Starr ER, Cole S, Margiotta JF (2014) PACAP induces plasticity at autonomic synapses by nAChR-dependent NOS1 activation and AKAP-mediated PKA targeting. Mol Cell Neurosci 63:1–12
Jentsch TJ, Pusch M (2018) CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol Rev 98:1493–1590
Jentsch TJ, Stein V, Weinreich F, Zdebik AA (2002) Molecular structure and physiological function of chloride channels. Physiol Rev 82:503–568
Jeub M, Herbst M, Spauschus A, Fleischer H, Klockgether T, Wuellner U, Evert BO (2006) Potassium channel dysfunction and depolarized resting membrane potential in a cell model of SCA3. Exp Neurol 201:182–192
Jeworutzki E, López-Hernández T, Capdevila-Nortes X, Sirisi S, Bengtsson L, Montolio M, Zifarelli G, Arnedo T, Müller CS, Schulte U, Nunes V, Martínez A, Jentsch TJ, Gasull X, Pusch M, Estévez R (2012) GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl− channel auxiliary subunit. Neuron 73:951–961
Jimenez-Garduño AM, Juarez-Hernandez LJ, Polanco MJ, Tosatto L, Michelatti D, Arosio D et al (2017) Altered ionic currents and amelioration by IGF-1 and PACAP in motoneuron-derived cells modelling SBMA. Biophys Chem 229:68–76
Kajita H, Omori K, Matsuda H (2000) The chloride channel ClC-2 contributes to the inwardly rectifying Cl− conductance in cultured porcine choroid plexus epithelial cells. J Physiol 523:313–324
Kennedy WR, Alter M, Sung JH (1968) Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology 18:671–680
Korkmaz O, Ay H, Ulupinar E, Tunçel N (2012) Vasoactive intestinal peptide enhances striatal plasticity and prevents dopaminergic cell loss in Parkinsonian rats. J Mol Neurosci 48:565–573
Kumar P, Kumar D, Jha SK, Jha NK, Ambasta RK (2016) Ion channels in neurological disorders. Adv Protein Chem Struct Biol 103:97–136
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352:77–79
Lieberman AP, Shakkottai VG, Albin RL (2019) Polyglutamine repeats in neurodegenerative diseases. Annu Rev Pathol 14:1–27
Mahadevan V, Woodin MA (2016) Regulation of neuronal chloride homeostasis by neuromodulators. J Physiol 594:2593–2605
Matsuyama Z, Wakamori M, Mori Y, Kawakami H, Nakamura S, Imoto K (1999) Direct alteration of the P/Q-type Ca2+ channel property by polyglutamine expansion in spinocerebellar ataxia 6. J Neurosci 19:RC14
Milioto C, Malena A, Maino E, Polanco MJ, Marchioretti C, Borgia D, Pereira MG, Blaauw B, Lieberman AP, Venturini R, Plebani M, Sambataro F, Vergani L, Pegoraro E, Sorarù G, Pennuto M (2017) Beta-agonist stimulation ameliorates the phenotype of spinal and bulbar muscular atrophy mice and patient-derived myotubes. Sci Rep 7:41046
Narayanan G, Sheila M, Chai J, Stanton LW (2017) Generation of sibling-matched induced pluripotent stem cell lines from spinal and bulbar muscular atrophy patients. Stem Cell Res 20:30–33
Niemeyer MI, Cid LP, Sepulveda FV, Blanz J, Auberson M, Jentsch TJ (2010) No evidence for a role of CLCN2 variants in idiopathic generalized epilepsy. Nat Genet 42:3
Nighot PK, Blikslager AT (2012) Chloride channel ClC-2 modulates tight junction barrier function via intracellular trafficking of occludin. Am J Phys Cell Physiol 302:C178–C187
Oki K, Halievski K, Vicente L, Xu Y, Zeolla D, Poort J, Katsuno M, Adachi H, Sobue G, Wiseman RW, Breedlove SM, Jordan CL (2015) Contractile dysfunction in muscle may underlie androgen-dependent motor dysfunction in spinal bulbar muscular atrophy. J Appl Physiol 118:941–952
Onodera K, Shimojo D, Ishihara Y, Yano M, Miya F, Banno H, Kuzumaki N, Ito T, Okada R, de Araújo Herculano B, Ohyama M, Yoshida M, Tsunoda T, Katsuno M, Doyu M, Sobue G, Okano H, Okada Y (2020) Unveiling synapse pathology in spinal bulbar muscular atrophy by genome-wide transcriptome analysis of purified motor neurons derived from disease specific iPSCs. Mol Brain 13:18
Palazzolo I, Burnett BG, Young JE, Brenne PL, La Spada AR, Fischbeck KH et al (2007) Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum Mol Genet 16:1593–1603
Palazzolo I, Stack C, Kong L, Musaro A, Adachi H, Katsuno M, Sobue G, Taylor JP, Sumner CJ, Fischbeck KH, Pennuto M (2009) Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 63:316–328
Parodi S, Pennuto M (2011) Neurotoxic effects of androgens in spinal and bulbar muscular atrophy. Front Neuroendocrinol 32:416–425
Pennuto M, Basso M (2016) In vitro and in vivo modeling of spinal and bulbar muscular atrophy. J Mol Neurosci 58:365–373
Pilzer I, Gozes I (2006) VIP provides cellular protection through a specific splice variant of the PACAP receptor: a new neuroprotection target. Peptides 27:2867–2876
Polanco MJ, Pennuto M (2018) Pituitary adenylyl Cyclase activating polypeptide (PACAP) signaling and the cell cycle machinery in neurodegenerative diseases. Curr Pharm Des 24:3878–3891
Polanco MJ, Parodi S, Piol D, Stack C, Chivet M, Contestabile A et al (2016) Adenylyl cyclase activating polypeptide reduces phosphorylation and toxicity of the polyglutamine-expanded androgen receptor in spinobulbar muscular atrophy. Sci Transl Med 8:370ra181
Poroca DR, Pelis RM, Chappe VM (2017) ClC channels and transporters: structure, physiological functions, and implications in human chloride channelopathies. Front Pharmacol 8:151
Price TJ, Cervero F, Gold MS, Hammond DL, Prescott SA (2009) Chloride regulation in the pain pathway. Brain Res Rev 60:149–170
Querin G, Bede P, Marchand-Pauvert V, Pradat PF (2018) Biomarkers of spinal and bulbar muscle atrophy (SBMA): a comprehensive review. Front Neurol 9:844
Ranganathan S, Harmison GG, Meyertholen K, Pennuto M, Burnett BG, Fischbeck KH (2009) Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum Mol Genet 18:27–42
Ratté S, Prescott SA (2011) ClC-2 channels regulate neuronal excitability, not intracellular chloride levels. J Neurosci 31:15838–15843
Reglodi D, Kiss P, Lubics A, Tamas A (2011) Review on the protective effects of PACAP in models of neurodegenerative diseases in vitro and in vivo. Curr Pharm Des 17:962–972
Rinaldi C, Malik B, Greensmith L (2016) Targeted molecular therapies for SBMA. J Mol Neurosci 58:335–342
Rinke I, Artmann J, Stein V (2010) ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J Neurosci 30:4776–4786
Roncador A, Jimenez-Garduño AM, Pasquardini L, Giusti G, Cornella N, Lunelli L, Potrich C, Bartali R, Aversa L, Verucchi R, Serra MD, Caponi S, Iannotta S, Macchi P, Musio C (2017) Primary cortical neurons on PMCS TiO2 films towards bio-hybrid memristive device: a morpho-functional study. Biophys Chem 229:115–122
Salazar-Grueso EF, Kim S, Kim H (1991) Embryonic mouse spinal cord motor neuron hybrid cells. Neuroreport 2:505–508
Scaramuzzino C, Casci I, Parodi S, Lievens PMJ, Polanco MJ, Milioto C, Chivet M, Monaghan J, Mishra A, Badders N, Aggarwal T, Grunseich C, Sambataro F, Basso M, Fackelmayer FO, Taylor JP, Pandey UB, Pennuto M (2015) Protein arginine methyltransferase 6 enhances polyglutamine-expanded androgen receptor function and toxicity in spinal and bulbar muscular atrophy. Neuron 85:88–100
Sculptoreanu A, Abramovici H, Abdullah AA, Bibikova A, Panet-Raymond V, Frankel D et al (2000) Increased T-type Ca2+ channel activity as a determinant of cellular toxicity in neuronal cell lines expressing polyglutamine-expanded human androgen receptors. Mol Cell Biochem 203:23–31
Smith RL, Clayton GH, Wilcox CL, Escudero KW, Staley KJ (1995) Differential expression of an inwardly rectifying chloride conductance in rat brain neurons: a potential mechanism for cell-specific modulation of postsynaptic inhibition. J Neurosci 15:4057–4067
Starr ER, Margiotta JF (2017) Pituitary adenylate cyclase activating polypeptide induces long-term, transcription-dependent plasticity and remodeling at autonomic synapses. Mol Cell Neurosci 85:170–182
Stölting G, Teodorescu G, Begemann B, Schubert J, Nabbout R, Toliat MR, Sander T, Nürnberg P, Lerche H, Fahlke C (2013) Regulation of ClC-2 gating by intracellular ATP. Pflugers Arch - Eur J Physiol 465:1423–1437
Stölting G, Fischer M, Fahlke C (2014) CLC channel function and dysfunction in health and disease. Front Physiol 5:378
Sulis Sato S, Artoni P, Landi S, Cozzolino O, Parra R, Pracucci E, Trovato F, Szczurkowska J, Luin S, Arosio D, Beltram F, Cancedda L, Kaila K, Ratto GM (2017) Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc Natl Acad Sci U S A 114:E8770–E8779
Thiemann A, Gründer S, Pusch M, Jentsch TJ (1992) A chloride channel widely expressed in epithelial and non-epithelial cells. Nature 356:57–60
Thompson CH, Olivetti PR, Fuller MD, Freeman CS, McMaster D, French RJ, Pohl J, Kubanek J, McCarty NA (2009) Isolation and characterization of a high affinity peptide inhibitor of ClC-2 chloride channels. J Biol Chem 284:26051–22662
Tomimatsu N, Arakawa Y (2008) Survival-promoting activity of pituitary adenylate cyclase-activating polypeptide in the presence of phosphodiesterase inhibitors on rat motoneurons in culture: cAMP–protein kinase A-mediated survival. J Neurochem 107:628–635
Tripathy D, Migazzi A, Costa F, Roncador A, Gatto P, Fusco F, Boeri L, Albani D, Juárez-Hernández JL, Musio C, Colombo L, Salmona M, Wilhelmus MMM, Drukarch B, Pennuto M, Basso M (2020) Increased transcription of transglutaminase 1 mediates neuronal death in in vitro models of neuronal stress and Aβ1-42-mediated toxicity. Neurobiol Dis 140:104849. https://doi.org/10.1016/j.nbd.2020.104849
Tsuchikawa D, Nakamachi T, Tsuchida M, Wada Y, Hori M, Farkas J, Yoshikawa A, Kagami N, Imai N, Shintani N, Hashimoto H, Atsumi T, Shioda S (2012) Neuroprotective effect of endogenous pituitary adenylate cyclase-activating polypeptide on spinal cord injury. J Mol Neurosci 48:508–517
Yu Z, Dadgar N, Albertelli M, Gruis K, Jordan C, Robins DM, Lieberman AP (2006) Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Invest 116:2663–2672
Zawilska JB, Dejda A, Niewiadomski P, Gozes I, Nowak JZ (2005) Receptors for VIP and PACAP in guinea pig cerebral cortex: effects on cyclic AMP synthesis and characterization by 125I-VIP binding. J Mol Neurosci 25:215–224
Zúñiga L, Niemeyer MI, Varela D, Catalán M, Cid LP, Sepúlveda FV (2004) The voltage-dependent ClC-2 chloride channel has a dual gating mechanism. J Physiol 555:671–682
Acknowledgments
We are grateful to Prof. Walter Stühmer (MPI for Experimental Medicine, Göttingen, D) for the illuminating suggestions about the Cl− conductances analysis, and to Prof. Alessio Accardi (Weill Medical College, New York, USA) for insightful advices about the functional characterization of the ClC-2 channel. We thank Dr. Michael Whalen (IBF-CNR, Trento, I) for the accurate revision of the English and comments on the final version of the manuscript.
Funding
This work has been performed in the framework of Bando Progetti Strategici di Ateneo–University of Trento (to M.P.); Telethon-Italy (GGP19128 to M.P.); Muscular Dystrophy Association (479363 to M.P.); CNCCS Scarl Pomezia (to M.P.), PRIN-MIUR Italy (to M.P.), NIH-R21 (1R21NS111768-01 to M.P.), and Association Française contre les Myopathies (22221 to M.B. and M.P.). LT was supported by Dulbecco Telethon Institute International Post-Doctoral Programme, Fondazione Umberto Veronesi and by Akira Arimura Foundation 2016 PACAP Research Award. AMJG and CM acknowledge the partial support from the Autonomous Province of Trento (PAT), Italy, “Grandi Progetti 2012-MaDEleNA Project”. LT and VAMR are currently granted by the Fondazione CARITRO, Trento, Italy, whose support CM and DA too gratefully recognize.
Author information
Authors and Affiliations
Contributions
Conceptualization: CM, AMJG, VAMR. Data Curation: AMJG, VAMR, CM. Formal Analysis: VAMR, AMJG, CM, DM, DA. Funding Acquisition: CM, MP. Investigation: VAMR, AMJG, DM, MM, LT. Methodology: VAMR, AMJG, CM, DA. Project Administration: CM. Resources: CM, MB, MP, DA. Supervision: CM. Writing—Original Draft Preparation: CM, VAMR, AMJG. Writing—Review & Editing. All authors read, revised, and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Martínez-Rojas, V.A., Jiménez-Garduño, A.M., Michelatti, D. et al. ClC-2-like Chloride Current Alterations in a Cell Model of Spinal and Bulbar Muscular Atrophy, a Polyglutamine Disease. J Mol Neurosci 71, 662–674 (2021). https://doi.org/10.1007/s12031-020-01687-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-020-01687-5