
Abstract
PTEN-induced kinase 1 (PINK1) mutations can cause early-onset Parkinson’s disease and patients are likely to develop cognitive decline, depression, and dementia. Several neurophysiological studies have demonstrated PINK1 deficiency impairs striatal and hippocampal presynaptic plasticity. Dendritic spine postsynaptic abnormalities are common in neurological diseases; however, whether PINK1 silencing modifies dendritic spine dynamics of hippocampal neurons is unclear. To address this question, confocal images of mouse cultured hippocampal neurons transfected with plasmids to silence PINK1 were analyzed. These studies revealed that PINK1 silencing increased density of thin spines and reduced head size of stubby spines. Immunoblotting analysis uncovered that PINK1 silencing decreased expression of postsynaptic density proteins (PSD95 and Shank) and glutamate receptors (NR2B and mGluR5). We also found PINK1 silencing regulated dendritic spine morphology by actin regulatory proteins (RhoGAP29 and ROCK2) and regulated neuronal survival by decreased Akt activation. These results suggest PINK1 may regulate postsynaptic plasticity in hippocampal neurons generating presymptomatic alterations in dendritic spines that eventually could lead to the neurodegeneration and cognitive decline often seen in Parkinson’s disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease characterized by motor symptoms such as resting tremor, rigidity, and bradykinesia and psychiatric symptoms at late stages of the disease such as mild cognitive decline, depression, and dementia (Meissner et al. 2011; Reetz et al. 2008). PD is largely idiopathic, but familial PD types resembling sporadic PD are caused by particular gene mutations (Valente et al. 2004). Among these, PTEN-induced kinase 1 (PINK1) mutations, which are associated with the loss of a protection mechanism, are the second most frequent cause of autosomal recessive early-onset PD (Davie 2008; Gautier et al. 2008; Valente et al. 2004). Recently, PINK1 deficiency has been studied as a model of PD that affects synaptic function (Feligioni et al. 2016; Madeo et al. 2014).
On one hand, in PINK1 knockout mice, nigrostriatal dopaminergic neurons decrease evoked dopamine release that impairs corticostriatal synaptic plasticity of striatal medium spiny neurons (Kitada et al. 2007). These findings were recapitulated in heterozygous PINK1 knockout mice, which, despite normal motor behavior, corroborate that PINK1 regulates dopamine release and striatal synaptic plasticity in the nigrostriatal circuit (Madeo et al. 2014). These synaptic abnormalities can have begun as subtle changes in excitability of substantia nigra dopaminergic neurons mediated by glutamatergic synapses (Pearlstein et al. 2016).
On other hand, in 6-month-old PINK1 knockout mice, hippocampal neurons increase spontaneous synaptic frequency and enhance glutamate release, which supports that PINK1 contributes to maintaining not only dopaminergic physiology and corticostriatal synaptic plasticity but also hippocampal synapses (Feligioni et al. 2016). In a preceding study, we showed that in mouse hippocampal neurons, when PINK1 expression level is increased using GW3965, a liver X receptor agonist, dendritic spine number increases (Báez-Becerra et al. 2018). These findings suggest that PINK1 plays a role in hippocampal synapses contributing to maintain not only presynaptic plasticity of axonal boutons but also postsynaptic plasticity of dendritic spines (Báez-Becerra et al. 2018; Feligioni et al. 2016). However, PINK1-dependent dendritic spine dynamics of hippocampal neurons have not been extensively determined.
Therefore, in this study, we asked whether PINK1 silencing modifies dendritic spine dynamics of hippocampal neurons. To answer this question, we assessed the number and morphology of dendritic spines in cultured mouse hippocampal neurons that were PINK1 silenced. We studied neuron images of fluorescence experiments. To search possible postsynaptic mechanisms underlying dendritic spine modifications, we evaluated expression levels of postsynaptic proteins.
Materials and Methods
Experimental Design
To determine whether PINK1 silencing modifies dendritic spine dynamics of hippocampal neurons, we studied mouse hippocampal neurons cultured by 13 days in vitro (DIV) of three different litters. This culture time allowed hippocampal neurons to differentiate and form well-distinguished dendritic spines. To reduce the expression of PINK1, neurons of 10 DIV were transfected with a lentiviral plasmid PINK1 shRNA, which encodes a RNA interference (RNAi). As control, neurons were transfected with a non-specific plasmid control shRNA, which encodes a random recognition sequence. Neurons were also transfected with a GFP plasmid vector, which encodes green fluorescent protein linked to an actin-binding recognition sequence. Next, for assessment of dendritic spines, we fixed and mounted neurons of 13 DIV on coverslips, then acquired confocal images, and then calculated number and type of dendritic spines. During each experiment, we extracted protein both for verification of PINK1 silencing and measurement of expression level of postsynaptic proteins by western blot. We also measured lactate dehydrogenase (LDH) as a check of neuronal survival.
Hippocampal Neuronal Culture
Primary culture of mouse hippocampal neurons has been described previously (Báez-Becerra et al. 2018; Beaudoin et al. 2012). Briefly, postnatal P0 hippocampus from C57BL/6 mice were isolated and then treated with trypsin (2.5% wt/vol, Lonza, CC-5012) in Hank’s Balanced Salt Solution (HBSS, Lonza, 10-547F) with no calcium, nor magnesium for 30 min at 37 °C. Hippocampal dissociated cells were cultured on poly-l-lysine-coated (1 mg/mL, Sigma, P1274) coverslips in plating medium (Neurobasal medium (Invitrogen, 21103049) supplemented with B27 (Invitrogen, 17504044), 2 mM GlutaMAX (Invitrogen, 25030081), and 1% penicillin/streptomycin (Invitrogen, 15140122) and containing 10% BFS (Gibco, 16000-044)). We plated 90,000 cells on 18-mm coverslips (Ilmglas) in 6-well plates. Three hours later, maintaining medium (Neurobasal medium supplemented) was added. Cytosine arabinoside (1 μM, 1-beta-d-arabinofuranosyl-cytosine, Calbiochem) was added to the culture 2 days after plating for 24 h to inhibit glial cell proliferation. Neurons were cultured by 13 DIV adding maintaining medium two times per week. All experiments were conducted in compliance with the National Institutes of Health Guidelines for Care and Use of Experimental Animals and approved by the Institutional Committee of Animal Care and Use at Universidad Nacional de Colombia-Bogotá.
Plasmids
PINK1 shRNA and control shRNA plasmids were kindly provided by Dr. Mark Cookson (National Institutes of Health, USA). This kind of plasmids encodes a RNAi. Primary hippocampal neurons were transfected with the following constructs: PINK1 shRNA (pLenti6), 5′-GCTGGAGGAGTATCTGATAGG-3′; control shRNA (pLenti6), 5′-CCTAGACGCGATAGTATGGAC-3′; and GFP (pEGFP-N1).
Transfection
Hippocampal neurons cultured by 10 DIV were transfected with 3 mg of either shPINK1 plasmid or shCTL plasmid and 2 mg of GFP plasmid per well using Lipofectamine 2000 (Invitrogen, 11668030). Briefly, maintaining medium was recycled and 1 mL of transfection mix was added to each well incubating at 37 °C for 3 h. Transfection mix was then removed and recycled medium was added. Immunofluorescence was performed 3 days after transfection.
Lactate Dehydrogenase Release Assay
To assess cell death percentage, we measure LDH release level in hippocampal neurons cultured by 13 DIV using CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, Madison, USA) in accordance with the manufacturer’s instructions.
Immunofluorescence
Neurons were fixed with 4% paraformaldehyde and 4% sucrose in HBSS for 15 min at room temperature. After 3 washings with phosphate-buffered saline (PBS), 3 washings with high-saline solution (20 mM PB and 500 mM NaCl), and 3 low-saline solution (10 mM PB and 150 mM NaCl) for 5 min each, coverslips were mounted with mounting solution (7 parts of glycerol and 3 parts PBS 10X and 10 μg/mL Hoechst). Representative images of control and silenced hippocampal neurons and high magnifications of their dendrites can be found in Figs. 2a, c.
Image Acquisition and Dendritic Spine Analysis
Confocal images were collected using a Nikon C1 plus Eclipse TI confocal laser scanning microscope with a × 60 objective (PLAN APO VC × 60 OIL DIC N2) at 0.2 μm/step through 18 z-dimension steps averaged 3 times each. Image resolution was 1024 × 1024 pixels with a laser exposure time of 1.68 μs. Laser powers were as follows: laser 408, 6.2%; laser 488, 16.3%; and laser 568, 17.3%; photomultiplicating gains were as follows: 515/30, 6; 590/50, 8.5; and 650 LP, 6; and pinhole was 30 μm. Image analyses were performed as previously described (Báez-Becerra et al. 2018). Briefly, dendritic spines were manually counted and classified as the number of spine total and spine types along 80–100 μm length of dendritic processes using NeuronStudio software (Rodriguez et al. 2008) and presented as mean + SEM. Spine-type classification was according to Peters and Kaiserman-Abramof (1970): filopodia, thin, stubby, and mushroom. Each experiment has been done two times and a total of 18–21 neurons were randomly selected. Two to five dendritic segments were selected for quantification. All analyses were done in a manner blind to the conditions. Representative high magnification images of dendrites from control and silenced groups analyzed with NeuronStudio software can be found in Fig. 2c.
Western Blot
Cells were lysed at 4 °C for 10 min using RIPA lysis buffer (Sigma-Aldrich, R0278) containing 1% protease inhibitor cocktail (Roche, 04693159002) and 1% phosphatase inhibitor cocktail (Roche, 04906837001). Protein concentration was determined using BCA protein assay kit (Fisher Scientific, 23225). Fifty micrograms of protein samples was run in a polyacrylamide gel at 100 V. After electrophoresis, proteins were transferred to a hydrophobic PVDF membrane (GE Healthcare Life Sciences Hybond ECL) and incubated in blocking buffer (5% powdered skim milk in TTBS) for 90 min at room temperature. PVDF membrane was then incubated overnight at 4 °C with monoclonal primary antibodies: PINK1 (Abcam, ab75487), PSD (Abcam, ab2723), Shank (Santa Cruz, sc-393963), NR2B (Biosciences, 610416), mGluR5 (Abcam, ab76316), RhoAGAP29 (Abcam, ab85853), ROCK2 (Abcam, ab71598), Akt (Cell Signaling, C67E7), and pAkt (Cell Signaling, D25E6), everyone at 1:1000 dilution factor in blocking buffer. The next day, membranes were washed 3 times in TTBS for 5 m each. This was followed by incubation with peroxidase-conjugated secondary anti-mouse (Cell Signaling, 7076S) or anti-rabbit (Cell Signaling, 7074S) at 1:2000 dilution factor in blocking buffer for 1 h, followed by 5 washings with TTBS. Secondary antibodies were detected with ECL system (Invitrogen, WP20005) using imaging documentation system (ChemiDocMP System Bio-Rad Laboratories). Quantification was performed by densitometry using ImageLab software (Bio-Rad).
Data Analysis
Data statistical analysis and graphical representation were done using Prism6 software (GraphPad, 92037, La Jolla, California, USA) and R software (Foundation for Statistical Computation, Vienna, Austria). Data obtained correspond to three independent experiments. We analyzed the data from each experiment by a two-way ANOVA followed by Tukey honest significant difference (TukeyHSD) test for multiple experimental group comparisons. And Student’s t test was used for comparisons between two experimental groups. We considered statistical significance present when the P value was < 0.05 and was represented as follows: *P < 0.05, **P < 0.01, and ***P < 0.001. All data are presented as means + standard error of the mean (SEM).
Results
Neuronal Survival Is Normal in PINK1-Silenced Hippocampal Neurons
In hippocampal neurons transfected using PINK1 shRNA (shPINK1), mean expression level of PINK1 protein was lower than in untransfected controls (CTL = 0.96 ± 0.02 vs. shPINK1 = 0.48 ± 0.06) (Fig. 1a). SH-SY5Y and HEK cells were used as positive controls of PINK1 expression. Untransfected and control shRNA (shCTL)-transfected hippocampal neurons had no difference in PINK1 expression level, while shPINK1-transfected hippocampal neurons reduced about 50% PINK1 protein expression level (NoTrans = 0.96 ± 0.05 vs. shCTL = 0.91 ± 0.09 vs. shPINK1 = 0.53 ± 0.37) (Fig. 1b). PINK1 silencing had no effect on cell death as measured by LDH release (LysisCTL = 97.3 ± 6.45 vs. NoTrans = 14.8 ± 1.24 vs.shCTL = 21.1 ± 2.11 vs. shPINK1 = 17.5 ± 3.76) (Fig. 1c). Phase contrast images of hippocampal neurons transfected with shCTL or shPINK1 display a healthy morphology (Fig. 1d).

PINK1 silencing in primary cultures of mouse hippocampal neurons. a shPINK1 plasmid effect on PINK1 protein expression in SH-SY5Y and HEK cells and mouse hippocampal neurons. b Vehicle and shCTL and shPINK1 plasmid effects on PINK1 protein expression in mouse hippocampal neurons. c LDH test. d Phase contrast microscopy image of 13 DIV mouse hippocampal neurons. Bar scale = 200 μm. Data are from three different experiments and represent mean + SEM. **P < 0.01, ***P < 0.001, ns P > 0.05 vs. control for TukeyHSD
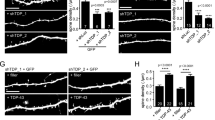
PINK1 Silencing Promotes Thin Spine Increment
This PINK1 silencing in hippocampal neurons increases the number of dendritic spines. Mean spine density was 40% higher than in shCTL hippocampal neurons (shCTL = 3.28 ± 0.27 vs. shPINK1 = 4.62 ± 0.21 spines/10 μm) (Fig. 2b). In shPINK1 hippocampal neurons, thin spine density mean was 90% higher than in shCTL hippocampal neurons (shCTL = 0.88 ± 0.11 vs. shPINK1 = 1.70 ± 0.13 thin spines/10 μm). The rest of spine types had no change, even though there was a tendency to increase (filopodia (shCTL = 0.14 ± 0.04 vs. shPINK1 = 0.21 ± 0.04); stubby (shCTL = 1.29 ± 0.15 vs. shPINK1 = 1.75 ± 0.11); mushroom (shCTL = 0.96 ± 0.09 vs. shPINK1 = 1.00 ± 0.07 spines/10 μm)) (Fig. 2d).

PINK1 silencing modifications on dendritic spine density and morphology. a, c Hippocampal neurons were transfected with the lentiviral plasmid shCTL or shPINK1, along with the reporter plasmid GFP. a Neuron images of superimposed z-dimension steps. Bar scale = 50 μm. b Spine average number + SEM per 10 μm of dendrite. **P < 0.01 for Student’s t test. c Image magnifications of dendrite segments analyzed using NeuronStudio software. Spine-type classification: filopodium (gray), thin (yellow), stubby (magenta), and mushroom (orange). Bar scale = 10 μm. d Mean spine number + SEM per 10 μm of dendrite by spine type. Data are from two different experiments. **P < 0.01 vs. control for TukeyHSD
PINK1 Silencing Shrinks Stubby Spine Head
PINK1 silencing in hippocampal neurons reduces head size of stubby spines as well. Mean head size of spines had no difference between shPINK1 hippocampal neurons and shCTL hippocampal neurons (shCTL = 0.37 ± 0.01 vs. shPINK1 = 0.35 ± 0.01 μm) (Fig. 3a). Figure 3b shows shCTL and shPINK1 neuron distributions of spine head diameter. shPINK1 neuron distribution had more spines with small heads than shCTL neuron distribution. In contrast, shCTL neuron distribution had more spines with large heads than shPINK1 neuron distribution. Mean stubby spine head size was 13% smaller in shPINK1 hippocampal neurons than in shCTL hippocampal neurons, while the rest of spine head sizes had no change (filopodia (shCTL = 0.32 ± 0.02 vs. shPINK1 = 0.36 ± 0.02); thin (shCTL = 0.28 ± 0.01 vs. shPINK1 = 0.29 ± 0.01); stubby (shCTL = 0.39 ± 0.01 vs. shPINK1 = 0.34 ± 0.01); mushroom (shCTL = 0.43 ± 0.01 vs. shPINK1 = 0.44 ± 0.01 spines/10 μm)) (Fig. 3c).

The silencing of PINK1 decreases the size or head of the spines of the hippocampal neurons. a Mean spine head diameter + SEM. P > 0.05 vs. control for Student’s t test. b Control and silenced neuron density distributions of spine head diameter. c Mean spine head diameter + SEM by spine type. **P < 0.01 vs. control for TukeyHSD
PINK1 Silencing Decreases Expression Level of Postsynaptic Density Proteins and Glutamate Receptors
These changes of dendritic spines in shPINK1 hippocampal neurons are accompanied by biochemical changes of postsynaptic proteins and receptors. In shPINK1 hippocampal neurons, PSD95 and Shank mean expression levels were each 30% lower than in shCTL hippocampal neurons (PSD95 (shCTL = 0.89 ± 0.11 vs. shPINK1 = 0.59 ± 0.08); Shank (shCTL = 0.957 ± 0.043 vs. shPINK1 = 0.662 ± 0.05)) (Fig. 4a). Additionally, in shPINK1 hippocampal neurons, NR2B mean expression level was 25% lower than in shCTL hippocampal neurons (shCTL = 0.96 ± 0.04 vs. shPINK1 = 0.71 ± 0.08) (Fig. 4b). Similarly, in shPINK1 hippocampal neurons, mGluR5 mean expression level was 27% lower than in shCTL hippocampal neurons (shCTL = 0.97 ± 0.02).

PINK1 silencing decreases the expression of PSD95, Shank, NR2B, and mGluR5. Representative western blots of PINK1, PSD95, Shank, NR2B, and mGluR5 mean expression levels. Bar graph summarizes densitometry data. Data represent mean + SEM. n = 3. *P < 0.05 vs. control for Student’s t test
PINK1 Silencing Regulates Expression Level of Actin Polymerization Proteins and Akt Phosphorylation
These biochemical changes of postsynaptic proteins and receptors affect actin polymerization and increase the vulnerability to neuronal death. In shPINK1 hippocampal neurons, RhoGAP29 mean expression level was 24% higher than in shCTL hippocampal neurons (shCTL = 0.93 ± 0.05 vs. shPINK1 = 1.17 ± 0.09) (Fig. 5a). In shPINK1 hippocampal neurons, ROCK2 mean expression level was 33% higher than in shCTL hippocampal neurons (shCTL = 0.93 ± 0.05 vs. shPINK1 = 1.17 ± 0.09) (Fig. 5a). Finally, Akt mean expression level had no change in shPINK1 and shCTL hippocampal neurons (shCTL = 1.01 ± 0.09 vs. shPINK1 = 1.04 ± 0.07) (Fig. 5b). However, in shPINK1 hippocampal neurons, Akt mean phosphorylation level was 13% lower than in shCTL hippocampal neurons (shCTL = 0.97 ± 0.03 vs. shPINK1 = 0.84 ± 0.04) (Fig. 5b).

PINK1 silencing regulates the expression of RhoGAP29 and ROCK2 and decreases phosphorylated Akt. Representative western blots of PINK1, RhoGAP29, ROCK2, Akt, and pAkt mean expression levels. Bar graph summarizes densitometry data. Data represent mean + SEM. n = 3. *P < 0.05 vs. control for Student’s t test
Discussion
In the present study, we demonstrated that PINK1 silencing modifies dendritic spine morphology and causes several biochemical changes of hippocampal neurons. PINK1 silencing increased dendritic spine density (by 40%) generating a greater number of thin spines, and reduced the head size of stubby spines. These changes were associated with decreased expression levels of postsynaptic density proteins such as PDS95 and Shank and glutamate receptors NR2B and mGluR5, increased expression levels of the cytoskeleton regulatory proteins RhoGAP29 and ROCK2, and decreased Akt phosphorylation (Fig. 6).

Model of PINK1 silencing modifications on dendritic spines. Top, PINK1 silencing increases thin dendritic spine density. Down, black arrows represent postsynaptic signaling pathways. Red arrows and T’s represent PINK1 silencing activation and inhibition, respectively. PINK1 silencing weakens synaptic strength reducing postsynaptic density proteins and glutamate receptors and inhibiting actin polymerization and Akt neuroprotection
These findings are in agreement with, as far as we know, the only study that has shown that PINK1 silencing increases dendritic spine density of hippocampal neurons (Yu et al. 2011) and particularly of thin dendritic spines (Fig. 2d). Thin spines have a thin, long neck and a small bulbous head, which suggests a small synapse has been established (Peters and Kaiserman-Abramof 1970). This finding suggests that PINK1 silencing neurons have increased their synapses generating new immature thin spines, which can increase neuronal vulnerability to cell death. Eventually, after this new thin spines mature, they may be associated with a higher susceptibility to generate excitotoxicity (Yu et al. 2011).
PINK1 silencing also reduced the head of stubby spines (Fig. 3). Stubby spines are devoid of a neck and have a big head (Peters and Kaiserman-Abramof 1970). This finding suggests that PINK1 silencing neurons have decreased synapse size of stubby spines. We believe this decreased synapse size might be the product of a synaptic scaling triggered by the increased excitatory input produced by the new thin spines (Turrigiano 1999). Therefore, this synaptic scaling could be considered a compensatory response against increased excitatory input and might therefore contribute to separate PINK1-depended synaptic abnormalities into a stable phenotype and a presymptomatic stage (Madeo et al. 2014).
The reason PINK1 silencing could contribute to a stable phenotype might be that despite the synaptic changes, it had no effect on neuronal survival (Fig. 1c). This finding agrees with some studies where PINK1 deficiency does not develop neurodegeneration (Valente et al. 2004; Zhou et al. 2011). However, when PINK1-deficient neurons are exposed to stressful stimuli, cell death increases, highlighting the neuroprotective mechanism of PINK1 (Contreras-Zárate et al. 2015; Mills et al. 2008). The neuroprotective mechanism could be mediated by Akt activation (Brunet et al. 2001), since several authors have shown that Akt is downstream of PINK1 (Contreras-Zárate et al. 2015; Shan et al. 2009), which is one of the main survival signaling pathways that could therefore confer susceptibility to neurodegeneration and cell death (Contreras-Zárate et al. 2015).
It is an interesting fact that although PINK1 silencing increased the number of dendritic spines, there are changes in excitatory postsynaptic machinery: decreased in postsynaptic density proteins PSD95 and Shank. Dagda et al. (2014) showed that in SH-SY5Y cells, PINK1 overexpression promotes the expression of MAP2B and PSD95 proteins, so it is plausible that PINK1 silencing has the opposite effects. Likewise, Shank deficiency has mainly been associated with autistic spectrum disorders and more recently with Alzheimer’s (Guilmatre et al. 2014), but it has not been associated with PINK1. We believe that the reduction of PSD95 and Shank probably reflects an overall reduction of synaptic strength (Venkatesh 1998), despite the generation of new thin spines.
In addition, there was also decrease in glutamate receptors NR2B and mGluR5 upon PINK1 silencing (Fig. 4b). Activation of synaptic NR2B triggers signaling pathways of development and synaptic plasticity (Chang et al. 2010). Activation of extra-synaptic NR2B due to oxygen and glucose deprivation reduces PINK1 expression and inhibits Akt phosphorylation contributing to neuronal death, while PINK1 overexpression neutralizes this inhibition, suggesting that Akt may be downstream of PINK1 (Shan et al. 2009). It has been reported that mGluR5 antagonists activate the signaling pathway of MAPK/ERK that modulates cell viability and stress responses in PD models (Ambrosi et al. 2014). In addition, NR2B interacts with mGluR through the PSD95/GKAP/Shank/Homer complex increasing cytosolic Ca2+ through NR2B and IP3R generating spine maturation associated with enhanced synaptic plasticity (Tu et al. 1999). Thus, decreased expression of NR2B and mGluR5 might restrict the excitatory input supporting that PINK1 deficiency can reduce synaptic strength.
How might PINK1 silencing modify dendritic spines? On the one hand, Yu et al. (2011) suggest that the increased dendritic spines may be a result of the fact that PINK1 deficiency maintains a mitochondrial network fused. This means that dendritic arborizations have more mitochondrial content that may result in more energy availability, but at the same time, PINK1 deficiency impairs mitochondrial quality control by means of an impaired mitophagy or impaired selective mitochondrial degradation (Yu et al. 2011). Thus, though neurons with a different phenotype remain healthy, when they are depolarized, cellular death increases highlighting that uncontrolled synaptic modifications can result in adverse excitability levels and increased vulnerability to cellular death.
On the other hand, Feligioni et al. (2016) have proposed that the increased dendritic spines may result from the specific enhancement in the presynaptic excitatory plasticity caused by PINK1 deficiency. PINK1 deficiency increases both spontaneous synaptic activation and glutamate release in hippocampal neurons of 6-month-old mice, which may stimulate increased synaptic density and dendritic spines (Feligioni et al. 2016). Although it is not yet clear whether the PINK1 effects on presynaptic boutons are directly responsible for the dendritic spine modifications, these increased dendritic spines eventually might confer neuronal vulnerability to excitotoxicity by increased excitatory synapses and enhanced synaptic strength.
In this study, PINK1 silencing increased the expression of actin polymerization regulating proteins (Fig. 5a), which partially agrees with spine changes due to RhoA inhibition. On one hand, PINK1 silencing increased the expression of RhoGAP29. RhoGAP29 inhibits RhoA and decreases ROCK2 activation, a major downstream effecter of RhoA (Truebestein et al. 2015). We believe that the increased expression of RhoGAP29 may inhibit RhoA creating new spines with long necks and small heads such as thin spines. This result suggests that PINK1 might be regulating actin polymerization in dendritic spines. On the other hand, PINK1 silencing increased the expression of ROCK2. This result was unexpected for previous studies that reported that RhoA inhibition decreases ROCK2 activation (Truebestein et al. 2015). We expected that as RhoGAP29 increased, its downstream effector ROCKII would decrease. Thus, either RhoGAP29 might not be specific of RhoA, so RhoGAP29 increase would not associate with a RhoA decrease, or generation of new spines might depend on Rac, which regulates spine maturation (Stankiewicz and Linseman 2014). Other authors have reported that RhoA is not needed for ROCK2 to be activated because ROCK2 is constitutively active, and if ROCK2 phosphorylates its substrates, it would depend ultimately of its subcellular location (Truebestein et al. 2015). Consequently, the increased expression of ROCK2 could be caused by a self-regulatory mechanism independent of RhoA.
However, pathological dendritic spine density and morphology can be triggered by different proteins associated with several brain diseases (Forrest et al. 2018). For instance, ADNP mutations could lead to decreased dendritic spines and synaptic abnormalities impairing spine maturation through interaction with microtubule system that model autism spectrum disorder (Hacohen-Kleiman et al. 2018). Another kind of protein associated with a pathological dendritic spine density is neurotrophins such as BDNF. It is believed that some forms of depression are due to unbalanced production and release of BDNF, which decreases dendritic spine density along with postsynaptic density proteins (Qiao et al. 2017). Thus, we cannot discard that other proteins may be involved in the dendritic spine modifications we observed here.
PINK1-dependent synaptic modifications are a common feature of both Parkinson’s and Alzheimer’s diseases (Du et al. 2017; Kitada et al. 2007; Madeo et al. 2014). On one hand, PINK1 deficiency impairs striatal synaptic plasticity decreasing stimulus-evoked dopamine release from nigral dopaminergic neurons and impairing synaptic plasticity of striatal medium spiny neurons (Kitada et al. 2007, Madeo et al. 2014). On the other hand, PINK1 is downregulated in Alzheimer’s patient brains (George et al. 2010), and a research has recently showed PINK1 deficiency exacerbates synaptic amyloid-β accumulation which increases impaired synaptic plasticity in hippocampal neurons of mAPP transgenic mice (Du et al. 2017). Moreover, hippocampal neurons exposed to amyloid-β or transfected with human mAPP decrease PINK1 expression (Reddy et al. 2018), as well as neurons of 12-month-old APP mice also reduce PINK1 expression (Manczak et al. 2018). In these Alzheimer’s models, both dendritic spines and postsynaptic proteins decreased due to the neurotoxic effect of synaptic amyloid-β accumulation, and as the PINK1 expression is reduced, neurons are more vulnerable to mitochondrial damage since PINK1-dependent mitophagy machinery is reduced as well (Manczak et al. 2018; Reddy et al. 2018).
A plausible picture can be developed based on the work of Fiala et al. (2002), who have suggested that spine pathology in neurons may result from a compensatory response to an enhancement of excitatory input. One might picture that an increase in the number of releasable synaptic vesicles or vesicle-docking sites may generate new spines increasing the excitatory input, which has already been suggested (Feligioni et al. 2016). This enhanced excitatory input due to the new spines might contribute to trigger a compensatory response that would help suppress the overactivation (Turrigiano 1999). In this study, we believe that the compensatory response might be reflected in both the adjusted morphology of stubby spines and the decreased excitatory postsynaptic machinery. However, it is not yet clear whether PINK1 effects on axonal terminals are directly responsible for modifications of dendritic spines.
In conclusion, this study indicates that PINK1 silencing modifies dendritic spine dynamics of hippocampal mouse neurons. Therefore, PINK1 regulation of postsynaptic morphology reported here provides a new pharmacological alternative that can be used to strengthen overall synaptic function to achieve adequate neuroprotection.
References
Ambrosi G, Cerri S, Blandini F (2014) A further update on the role of excitotoxicity in the pathogenesis of Parkinson’s disease. J Neural Transm 121(8):849–859. https://doi.org/10.1007/s00702-013-1149-z
Báez-Becerra C, Filipello F, Sandoval-Hernández A, Arboleda H, Arboleda G (2018) Liver X receptor agonist GW3965 regulates synaptic function upon amyloid beta exposure in hippocampal neurons. Neurotox Res 33(3):569–579. https://doi.org/10.1007/s12640-017-9845-3
Beaudoin GMJ, Lee S-H, Singh D, Yuan Y, Ng Y-G, Reichardt LF, Arikkath J (2012) Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc 7(9):1741–1754. https://doi.org/10.1038/nprot.2012.099
Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11(3):297–305. https://doi.org/10.1016/S0959-4388(00)00211-7
Chang N, Li L, Hu R, Shan Y, Liu B, Li L, Wang H, Feng H, Wang D, Cheung C, Liao M, Wan Q, (2010) Differential regulation of NMDA receptor function by DJ-1 and PINK1. Aging Cell 9 (5):837–850. https://doi.org/10.1111/j.1474-9726.2010.00615.x
Contreras-Zárate MJ, Niño A, Rojas L, Arboleda H, Arboleda G (2015) Silencing of PINK1 inhibits insulin-like growth factor-1-mediated receptor activation and neuronal survival. J Mol Neurosci 56(1):188–197. https://doi.org/10.1007/s12031-014-0479-0
Dagda RK, Pien I, Wang R, Zhu J, Wang KZQ, Callio J, Banerjee TD, Dagda RY, Chu CT (2014) Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J Neurochem 128(6):864–877. https://doi.org/10.1111/jnc.12494
Davie CA (2008) A review of Parkinson’s disease. Br Med Bull 86(1):109–127. https://doi.org/10.1093/bmb/ldn013
Du F, Yu Q, Yan S, Hu G, Lue LF, Walker DG et al (2017) PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 140(12):3233–3251. https://doi.org/10.1093/brain/awx258
Feligioni M, Mango D, Piccinin S, Imbriani P, Iannuzzi F, Caruso A, de Angelis F, Blandini F, Mercuri NB, Pisani A, Nisticò R (2016) Subtle alterations of excitatory transmission are linked to presynaptic changes in the hippocampus of PINK1-deficient mice. Synapse 70(6):223–230. https://doi.org/10.1002/syn.21894
Fiala JC, Spacek J, Harris KM (2002) Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Rev 39:29–54. https://doi.org/10.1016/S0165-0173(02)00158-3
Forrest MP, Parnell E, Penzes P (2018) Dendritic structural plasticity and neuropsychiatric disease. Nat Rev Neurosci 19:215–234. https://doi.org/10.1038/nrn.2018.16
Gautier CA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci 105(32):11364–11369. https://doi.org/10.1073/pnas.0802076105
George AJ, Gordon L, Beissbarth T, Koukoulas I, Holsinger RMD, Perreau V, Cappai R, Tan SS, Masters CL, Scott HS, Li QX (2010) A serial analysis of gene expression profile of the Alzheimer’s disease Tg2576 mouse model. Neurotox Res 17(4):360–379. https://doi.org/10.1007/s12640-009-9112-3
Guilmatre A, Huguet G, Delorme R, Bourgeron T (2014) The emerging role of SHANK genes in neuropsychiatric disorders. Developmental Neurobiology 74(2):113–122. https://doi.org/10.1002/dneu.22128
Hacohen-Kleiman G, Sragovich S, Karmon G, Gao A, Grigg I, Pasmanik-Chor M, Le A, Korenková V, McKinney R, Gozes I (2018) Activity-dependent neuroprotective protein deficiency models synaptic and developmental phenotypes of autism-like syndrome. J Clin Invest 128(11):4956–4969. https://doi.org/10.1172/JCI98199
Kitada T, Pisani A, Porter D, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos E, Shen J (2007) Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S 104:11441–11446 https://www.pnas.org/content/104/27/11441
Madeo G, Schirinzi T, Martella G, Latagliata EC, Puglisi F, Shen J, Valente EM, Federici M, Mercuri NB, Puglisi-Allegra S, Bonsi P, Pisani A (2014) PINK1 heterozygous mutations induce subtle alterations in dopamine-dependent synaptic plasticity. Mov Disord 29(1):41–53. https://doi.org/10.1002/mds.25724
Manczak M, Kandimalla R, Yin X, Reddy PH (2018) Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Human Molecular Genetics 27(8, 15):1332–1342. https://doi.org/10.1093/hmg/ddy042
Meissner WG, Frasier M, Gasser T, Goetz CG, Lozano A, Piccini P, Obeso JA, Rascol O, Schapira A, Voon V, Weiner DM, Tison F, Bezard E (2011) Priorities in Parkinson’s disease research. Nat Rev Drug Discov 10(5):377–393. https://doi.org/10.1038/nrd3430
Mills RD, Sim CH, Mok SS, Mulhern TD, Culvenor JG, Cheng HC (2008, April) Biochemical aspects of the neuroprotective mechanism of PTEN-induced kinase-1 (PINK1). J Neurochem 105:18–33. https://doi.org/10.1111/j.1471-4159.2008.05249.x
Pearlstein E, Michel FJ, Save L, Ferrari DC, Hammond C (2016) Abnormal development of glutamatergic synapses afferent to dopaminergic neurons of the Pink1−/− mouse model of Parkinson’s disease. Front Cell Neurosci 10(168). https://doi.org/10.3389/fncel.2016.00168
Peters A, Kaiserman-Abramof IR (1970) The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat 127:321–355. https://doi.org/10.1002/aja.1001270402
Qiao H, An S, Xu C, Ma X (2017) Role of proBDNF and BDNF in dendritic spine plasticity and depressive-like behaviors induced by an animal model of depression. Brain Res 1663:29–37. https://doi.org/10.1016/j.brainres.2017.02.020
Reddy PH, Yin XL, Manczak M, Kumar S, Pradeepkiran JA, Vijayan M, Reddy AP (2018) Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum Mol Genet 27(14):2502–2516. https://doi.org/10.1093/hmg/ddy154
Reetz K, Lencer R, Steinlechner S, Gaser C, Hagenah J, Büchel C, Petersen D, Kock N, Djarmati A, Siebner HR, Klein C, Binkofski F (2008) Limbic and frontal cortical degeneration is associated with psychiatric symptoms in PINK1 mutation carriers. Biol Psychiatry 64(3):241–247. https://doi.org/10.1016/j.biopsych.2007.12.010
Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL (2008) Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One 3(4):e1997. https://doi.org/10.1371/journal.pone.0001997
Shan Y, Liu B, Li L, Chang N, Li L, Wang H, Wang D, Feng H, Cheung C, Liao M, Cui T, Sugita S, Wan Q (2009) Regulation of PINK1 by NR2B-containing NMDA receptors in ischemic neuronal injury. J Neurochem 111(5):1149–1160. https://doi.org/10.1111/j.1471-4159.2009.06398.x
Stankiewicz TR, Linseman DA (2014) Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front Cell Neurosci 8. https://doi.org/10.3389/fncel.2014.00314
Truebestein L, Elsner DJ, Fuchs E, Leonard TA (2015) A molecular ruler regulates cytoskeletal remodelling by the Rho kinases. Nat Commun 6. https://doi.org/10.1038/ncomms10029
Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF (1999) Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23:583–592. https://doi.org/10.1016/S0896-6273(00)80810-7
Turrigiano GG (1999) Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci 22:221–227. https://doi.org/10.1016/S0166-2236(98)01341-1
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S et al (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304(5674):1158–1160. https://doi.org/10.1126/science.1096284
Venkatesh NM (1998) Synaptic plasticity: step-wise strengthening. Curr Biol 8(18):R650–R653. https://doi.org/10.1016/S0960-9822(07)00414-9
Yu W, Sun Y, Guo S, Lu B (2011) The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet 20(16):3227–3240. https://doi.org/10.1093/hmg/ddr235
Zhou H, Huang C, Tong J, Xia XG (2011) Early exposure to paraquat sensitizes dopaminergic neurons to subsequent silencing of PINK1 gene expression in mice. Int J Biol Sci 7(8):1180–1187. https://doi.org/10.7150/ijbs.7.1180
Funding
This work was supported by Research Division Bogotá-Universidad Nacional de Colombia (DIB-UNAL, Project No. 37405).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All experiments were conducted in compliance with the National Institutes of Health Guidelines for Care and Use of Experimental Animals and approved by the Institutional Committee of Animal Care and Use at Universidad Nacional de Colombia-Bogotá.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hernández, C.J., Báez-Becerra, C., Contreras-Zárate, M.J. et al. PINK1 Silencing Modifies Dendritic Spine Dynamics of Mouse Hippocampal Neurons. J Mol Neurosci 69, 570–579 (2019). https://doi.org/10.1007/s12031-019-01385-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-019-01385-x