
Abstract
Carbon monoxide-releasing molecule (CORM-2) acts as a carbon monoxide (CO) deliverer in a more controlled manner without altering carboxyhemoglobin level and exerts potential function in inhibiting inflammation and/or acute nociception. However, the regulatory mechanism of CORM-2 on spinal nerve ligation (SNL)-induced neuropathic pain is not currently clear. Our study aims to investigate the role of CORM-2 in neuropathic pain and the underlying mechanism. We found that spinal cord astrocytes were dramatically activated on day 7 after SNL. L-α-aminoadipate (L-α-AA), an astroglial toxin, reversed SNL-induced astrocyte activation at sub-toxic dose. Intrathecal administration of CO donor CORM-2 induced antiallodynic and antihyperalgesic effects in neuropathic animals induced by SNL and suppressed SNL-induced spontaneous excitatory postsynaptic current (EPSC) frequency in lamina II neurons of spinal cord slices. CORM-2 administration markedly inhibited SNL-induced connexin 43 (Cx43) expression, hemichannel function, and gap junction function on spinal astrocyte membranes. Moreover, exogenous CORM-2 could attenuate HO-1 expression, while overexpressed heme oxygenase-1 (HO-1) increased intracellular CO production, attenuated Cx43 expression, hemichannel function, and gap junction function on spinal astrocyte membranes. Additionally, Cx43 over-expression markedly reduced CORM-2-induced mechanical threshold and thermal hyperalgesia and elevated CORM-2-induced spontaneous EPSC frequency. In conclusion, CORM-2 attenuated SNL-induced neuropathic pain via suppressing Cx43-hemichannel function, which may contribute to understanding of the pathology of neuropathic pain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pain is still an open clinical issue, and there is a lack of effective analgesics in the world today. Pain can be categorized as physiological pain and pathological pain. Physiological pain induces body’s self-defense reaction to avoid further injuries within short durations. While pathological pain is triggered by pathological changes and dysfunctions in multiple sites of nervous system, which is associated with injury, infection, and metabolic disorders of peripheral and central nervous system (CNS), neuropathic pain is induced by peripheral nerve and CNS injuries, leading to long-lasting activation of sensory nerves (Polusani et al. 2011). However, the underlying mechanisms of neuropathic pain have not been fully understood up to now.
Spinal cord acts as an integrator for receiving signal inputs from nociceptors and projecting them to the brain, playing an important role in the modulation of pain-related signals (Leon-Paravic et al. 2014). Accumulated evidence suggests that nerve injury converts resting spinal cord astrocytes into an activated state, which is required for the development and maintenance of neuropathic pain (Wilkinson and Kemp 2011). Spinal cord astrocytes play crucial roles in the onset of neuropathic pain, which are frequently activated after nerve injury. Based on the critical role of astrocytes in neuropathic pain initiation and propagation, development of new therapeutic approaches in regulating astrocyte activity is promising strategies for pain management.
Carbon monoxide-releasing molecules (CORM-2), a new class of chemical agents able to reproduce several biological effects of carbon monoxide (CO), inhibits inflammation and/or acute nociception (Jurga et al. 2016). CO has been recognized as a neurotransmitter or neuromodulator in nervous system (Liu et al. 2016). A compelling study has documented that exogenous delivery of CO using CO-releasing molecules or increasing endogenous CO production may represent a novel stratagem in the management of neuropathic pain (Hervera et al. 2013). Endogenous CO is synthesized by cleavage of heme molecule, a process catalyzed by enzyme heme oxygenase (HO) (Araujo et al. 2012).
Hemichannels are ion channels composed of six connexins, which share a common plasma membrane topology: four transmembrane domains, two extracellular loops, one intracellular loop, and both the C- and N-termini located on the cytoplasmic side. These hemichannels have the peculiarity to be permeable not only to ions, but also to molecules such as ATP and glutamate. Emerging evidence showed that CO can cross the plasma membrane and directly and indirectly modulate the function of ion channels, which in turn have important repercussions in the cellular behavior (Retamal 2016). Moreover, León-Paravic et al. reported that CO was a potential and novel inhibitor of connexin hemichannels, including Cx43 and Cx46 in HeLa and MCF-7 cells (Leon-Paravic et al. 2014). However, whether CORM-2 could target to Cx43 to suppress neuropathic pain is currently unknown. Astrocytic Cx43 has been implicated in gap junction and hemichannel communication of cytosolic contents through the glial syncytia and to the extracellular space, respectively (Jurga et al. 2016). Cx43 also plays an essential role in facilitating the development of neuropathic pain; yet, the mechanism for this contribution remains unknown (Chen et al. 2012). In the current study, we aim to identify whether pharmacological elevation of CO using exogenous CORM-2 will exert an inhibitory effect on neuropathic pain involving Cx43 in SNL animal model.
Materials and Methods
Animal Model of Spinal Nerve Ligation and Drug Treatment
Sprague-Dawley rats (150-200 g) were provided by the experimental animal center of Xi’an Jiaotong University. All rats were housed in specific pathogen-free conditions, and all experiments were performed with approval of the institutional animal care and use committee of Xi’an Jiaotong University. Spinal nerve ligation (SNL) surgery on the animals (n = 90) was performed as previously described (Kim and Chung 1992; Lau et al. 2008). In brief, after rats were anesthetized with pentobarbital sodium (60 mg/kg, intraperitoneally) (Sigma, St. Louis, MO, USA), the left L5 spinal nerve was ligated tightly. Penicillin was applied at surgical site, and the incision was sutured layer by layer. After postoperative palinesthesia, rats were transferred to a clean cage. All surgeries were performed by the same person. The sham surgery operation (n = 70) only exposed spinal nerve without ligation. The timings of the treatments performed after SNL are shown in Fig. 1. Intrathecal catheter implantation was performed as previously described (Zhuang et al. 2006). A PE-10 silastic tube was implanted into the intrathecal space around the lumbar enlargement of the rat spinal cord, ending at spinal L4-L5 level, and after implantation of intrathecal catheter, animals were allowed to recover for 24 h. CORM-2 (Sigma, St. Louis, MO, USA) was dissolved in distilled water with 1% of DMSO and intrathecally injected into sham and SNL animals once per day for 20 days at dose of 10 μg dissolved in 10 μL of vehicle. The scheme of intrathecal CORM-2 administration is shown in Fig. 1. In addition, L-AA (150 nM) was purchased from Sigma (St. Louis, MO, USA) and dissolved in 10 μL of saline and injected intrathecally by the way of a single acute administration. L-AA was injected 7 days after SNL.

Scheme of CORM-2 administration and behavioral assessment in SNL rats. SNL and sham-operated rats model were used to evaluate effects of CORM administration on neuropathic pain. Behavioral tests and molecular examinations were performed 3 days before SNL as baseline (BL), and 1, 3, 7, and 14 days after SNL. CORM-2 was intrathecally injected (10 μg/10 μL) into SNL or sham rats 7 days post-surgeries. The injection was repeated once per day and lasted for 1–20 days. Behavioral tests were performed 2 h after each CORM injection
Behavioral Tests
Mechanical withdrawal threshold (MWT) and thermal withdrawal latency (TWL) of the animals were examined 3 days before SNL and averaged from three replicates as baseline. Behavioral analysis was performed at 1, 3, 7, and 14 days after SNL or at 1, 5, 10, 15, and 20 days approximately 2 h after each CORM-2 injection, as described previously (Kawasaki et al. 2008; Makuch et al. 2013). For MWT analysis, animal was put in a box on an elevated metal mesh floor and allowed a 30-min habituation period before examination. The plantar surface of each hindpaw was stimulated with a series of von Frey hairs with logarithmically incrementing stiffness (0.02–2.56 g; Stoelting, USA), presented perpendicular to the plantar surface (3–5 s for each hair). Based on Dixon’s up-down method (Dixon 1980), six von Frey tests were performed in each animal, and the 50% paw-withdrawal threshold (PWT) was determined.
Thermal withdrawal latency was assessed using a hot sting instrument (Beijing Zhishuduobao biological technology CO., LTD, Beijing, China) as described previously (Mika et al. 2007). Animal was put in a vitreous test box and allowed a 10-min habituation period before examination. Opening the hot sting instrument, thermal radiation light irradiated the middle part of the right hind foot of rats. The light was cut and then the automatic timer record hot-stimulated paw withdrawal latency (PWL) when the hind foot of the rat was lifted because of hot stimulation. PWL was measured for three times, with a 5 min interval for each test. The timings of behavioral tests are shown in Fig. 1.
Analysis of Astrocyte Activation
Ipsilateral dorsal lumbar (L4-L6) spinal cords were collected and ground to form single-cell suspension. Cells were plated onto a 100-mm Petri dish and incubated at 37 °C in a 5% CO2 humidified atmosphere for 2 h. Cells were washed and then stained using anti-glial fibrillary acid protein (GFAP) antibody (Abcam, Cambridge, UK). Cells were washed and analyzed by a FACSCalibur with CELLQuest software version 3.1 (Becton Dickinson).
Primary Astrocyte Cultures
Primary cultures of astrocytes were prepared as described previously (Gao et al. 2009). In brief, astrocyte cultures were prepared from the spinal cords of rats undergoing sham or SNL surgery at 7 days and cultured in a 75-cm2 flask at a density of 2.5 × 105 cells/cm2 in low-glucose Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum. The medium was replaced twice a week. For drug administration, isolated astrocytes were treated with 50, 100, or 200 μM CORM-2 dissolved in distilled water with 1% of DMSO for 1 h (Garcia-Arnandis et al. 2011). In addition, freshly prepared HO-1 inductor cobalt protoporphyrin IX (CoPP) (5 μM) and HO-1 inhibitor zinc protoporphyrin IX (ZnPP) (5 μM) were dissolved in DMSO (1% solution in saline) and administrated to the astrocyte cultures approximately 4 h before evaluations of Cx43 expression and activity as well as CO production. Isolated astrocytes from sham group were treated with the same volume of DMSO to be used as vehicles.
The HO Vector and siRNA Transfection
The pcDNA3.1(+)/HO-1 expression vector was constructed by cloning HO-1 fragment from normal rat cDNA into pcDNA3.1(+) (Invitrogen, Carlsbad, CA, USA) between BamH I and EcoR I sites to express HO-1 in abundance in E. coli DH5α cells. The primers for HO-1 were as follows: forward primer: 5′-GGA GGA TCC GGC TGT GAA CTC TGT CTC-3′, and reverse primer: 5′-ctc GAA TTC GGC ATC TCC TTC CAT TCC-3′. The recombinant plasmid was identified by endonuclease digestion and DNA sequencing. Thereafter, the pcDNA3.1(+)/HO-1 vector was transfected into the sham and SNL-induced astrocytes mediated by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) manually. The stably transfected clones were screened by G418 and identified by Western blot. Preparation and transfection of the pcDNA3.1(+)/Cx43 expression vector were similar to the above-mentioned methods.
HO-1 siRNA and control siRNA were transfected into cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) manually. The siRNA primer sequences were designed by Invitrogen Block-iT RNAi Designer. HO-1 siRNA_445: 5′-CCA CAC AGC ACU ACG UAA AdTdT-3′ (F), 5′-UUU ACG UAG UGC UGU GUG GdTdT-3′ (R). HO-1 siRNA_740: 5′-GCU CAA CAU UGA GCU GUU UdTdT-3′ (F), 5′-AAA CAG CUC AAU GUU GAG CdTdT-3′ (R). HO-1 siRNA_837: 5′-GCU AGC CUG GUU CAA GAU AdTdT-3′ (F), 5′-UAU CUU GAA CCA GGC UAG CdTdT-3′ (R).
Determination of Intracellular CO
A hemoglobin (Hb)-based assay was used to detect intracellular CO release, as described previously (Leon-Paravic et al. 2014). In brief, Hb (1 mg) was dissolved in 1 mL of ethanol solution, and then, Hb solution was supplemented with 0.1% sodium dithionite to deoxygenate them, to allow CO binding to Hb to form carboxyhemoglobin. The content of carboxyhemoglobin was assessed from the changes in absorbance at 540 nm.
Gap Junction and Hemichannel Function Determination
Gap junction function of Cx43 in astrocytes was examined based on the previously described (Polusani et al. 2011). In brief, lucifer yellow (5% in 1 M lithium chloride, Sigma) was microinjected to isolated astrocytes via a glass pipette (diameter of 2–4 μm). Diffusion of the dye to the neighboring astrocytes was observed for 10 min. Astrocytes were stimulated with CORM-2 for 1 h before the dye delivery. The images of labeled astrocytes were captured with a CCD Spot camera, and the number of labeled astrocytes was quantified with the NIH ImageJ software.
As hemichannels are permeable to the dye ethidium bromide, we use ethidium bromide uptake to indicate the function of hemichannels. Astrocytes were stimulated with CORM-2 for 1 h and exposed to 0.5 mM ethidium bromide (Sigma) for 10 min at 37 °C. Then, cells were washed with Hank’s balanced salt solution (HBSS) and supplemented with 1.2 mM CaCl2 (HBSS-Ca2+, Gibco). Astrocytes were examined with a Nikon fluorescence microscope, and images were captured with a CCD Spot camera. The positive staining of ethidium bromide was analyzed with NIH ImageJ software.
Patch-Clamp Techniques
The whole cell patch-clamp recordings were made from lamina IIo neurons which receive C-fiber nociceptive input and also make synapses with lamina I projection neurons (Park et al. 2011; Todd 2010) in the voltage-clamp mode (Kawasaki et al. 2008; Park et al. 2011). Briefly, under a dissecting microscope with transmitted illumination, the substantia gelatinosa (lamina II) is clearly visible as a relatively translucent band across the dorsal horn. Patch pipettes were fabricated from thin-walled, borosilicate, glass-capillary tubing (1.5-mm outer diameter; World Precision Instruments). After establishing the whole cell configuration, neurons were held at their holding potentials at 70 mV to record spontaneous excitatory postsynaptic currents (EPSCs). The resistance of a typical patch pipette was 5–10 MΩ. The internal solution contained the following (in mM): 135 potassium gluconate, 5 KCl, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES, and 5 ATP-Mg. Membrane currents were amplified with an Axopatch 200 A amplifier (Molecular Devices) in voltage-clamp mode. Signals were filtered at 2 kHz and digitized at 5 kHz. Data were stored with a personal computer using pClamp 6 software and analyzed with Mini Analysis (Synaptosoft). Those cells that showed 45% changes from the baseline levels were regarded as responsive (Kawasaki et al. 2008).
Western Blot
Total protein was extracted using the Tissue or Cell Total Protein Extraction Kit (Amresco, USA) from spinal cord or astrocytes. All primary antibodies were purchased from Abcam (Cambridge, UK). The proteins were separated by SDS-PAGE followed by electrotransfer to NC membrane; the membranes were probed using antibodies against GFAP (1:5000) (ab4674), HO-1 (1:200) (ab13248), and Cx43 (1:5000) (ab170190) followed by a horseradish peroxidase (HRP)-conjugated secondary antibody (1:2000) (ab6734). Bands were revealed with ECL reagent (Millipore, Boston, MA, USA) and recorded on X-ray films (Kodak, China). Densitometry of each band was quantified by Gel imaging system and Quantity One 4.62 software (Bio-Rad, Hercules, CA, USA).
RT-PCR
Total RNA was extracted using TRIzol reagents (Invitrogen, Carlsbad, CA, USA) from astrocytes. Isolated RNA was electrophoresed on 1% agarose gel to detect the purity of total RNA. The first-strand cDNA was synthesized using 1 μg total RNA and SuperScript® III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). PCR amplification was performed using the PCR amplification kit (Takara Biotechnology, Dalian, China). The specific primers were designed using Primer Premier 6.0 software and synthesized by Sangon Biotech (Shanghai, China). The primers for Cx43 were 5′-GGC TGT GAA CTC TGT CTC-3′ (forward) and 5′-GGC ATC TCC TTC CAT TCC-3′ (reverse). The primers for GAPDH as an internal control were 5′-ACC ACA GTC CAT GCC ATC AC-3′ (forward) and 5′-TCC ACC ACC CTG TTG CTG TA-3′ (reverse). PCR products were electrophoresed on 1% agarose gel and visualized by Gel Imaging System of Bio-Rad Corp (Bio-Rad, Hercules, CA, USA). Each band was analyzed by Quantity One 4.62 software (Bio-Rad, Hercules, CA, USA).
Statistical Analysis
Data are reported as means ± SD in at least three replicates per group. Data were analyzed by SPSS19.0 software (IBM, USA). Differences between groups were compared using Student’s t test or ANOVA, followed by least significant difference (LSD) multiple comparison tests. Repeated measures ANOVA were performed for the analysis of time courses of the behavioral results. Differences were considered significant at p < 0.05.
Results
Astrocyte Activation Is Involved in the Development of Neuropathic Pain Following Spinal Nerve Ligation
In this study, we used a conventional spinal nerve ligation rat model to develop neuropathic pain in animals. The behavioral tests showed that compared with baseline measured 3 days before SNL, mechanical thresholds of neuropathic animals gradually decreased on days 1, 3, 7, and 14 after SNL (p < 0.001, Fig. 2a). Similar results were observed in the change of thermal hyperalgesia after SNL (Fig. 2b). To investigate the effect of spinal nerve injury on astrocyte activation, GFAP expression and GFAP-positive cells, which was a hallmark of astrocyte activation, were evaluated in ipsilateral spinal dorsal horn. The results showed that the number of GFAP-positive cells in spinal cord were dramatically increased on days 3, 7, and 14 after SNL compared with naïve animals without any operation (p < 0.001, Fig. 2c). Similar results were observed in the effect of spinal nerve injury on GFAP expression in spinal cord (Fig. 2c, d).

Effect of spinal nerve injury on astrocyte activation in ipsilateral spinal dorsal horn. Changes of mechanical allodynia (a) and thermal hyperalgesia (b) in sham and SNL rats 3 days before (BL) and 1, 3, 7, and 14 days after the operations. The differences between groups were analyzed by repeated measures ANOVA followed by LSD tests. ** p < 0.01, *** p < 0.001 vs BL. N = 6 rats/group. c Spinal cord isolated from sham and SNL rats at indicated days were grinded and prepared single-cell suspension, and then stained using anti-GFAP antibody. Numbers of GFAP-positive cells were counted by flow cytometry. Naïve spinal cords were isolated from rats without any treatment. d GFAP expression was analyzed by Western blot after SNL, N = 4 rats/group, * p < 0.05, ** p < 0.01,*** p < 0.001 vs BL. e–f Astroglial toxin L-α-AA was intrathecally injected (150 nM/10 μL) into sham and SNL rats 7 days after the surgeries, and mechanical allodynia and thermal hyperalgesia were examined 2 h post-injections. N = 4 rats/group. ** p < 0.01. g Flow cytometry determination of the number of GFAP-positive cells after L-α-AA injection in sham and SNL rats on postoperative day 7. ** p < 0.01. h–i Western blot analysis and quantification of GFAP expression. N = 4 rats/group, statistical significance was determined using ANOVA followed by LSD tests
Besides, to further evaluate whether activated spinal astrocytes were crucial for neuropathic pain, the astroglial toxin L-α-aminoadipate (L-α-AA) was intrathecally administrated at a concentration of 150 nM to sham and SNL rats on postoperative day 7. The effect of L-α-AA on neuropathic pain was determined 2 h after L-α-AA administration. L-α-AA reversed SNL-induced mechanical allodynia (p < 0.01, Fig. 2e) and thermal hyperalgesia (p < 0.01, Fig. 2f) compared with the SNL group. In addition, compared with the SNL group, L-α-AA markedly attenuated the SNL-induced number of GFAP-positive cells (p < 0.01, Fig. 2g) and GFAP expression (p < 0.01, Fig. 2h, i) in spinal cords.
CORM-2 Inhibits Neuropathic Pain and SNL-Induced Spontaneous EPSCs in Lamina II Neurons of Spinal Cord Slices
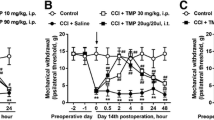
To explore the effect of CORM-2 on SNL-induced neuropathic pain, we intrathecally injected a CORM-2 into SNL-exposed rats, and then, the indices of mechanical response and thermal hyperalgesia were determined. Results showed that CORM-2 administration dramatically attenuated SNL-induced mechanical allodynia (Fig. 3a) and thermal hyperalgesia (Fig. 3b) in a dose-dependent manner, and the efficiency of CORM-2 reached a peak when CORM-2 was administered for 15 days after SNL. Furthermore, we examined whether CORM-2 was able to normalize EPSCs in spinal cord slices after nerve injury. Compared with the sham group, SNL induced a profound increase in spontaneous EPSC frequency, but not amplitude, demonstrating that nerve injury was connected with long-lasting increases in spinal cord synaptic transmission. After CORM-2 administration, SNL-induced spontaneous EPSC frequency was significantly reduced in a dose-dependent manner (Fig. 3c–e).

Effect of CORM-2 on SNL-induced neuropathic pain and spontaneous EPSCs. a, b CORM-2 injection was maintained for 5, 10, and 15 days, respectively, and mechanical and thermal response in SNL rats were tested at indicated days before and after injury. N = 6 rats/group, the differences between groups were analyzed by repeated measures ANOVA followed by LSD tests. * p < 0.05, ** p < 0.01, *** p < 0.001 vs vehicle. c Traces of spontaneous EPSCs determined at the end of the last injection. N = 8 neurons/group. d, e Frequency and amplitude of spontaneous EPSCs. N = 8 neurons/group, the differences between groups were analyzed using ANOVA followed by LSD tests. * p < 0.05, ** p < 0.01, *** p < 0.001 vs vehicle
CORM-2 Inhibits HO-1 Expression and Connexin 43-Hemichannel Activity in Ipsilateral Spinal Dorsal Horn
Generation of endogenous CO was mainly dependent on the expression and activity of heme oxygenase (HO). We therefore investigated change of HO-1 expression after SNL and the effect of CORM-2 on HO-1 expression. The data showed that HO-1 expression in spinal cord dramatically increased on days 3, 7, and 14 after SNL compared with the naïve value in animals without any operation and the matched sham animals (Fig. 4a, b), and L-α-AA reduced SNL-induced HO-1 expression (p < 0.01, Fig. 4c) compared with the SNL group. Furthermore, CORM-2 administration reduced SNL-induced HO-1 expression compared with the SNL group in a dose-dependent manner (Fig. 4d).

Effects of CORM-2 on HO-1 expression in spinal dorsal and Cx43-hemichannel function in spinal astrocytes. a, b Changes of HO-1 expression in the spinal cord isolated from sham and SNL rats determined using Western blot. N = 4 rats/group, the differences between groups were analyzed by repeated measures ANOVA followed by LSD tests ** p < 0.01, *** p < 0.001 vs BL. c HO-1 expression in spinal cords from sham and SNL rats in response to the stimulation of astroglial toxin L-α-AA at day 7 postoperativelly. d, e Effects of CORM-2 injection duration on SNL-induced HO-1 and Cx43 expression in spinal cords in SNL animals at the end of the last administration for 5, 10, and 15 days. HO-1 and Cx43 were examined in sham and SNL rats 7 days after the surgeries. * p < 0.05, ** p < 0.01, and *** p < 0.001 vs sham; ## p < 0.01, ### p < 0.001 vs SNL. f Identification of isolated astrocytes from sham and SNL rats. Student’s t test, *** p < 0.001 vs sham. Primary astrocytes were isolated from sham and SNL rats 7 days after the surgeries, and astrocytes were further treated with 50, 100, and 200 μM CORM-2 for 1 h. Then, Cx43 expression was evaluated using Western blot (g). Effect of CORM-2 on SNL-induced hemichannel function was tested by ethidium bromide staining in astrocytes (h). Effect of CORM-2 on SNL-induced gap junction function was tested after microinjection of lucifer yellow into astrocytes (i). The differences between groups were compared using ANOVA followed by LSD tests, * p < 0.05, ** p < 0.01, *** p < 0.001 vs sham; # p < 0.05, ## p < 0.01, ### p < 0.001 vs SNL
CORM-2 dose-dependently suppressed SNL-induced Cx43 expression in spinal cord compared to the SNL group (Fig. 4e). Moreover, we evaluated the hemichannel and gap junction function in response to the stimulation with 50–200 μM CORM-2 in spinal astrocytes of rats that underwent sham or SNL surgeries. As displayed in Fig. 4f, GFAP expression of astrocytes derived from SNL rats was dramatically elevated compared with the astrocytes derived from sham rats (p < 0.001). Besides, CORM-2 stimulation dose-dependently down-regulated SNL-induced Cx43 expression in astrocytes compared to the SNL group (Fig. 4g) and reduced SNL-induced hemichannel function of spinal astrocyte membranes (Fig. 4h) and gap junction function of cell-cell contact (Fig. 4i), suggesting that CORM-2 was implicated in Cx43-hemichannel function on spinal astrocyte membranes.
HO-1 Induces Connexin 43-Hemichannel Closure
To investigate whether endogenous HO-1 also inhibited the CX43-hemichannel function on spinal astrocyte membranes, we transfected the pcDNA3.1(+)/HO-1 or HO-1 siRNA into astrocytes that were isolated from sham or SNL rats. Besides, HO-1 inductor CoPP and HO-1 inhibitor ZnPP were applied to evaluate the effect of HO-1 on hemichannel function. As shown in Fig. 5a, intracellular CO production was significantly reduced after SNL compared to the sham control (P < 0.05). HO-1 overexpression or inductor CoPP elevated intracellular CO production, especially in SNL groups (P < 0.05). However, intracellular CO production had no further decrease in response to HO-1 inhibition either by siRNA or ZnPP compared to their corresponding control in SNL groups (P > 0.05), and substantially lowered CO after SNL may contribute for this invalidation of HO-1 inhibition (Fig. 5a). Cx43 mRNA level substantially increased following SNL damage. However, either HO-1 overexpression or activation using CoPP significantly suppressed the Cx43 mRNA level compared with their matched controls. HO-1 siRNA transfection and inhibitor administration significantly elevated Cx43 mRNA (Fig. 5b). We further evaluated the hemichannel function and gap junction function of Cx43 when HO-1 was manipulated. The results showed that both HO-1 upregulation and activation using CoPP significantly suppressed the functions of Cx43 compared with their matched controls, while HO-1 inhibition showed opposite effects (Fig. 5c, d).

HO-1 modulation on CX43 expression and function in astrocytes. Primary astrocytes were isolated from sham and SNL rats 7 days after the operations. Then, the cells were transfected with HO-1 overexpressing vectors or siRNA and their matched controls 24 h before further evaluations of CO production and Cx43 expression and function. In addition, freshly prepared HO-1 inductor CoPP (5 μM) and HO-1 inhibitor ZnPP (5 μM) were dissolved in DMSO and administrated to the astrocyte cultures approximately 4 h before further evaluations. a Intracellular CO production in astrocytes was evaluated using a hemoglobin (Hb)-based assay. b Cx43 mRNA levels in astrocytes after each treatment. c Hemichannel function in astrocytes evaluated using ethidium bromide staining. d The effect of HO-1 on gap junction function in astrocytes determined by lucifer yellow microinjection into astrocytes. The differences between groups were compared using ANOVA followed by LSD tests, * p < 0.05, ** p < 0.01 vs matched controls; # p < 0.05, ## p < 0.01 vs sham
Connexin 43 Is Implicated in CORM-2-Modulated Neuropathic Pain and Excitatory Synaptic Transmission
To confirm whether Cx43 was implicated in CORM-2-regulated neuropathic pain and excitatory synaptic transmission, we intrathecally injected the pcDNA3.1(+)/Cx43 into the rats on day 7 after SNL and treatment with CORM-2 after SNL. Results demonstrated that Cx43 overexpression had a proallodynic effect to mechanical stimulus (p < 0.001, Fig. 6a) and a prohyperalgesic effect to thermal stimulus (p < 0.001, Fig. 6b) in SNL animals treated with or without CORM-2 compared with their corresponding controls. Besides, Cx43 overexpression markedly elevated CORM-2-induced spontaneous EPSC frequency (p < 0.01, Fig. 6c), but not amplitude (Fig. 6d) compared with control.

Cx43 antagonized the effect of CORM-2 on neuropathic pain and excitatory synaptic transmission in spinal cord. SNL animals treated with or without CORM-2 were intrathecally injected with Cx43 overexpressing vectors; then, behavioral tests and spontaneous EPSCs were evaluated 2 or 24 h after the injections. a, b The effect of Cx43 overexpression and Cx43 activation on CORM-2-induced mechanical allodynia and thermal hyperalgesia. N = 4 rats/group. c, d Frequency and amplitude of spontaneous EPSCs. N = 8 neurons/group. The differences between groups were compared using ANOVA followed by LSD tests. ** p < 0.01, *** p < 0.001 vs control; ## p < 0.01 vs naïve
Discussion
In this study, we demonstrated a mechanism of CORM-2 in attenuating nerve injury-induced neuropathic pain by inhibiting gap junction protein Cx43 expression, hemichannel function, and gap junction function in SNL injury-activated astrocytes.
Neuropathic pain is caused by lesion or dysfunction of peripheral nerve and CNS injuries. It is one of the most intractable human complaints and is largely resistant to the current treatments (Dworkin et al. 2003). Astrocytes in CNS form an intimately associated network with neurons. Activation of spinal cord astrocytes plays an important role in the establishment and maintenance of neuropathic pain (Ji et al. 2013). Previous studies demonstrated that activation of astrocytes in anterior cingulate cortex promotes neuronal excitation and development of negative emotions during pain hypersensitivity after peripheral inflammation (Ikeda et al. 2013). In our study, we also found that astrocytes were markedly activated in spinal dorsal horn after SNL, as reflected by the increased GFAP expression. In addition, we found that L-α-AA, an inhibitor of astrocyte activation, markedly alleviated neuropathic pain induced by SNL. In our experiment, L-α-AA was administrated at a dose of 150 nM, which was largely lower than the reported sub-toxic dose range of less than 0.5 mM (Koyama et al. 1997). Meanwhile, Ohmichi et al. reported that L-α-AA, intrathecally administered at 150 nM, inhibited mechanical hyperalgesia in several body parts including the lower leg skin and muscles bilaterally, hindpaws, and tail in a chronic post-cast pain (CPCP) rat model (Ohmichi et al. 2014).
Currently, more previous studies and our findings indicated that alterations of CO content in activated astrocytes may represent another mechanism that contributed to the development of neuropathic pain. CO is produced endogenously by HO enzymes, which is highly expressed in many inflammatory disease states and broadly protective (Wilkinson and Kemp 2011). Currently, increasing evidence indicate that CO is a neurotransmitter associated with cytoprotection and maintenance of homeostasis in pathogenesis of cerebral ischemia, chronic neurodegenerative diseases, multiple sclerosis, and pain (Queiroga et al. 2015). HO-1/CO axis was shown to play roles in pain relief. Emerging studies demonstrated that HO-1 upregulation elicited potent analgesic effects, partially by inhibiting spinal glia activation dependent on its anti-inflammatory and anti-oxidant roles (Liu et al. 2016). In our study, we found that HO-1 was substantially increased after SNL insults. However, this increase was insufficient for overriding SNL-induced neuropathic pain, just like what was concluded by Araujo that the degree of HO-1 fold induction over the basal might reflect the level of stress induced by stimuli rather than an ability to protect against the injury (Araujo et al. 2012). Furthermore, we also found that there was a controversy that the expression of HO-1 increased for several folds after SNL while its catalyzed product CO reduced after SNL. We considered that there were multiple factors contributed to this phenomenon. HO-1 is a ubiquitous inducible stress response protein whose expression is upregulated by a variety of cellular insults. Indeed, by itself or by its enzymatic products, HO-1 acts in tissue homeostasis by suppressing oxidative stress and maintaining cellular integrity. Especially, under the condition of SNL insult, the acute and detrimental stress-induced increase of HO-1 expression may have certain effects against tissue damage. However, the increase of HO-1 after sensing the stress may not be a direct indicator or predictor of its linear increase of the molecular activity. For one hand, just as what inferred by Araujo et al., the basal HO-1 levels may be more important than the degree of upregulation or induction, which may only reflect the stress level in cells (Araujo et al. 2012). On the other hand, the activity of HO-1 was modulated or constricted by multiple complicated factors, such as free and available substrate level, energy supply as well as oxidative stress level, since the SNL insult is a complex process that involved various pathophysiologic mechanisms. Besides, the produced CO could bind avidly to the heme-iron contained enzymes like NADPH oxidase, cytochrome P450, and inducible nitric oxide synthase and also can be slowly oxidized to CO2, which may affect the detection of free CO (Gullotta et al. 2012; Srisook et al. 2006). Moreover, in this study, CO level was measured in isolated astrocytes in vitro. Thus, we considered that the isolate procedure may affect the content of CO.
CORM are ruthenium-containing compounds that are developed to create the opportunity to deliver CO in a more practical, controllable, and accurate way to the target site in comparison to CO gas inhalation (Babu et al. 2015). We found that intrathecal injection of CORM-2 could suppress neuropathic pain symptoms in rats induced by SNL injury. Hervera has reported that exogenous delivery of CO using CO-releasing molecules or increasing the endogenous CO production improved the main neuropathic pain symptoms induced by nerve injury in a time-dependent manner, involving the interaction between CO and NOS system (Hervera et al. 2013). Our finding demonstrated that intrathecal administration of CORM-2 suppressed HO-1 expression in astrocytes of SNL rats. This conclusion is not consistent with what is reported by Bijjem et al., who shows that subcutaneous injection of CORM-2 significantly improves the levels of HO-1 in CCI Wistar rats. We consider that different neuropathic pain rat model and drug administration route may contribute to this discrepancy. In addition, it was also documented that CORM-2 downregulated HO-1 expression in LPS-stimulated macrophages (Srisook et al. 2006). HO, also known as the heat shock protein 32, is expressed/activated in response to a wide range of different cellular stress conditions, such as oxidative stress, inflammation, or ischemia (Queiroga et al. 2015). It is well acknowledged that oxidative stress and inflammation are the principal determinants for the development of neuropathic pain (Kiasalari et al. 2017). Furthermore, CORM-2 has been shown to be beneficial in resolution of acute inflammation (Katada et al. 2010; Qureshi et al. 2016). Therefore, we think that the inhibition of CORM-2 on inflammation in response to neuropathic injury may suppress the induction of HO-1. Likewise, Srisook et al. concluded that administering CO gas in low dose was able to produce the anti-inflammatory and anti-apoptotic effects and could replace the protective effects of HO-1 induction (Srisook et al. 2006). Since vast CO enters astrocytes when CORM-2 is administrated, and higher CO concentration in astrocytes cause catalytic reactions of HO-1 to scarcely be in the ascendant. It is reported that the enhancement of HO-1 protein is often accompanied by a higher total gap junction protein Cx43 level (Lakkisto et al. 2009). Therefore, our study evaluated whether Cx43 was involved in HO-1/CO axis mediated analgesia effect after SNL. Astrocytes in CNS form interconnected networks coupled by gap junctions. The major structural components of gap junctions are connexins, and Cx43 subunit is the principal connexin expressed on astrocytes (Bennett et al. 2012). In current study, Cx43 expression was up-regulated, and the hemichannel function and gap junction function on spinal astrocyte membranes were enhanced after nerve injury, indicating that Cx43-hemichannel is implicated in development of neuropathic pain and opened after nerve injury. CO is known to directly and indirectly modulate the function of ion channels at the plasma membrane, which in turn have important repercussions in the cellular behavior (Retamal et al. 2015). It was reported that CORM-2 potentiated the anti-hyperalgesic and anti-allodynic properties of morphine and buprenorphine by non-competitive antagonizing purinergic P2X4 receptors and leading to the inhibition of glial activation in neuropathic pain model (Jurga et al. 2016). On the other hand, purinergic receptors are known to open up hemichannels in astrocytes (Baroja-Mazo et al. 2013). Therefore, CORM-2 may function to inhibit Cx43 indirectly via purinergic P2X4 receptors. Hemichannels are thought to be normally closed, preventing cell loss of important metabolites such as ATP and glutathione, as well as the massive entry of Ca2+ (Saez et al. 2010; Sanchez et al. 2010). After nerve injury, Cx43-hemichannels on astrocyte membranes are opened and excitatory transmitters, including ATP, glutamate and so on, are released from astrocytes. At the same time, K+ and Ca2+ in the extracellular space enter into the astrocyte, causing a rapid increase of intracellular Ca2+ concentration to trigger Ca2+-dependent processes such as AMPA/kainate and metabotropic glutamate receptors (mGluRs) on astrocytes to induce pain (Bezzi et al. 1998). Therefore, abundant CO, acting as a novel inhibitor of Cx 43-hemichannels, cross the membrane of astrocytes or not after CORM-2 injection and induce Cx43-hemichannel closure (Leon-Paravic et al. 2014). The closed Cx43-hemichannels maintain small molecules in astrocytes and block the massive entry of Ca2+, thereby preventing Ca2+-dependent processes for contributing to the treatment of neuropathic pain. Finally, it is worth noting that CORM-2 is a ruthenium (Ru)-containing compound and Ru has been shown to play potential roles in affecting vascular tone (Pauwels et al. 2015). Also, Freitas et al. showed that the Ru complexes containing nicotinic acid and isonicotinic acid inhibited mechanical hyperalgesia induced by prostaglandin E2. Besides, it was also evidenced that the control [Ru(NH3)6]Cl3 without the above ligands by themselves also play certain roles in reducing hyperalgesia induced by PGE2, in spite to a lower extent than other Ru-compounds. However, the mechanism is not clear yet (Freitas et al. 2015). Therefore, whether Ru functions in the anti-nociceptive activity of CORM-2 or synergistically functions with CO are currently unknown, which should be focused in further studies.
Conclusion
Our study demonstrated that activation of spinal cord astrocytes plays an important role in attenuating neuropathic pain induced by SNL. Intraspinal injection of CORM-2 after SNL could greatly alleviate neuropathic pain by elevating mechanical thresholds and thermal hyperalgesia, and reducing spontaneous EPSCs. Administration of exogenous CORM-2 and up-regulation of endogenous CO producer HO-1 both suppressed SNL-induced Cx43 expression, hemichannel function and gap junction function. Therefore, CORM-2 attenuated SNL-induced neuropathic pain via suppressing Cx43-hemichannel functioning. The current findings will contribute to the understanding of the underlying mechanisms of neuropathic pain. However, more details concerning the mechanism of CORM-2 in suppressing pain should be described.
References
Araujo JA, Zhang M, Yin F (2012) Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front Pharmacol 3:119. doi:10.3389/fphar.2012.00119
Babu D, Motterlini R, Lefebvre RA (2015) CO and CO-releasing molecules (CO-RMs) in acute gastrointestinal inflammation. Br J Pharmacol 172:1557–1573. doi:10.1111/bph.12632
Baroja-Mazo A, Barbera-Cremades M, Pelegrin P (2013) The participation of plasma membrane hemichannels to purinergic signaling. Biochim Biophys Acta 1828:79–93. doi:10.1016/j.bbamem.2012.01.002
Bennett MV, Garre JM, Orellana JA, Bukauskas FF, Nedergaard M, Saez JC (2012) Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Res 1487:3–15. doi:10.1016/j.brainres.2012.08.042
Bezzi P et al (1998) Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 391:281–285. doi:10.1038/34651
Chen MJ, Kress B, Han X, Moll K, Peng W, Ji RR, Nedergaard M (2012) Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. Glia 60:1660–1670. doi:10.1002/glia.22384
Dixon WJ (1980) Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 20:441–462. doi:10.1146/annurev.pa.20.040180.002301
Dworkin RH et al (2003) Advances in neuropathic pain: diagnosis, mechanisms, and treatment recommendations. Arch Neurol 60:1524–1534. doi:10.1001/archneur.60.11.1524
Freitas CS, Roveda AC Jr, Truzzi DR, Garcia AC, Cunha TM, Cunha FQ, Franco DW (2015) Anti-inflammatory and anti-nociceptive activity of ruthenium complexes with Isonicotinic and nicotinic acids (niacin) as ligands. J Med Chem 58:4439–4448. doi:10.1021/acs.jmedchem.5b00133
Gao YJ et al (2009) JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci 29:4096–4108. doi:10.1523/JNEUROSCI.3623-08.2009
Garcia-Arnandis I, Guillen MI, Gomar F, Castejon MA, Alcaraz MJ (2011) Control of cell migration and inflammatory mediators production by CORM-2 in osteoarthritic synoviocytes. PLoS One 6:e24591. doi:10.1371/journal.pone.0024591
Gullotta F, di Masi A, Coletta M, Ascenzi P (2012) CO metabolism, sensing, and signaling. Biofactors 38:1–13. doi:10.1002/biof.192
Hervera A, Leanez S, Motterlini R, Pol O (2013) Treatment with carbon monoxide-releasing molecules and an HO-1 inducer enhances the effects and expression of micro-opioid receptors during neuropathic pain. Anesthesiology 118:1180–1197. doi:10.1097/ALN.0b013e318286d085
Ikeda H, Mochizuki K, Murase K (2013) Astrocytes are involved in long-term facilitation of neuronal excitation in the anterior cingulate cortex of mice with inflammatory pain. Pain 154:2836–2843. doi:10.1016/j.pain.2013.08.023
Ji RR, Berta T, Nedergaard M (2013) Glia and pain: is chronic pain a gliopathy? Pain 154(Suppl 1):S10–S28. doi:10.1016/j.pain.2013.06.022
Jurga AM, Piotrowska A, Starnowska J, Rojewska E, Makuch W, Mika J (2016) Treatment with a carbon monoxide-releasing molecule (CORM-2) inhibits neuropathic pain and enhances opioid effectiveness in rats. Pharmacol Rep 68:206–213. doi:10.1016/j.pharep.2015.08.016
Katada K et al (2010) Carbon monoxide liberated from CO-releasing molecule (CORM-2) attenuates ischemia/reperfusion (I/R)-induced inflammation in the small intestine. Inflammation 33:92–100. doi:10.1007/s10753-009-9162-y
Kawasaki Y et al (2008) Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med 14:331–336. doi:10.1038/nm1723
Kiasalari Z, Rahmani T, Mahmoudi N, Baluchnejadmojarad T, Roghani M (2017) Diosgenin ameliorates development of neuropathic pain in diabetic rats: involvement of oxidative stress and inflammation. Biomed Pharmacother 86:654–661. doi:10.1016/j.biopha.2016.12.068
Kim SH, Chung JM (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50:355–363
Koyama Y, Kimura Y, Baba A (1997) Induction of glutamine synthetase by L-alpha-aminoadipate in developmental stages of cultured astrocytes. Neurosci Lett 223:65–68
Lakkisto P, Csonka C, Fodor G, Bencsik P, Voipio-Pulkki LM, Ferdinandy P, Pulkki K (2009) The heme oxygenase inducer hemin protects against cardiac dysfunction and ventricular fibrillation in ischaemic/reperfused rat hearts: role of connexin 43. Scand J Clin Lab Invest 69:209–218. doi:10.1080/00365510802474392
Lau WK, Chan WK, Zhang JL, Yung KK, Zhang HQ (2008) Electroacupuncture inhibits cyclooxygenase-2 up-regulation in rat spinal cord after spinal nerve ligation. Neuroscience 155:463–468. doi:10.1016/j.neuroscience.2008.06.016
Leon-Paravic CG et al (2014) Carbon monoxide (CO) is a novel inhibitor of connexin hemichannels. J Biol Chem 289:36150–36157. doi:10.1074/jbc.M114.602243
Liu X et al (2016) Spinal Heme Oxygenase-1 (HO-1) exerts Antinociceptive effects against neuropathic pain in a mouse model of L5 spinal nerve ligation. Pain Med 17:220–229. doi:10.1111/pme.12906
Makuch W, Mika J, Rojewska E, Zychowska M, Przewlocka B (2013) Effects of selective and non-selective inhibitors of nitric oxide synthase on morphine- and endomorphin-1-induced analgesia in acute and neuropathic pain in rats. Neuropharmacology 75:445–457. doi:10.1016/j.neuropharm.2013.08.031
ika J, Osikowicz M, Makuch W, Przewlocka B (2007) Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur J Pharmacol 560:142–149. doi:10.1016/j.ejphar.2007.01.013
Ohmichi M et al (2014) Activated spinal astrocytes are involved in the maintenance of chronic widespread mechanical hyperalgesia after cast immobilization. Mol Pain 10:6. doi:10.1186/1744-8069-10-6
Park CK, Lu N, Xu ZZ, Liu T, Serhan CN, Ji RR (2011) Resolving TRPV1- and TNF-alpha-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J Neurosci 31:15072–15085. doi:10.1523/JNEUROSCI.2443-11.2011
Pauwels B, Boydens C, Van de Voorde J (2015) The influence of ruthenium on vascular tone. J Pharm Pharmacol 67:1263–1271. doi:10.1111/jphp.12417
Polusani SR, Kar R, Riquelme MA, Masters BS, Panda SP (2011) Regulation of gap junction function and Connexin 43 expression by cytochrome P450 oxidoreductase (CYPOR). Biochem Biophys Res Commun 411:490–495. doi:10.1016/j.bbrc.2011.06.132
Queiroga CS, Vercelli A, Vieira HL (2015) Carbon monoxide and the CNS: challenges and achievements. Br J Pharmacol 172:1533–1545. doi:10.1111/bph.12729
Qureshi OS et al (2016) Enhanced acute anti-inflammatory effects of CORM-2-loaded nanoparticles via sustained carbon monoxide delivery. Eur J Pharm Biopharm 108:187–195. doi:10.1016/j.ejpb.2016.09.008
Retamal MA (2016) Carbon monoxide modulates Connexin function through a lipid peroxidation-dependent process: a hypothesis. Front Physiol 7:259. doi:10.3389/fphys.2016.00259
Retamal MA et al (2015) Carbon monoxide: a new player in the redox regulation of connexin hemichannels. IUBMB Life 67:428–437. doi:10.1002/iub.1388
Saez JC, Schalper KA, Retamal MA, Orellana JA, Shoji KF, Bennett MV (2010) Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp Cell Res 316:2377–2389. doi:10.1016/j.yexcr.2010.05.026
Sanchez HA, Mese G, Srinivas M, White TW, Verselis VK (2010) Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J Gen Physiol 136:47–62. doi:10.1085/jgp.201010433
Srisook K, Han SS, Choi HS, Li MH, Ueda H, Kim C, Cha YN (2006) CO from enhanced HO activity or from CORM-2 inhibits both O2- and NO production and downregulates HO-1 expression in LPS-stimulated macrophages. Biochem Pharmacol 71:307
Todd AJ (2010) Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci 11:823–836. doi:10.1038/nrn2947
Wilkinson WJ, Kemp PJ (2011) The carbon monoxide donor, CORM-2, is an antagonist of ATP-gated, human P2X4 receptors. Purinergic Signal 7:57–64. doi:10.1007/s11302-010-9213-8
Zhuang ZY et al (2006) A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci 26:3551–3560. doi:10.1523/JNEUROSCI.5290-05.2006
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All rats were housed in specific pathogen-free conditions, and all experiments were performed with approval of the institutional animal care and use committee of Xi’an Jiaotong University.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Wang, H., Sun, X. Carbon Monoxide-Releasing Molecule-2 Inhibits Connexin 43-Hemichannel Activity in Spinal Cord Astrocytes to Attenuate Neuropathic Pain. J Mol Neurosci 63, 58–69 (2017). https://doi.org/10.1007/s12031-017-0957-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-017-0957-2