
Abstract
The neuroprotective action of puerarin in Parkinson’s disease (PD) models has been well investigated. However, the mechanisms involved in protection have not been completely understood. G protein-coupled receptor 30 (GPR30) is a G protein-coupled estrogen receptor and considered a potential target in the neuroprotection against PD. In this study, we investigated whether puerarin prevented against 1-methyl-4-phenylpyridinium (MPP+)-induced cell death via GPR30. Our results showed that the GPR30 agonist, G1, exhibited puerarin-mediated neuroprotection against MPP+-induced cell death of SH-SY5Y cells. This protective action was reversed by the GPR30 antagonist. Moreover, a time- and concentration-dependent effect of puerarin on GPR30 expression was verified at the protein level but not at the mRNA level. Further, we showed that an mTor-dependent new GPR30 synthesis contributed to the protection conferred by puerarin. Finally, glial cell line-derived neurotrophic factor (GDNF) levels were enhanced by puerarin and G1 in both control and MPP+-lesioned cells via GPR30. Taken together, our data strongly suggest that puerarin prevents MPP+-induced cell death via facilitating GPR30 expression and GDNF release.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a severe neurodegenerative disease symptomized by both motor and non-motor dysfunctions. Dopaminergic neuronal death was considered the major pathological cause for these deficits (Surmeier et al. 2010). Mitochondrial membrane impairment, radical oxidative stress, and dysfunction of protein degradation systems are considered the three major causative factors for dopaminergic cell death (Malkus et al. 2009). Compounds with the potential to reverse the impairment of mitochondrial membrane potential, antagonize oxidative stress, or promote the degradation of accumulated proteins are effective in the prevention of neuronal death (Cheng et al. 2009; Javed et al. 2016; Ojha et al. 2016).
G protein-coupled receptor 30 (GPR30) is a G protein-coupled estrogen receptor mainly located on the cell membrane. GPR30 is different from the traditional estrogen receptors for its location and mechanism of action (Jacenik et al. 2016). The classical estrogen receptors (ERα and ERβ) mainly function through their genomic responses to 17β-estradiol (E2), whereas GPR30 activation is mediated by non-genomic responses (Tran et al. 2016). E2 is reportedly neuroprotective as per various experimental and clinical studies (Al Sweidi et al. 2012; Lebesgue et al. 2009). However, long-term clinical utility of E2 is hampered by elevated risk for breast and uterine cancers owing to its genomic nuclear response. G1, a GPR30 agonist, shows high selectivity in GPR30 activation, thus avoiding the genomic response and adverse effects. E2 along with some phytoestrogens are also known to activate GPR30 to exert their biological activities (Kim et al. 2016; Lee et al. 2014). Interestingly, GPR30 shows neuroprotective functions (Tang et al. 2014) as well as contributing to hippocampal synaptic plasticity (Briz et al. 2015a). In PD, GPR30 activation also prevented dopaminergic cell death likely through the activation of striatal Akt signaling and increase in Bcl-2 and brain-derived neurotrophic factor (BDNF) levels (Bourque et al. 2013; Bourque et al. 2015; Cote et al. 2015).
A variety of components extracted from Chinese herbs have neuroprotective action in PD (Cheng et al. 2009; Zhao et al. 2016). Puerarin (C21H20O9) is one such active component, which was purified from Pueraria lobata (Willd.) and Ohwi Pueraria thomsonii Benth. Puerarin-mediated neuroprotection has been well documented both in in vitro and in vivo PD models (Cheng et al. 2009; Zhu et al. 2010; Zou et al. 2013) likely through apoptosis inhibition and release of anti-oxidant and neurotropic factors (Zhu et al. 2014). However, the exact target involved in this mechanism is not well understood. With a chemical structure of isoflavone, puerarin also exhibits neuroprotection via its weak estrogen-like activity (Ji et al. 2013; Zou et al. 2013). Glial cell line-derived neurotrophic factor (GDNF) is a small protein that potently promotes the survival of many neuron types, especially dopaminergic neurons. We previously reported that puerarin facilitated GDNF expression in a 6-OHDA-induced PD model (Zhu et al. 2012). In this study, we further investigate the exact target involved in puerarin-mediated neuroprotection in 1-methyl-4-phenylpyridinium (MPP+)-induced cell death and GDNF decrease.
Materials and Methods
Reagents
The companies from which antibodies were purchased are listed as follows: anti-phospho-mTor (Ser-2448, Santa Cruz, USA), anti-mTor (Santa Cruz, USA), anti-phospho-p70S6 K (CST, USA), anti-p70S6 K (CST, USA), anti-GPR30 (Novus Biologicals, USA), and anti-actin (CST, USA). Puerarin, MPP+, and 3-(4,5-cimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma (St. Louis, MO, USA).
Groups and Drug Treatment
SH-SY5Y cell line (ATCC, USA) was cultured in an incubator with 5 % CO2 at 37 °C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum (Hyclone, Logan, UT), 100 U/ml penicillin, and 100 mg/ml streptomycin. The following study groups were included: control, 1 mM MPP+, 1–100 μM puerarin + MPP+, G1 + MPP+, GPR30 antagonist (G15) + 50 μM puerarin + MPP+, G1, G15, and puerarin groups. The cells were incubated with puerarin for 3 h before MPP+ insult; cells in the G15 group underwent pre-treatment with G15 prior to puerarin treatment. Puerarin, G1, and G15 were dissolved in dimethyl sulfoxide (DMSO; final concentration less than 0.1 %). All the experimental protocols were approved by the ethics committee of North China University of Science and Technology.
Cell Viability
SH-SY5Y cells in logarithmic growth phase were seeded in 96-well culture plates. After treatment with the indicated drugs for 48 h, 20 μl MTT (5 mg/ml) was added to the 200-μl culture medium of each well. Four hours later, the medium was removed, and 150-μl DMSO was added into each well in order to dissolve the precipitation. The absorbance (A) was measured at a wavelength of 490 nm using an automated microplate reader (Multiskan FC, Thermo Scientific). Cell viability was calculated by the following formula: cell viability (%) = average absorbance of treated group/average absorbance of control group × 100 %.
Total RNA Extraction and Semi-Quantitative PCR
After treatment with the indicated drugs, the cells were collected and total RNA was isolated using TRIzol Reagent according to the manufacturer’s instructions (Thermo Fisher Scientific, USA). Briefly, 2 μg RNA was reverse-transcripted using the reverse-transcription system (Promega Corporation, USA). The following primers (synthesized by Shenggong, Shanghai) were employed for GPR30 amplification:
-
Forward 5′-GACGACCTCAACGCACAGTA-3′;
-
Reverse 5′-AGGAGTCCCATGATGAGATTGT-3′.
Actin primers were as follows:
-
Forward 5′-AAGGACTCCTATAGTGGGTGACGA-3′
-
Reverse 5′-ATCTTCTCCATGTCGTCCCAGTTG-3′.
PCR amplification was carried out under the following conditions: an initial holding at 95 °C for 10 s followed by 40 cycles at 95 °C for 5 s and 60 °C for 45 s.
Immunoblot Analysis
The extraction of cytoplasmic protein was performed according to the Cytoplasmic Protein Extraction Kit (Beyotime, Jiangsu, China). After treatment with the indicated drugs, the cells were washed with 1-ml ice-cold PBS, collected, and centrifuged for 5 min at 1200 rpm at 4 °C. The pellet was dissolved with cytoplasmic protein extraction agent A supplemented with 1 mM PMSF. After a 5-s vortex, the tubes were incubated for 10–15 min on ice to promote cell lysis. Then, the cytoplasmic protein extraction agent B was added. The samples were then centrifuged for 5 min at 14,000g at 4 °C, and the supernatant was collected for further analysis.
Protein concentration was quantified by the BCA method (Beyotime, Jiangsu, China), and an equal amount of protein (20 μg) was loaded onto 10 % sodium dodecyl sulfate-polyacrylamide gels for electrophoresis (SDS-PAGE). After electrophoresis, proteins were transferred onto a nitrocellulose membrane. Non-specific protein binding was blocked by 4 % defat milk. The membranes were incubated with anti-phospho-mTor (Ser-2448, 1:1000), anti-mTor (1:1000), anti-phospho-S6 K (1:1000), anti-S6 K (1:1000), anti-GPR30 (1:1000), and anti-actin (1:5000) at 4 °C overnight. After washing, the membranes were incubated with a secondary antibody (peroxidase-labeled anti-mouse antibody; 1:5000 dilution). The signal was detected using an enhanced chemiluminescence detection kit (Amersham ECL RPN 2106 Kit, Amersham Pharmacia Biotech, QC, Canada).
De novo protein synthesis was determined by using metabolic labeling with Click-iT L-azidohomoalanine (AHA, Molecular Probes) as previous described, with minor modification (Wang et al. 2014). The cells were pre-incubated with 500 mM AHA in the absence and presence of 1 μM rapamycin for 20 min and then treated with 50 μM puerarin for 3 h. After rinsing with PBS, the protein was isolated and samples were incubated with phosphine-PEG3-Biotin (Thermo) for 4 h at 37 °C to conjugate biotin to AHA-containing proteins. Samples were then eluted on Zeba Spin desalting columns (7 K MWCO; Thermo) to remove the excess of phosphine-PEG3-biotin. Equal amounts of proteins were incubated with GPR30 antibody (1:100) overnight at 4 °C. Subsequently, 50-ml protein A-sepharose beads (1:1, Sigma) were added to each sample and incubated for 1 h at 4 °C with gentle rocking. After three washes in PBS, samples were processed for SDS-PAGE and western blotting. IRDye 800CW streptavidin (1:2000, LI-COR Biosciences) was used to detect biotin-conjugated (newly synthesized) GPR30. Anti-GPR30 antibody (1:1000; Novus Biologicals, USA) was used to detect total immunoprecipitated GPR30.
Immunochemical Staining
After treatment with indicated drugs, the cells were fixed in 4 % paraformaldehyde. Non-specific staining was blocked by 0.1 M PBS containing 10 % goat serum with 0.4 % Triton X-100. The cells were mounted on a coverslip and incubated with anti-GPR30 antibody (1:400) in 0.1 M PBS containing 5 % goat serum and 0.4 % Triton X-100 overnight at 4 °C. Sections were washed three times (15 min each) in PBS and incubated with Alexa Fluor 594 goat anti-rabbit IgG (Life Technologies, USA) for 2 h at room temperature. Fluorescent images were captured using the Olympus fluorescence microscopy (Olympus, Japan).
GDNF Assay
SH-SY5Y cells in logarithmic growth phase were seeded in a 48-well culture plate. After treatment with the indicated drugs for 48 h, a 100-μl aliquot of the supernatant was analyzed using enzyme-linked immunosorbant assay (Rat GDNF PicoKine™ ELISA Kit, Boster, CA, USA). The absorbance was determined in a microplate reader at 450-nm wavelength (Multiskan FC, Thermo Scientific).
Statistical Analysis
The values are presented as means ± standard error of mean (SEM). Statistical analyses of the data were performed using Student’s t test, one-way analysis of variance, and Newman–Keuls multiple comparisons. P < 0.05 was considered statistically significant for all analyses.
Results
Puerarin Prevented MPP+-Induced Cell Death via Activating GPR30
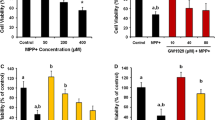
After treatment with 1 mM MPP+ for 48 h, the cell viability was about 60 %. Puerarin at concentrations of 50 and 100 μM significantly decreased MPP+-induced cell loss. Interestingly, we found that the effect of 50 μM puerarin was blocked by G15 (1 μM) (Fig. 1a). Moreover, pre-treatment with the GPR30 agonist G1 (2 μM) also decreased MPP+-induced cell death. No difference in cell viability was observed in the control, 50-μM puerarin, G1, or G15 alone groups (Fig. 1b). These data showed that puerarin moderated MPP+-induced cell death via GPR30 activation.

Puerarin prevents against MPP+-induced SH-SY5Y cell death via activating GPR30. a Puerarin (1–100 μM) prevents against MPP+-induced SH-SY5Y cell death in a concentration-dependent manner. G1 prevents against MPP+-induced SH-SY5Y cell death, while G15 blocks the effects of puerarin on MPP+-induced SH-SY5Y cell death. b Puerarin (50 μM), G1, or G15 alone did not affect the cell viability. Data are presented as mean ± SEM with six repeats. *p < 0.05 compared with control, # p < 0.05 compared with MPP+, $ p < 0.05 compared with 50 μM puerarin + MPP+. Pue puerarin, G1 GPR30 agonist, G15 GPR30 antagonist
Puerarin Activated mTor Signaling Pathway to Increase GPR30 Expression
As shown in Fig. 2a, puerarin promoted GPR30 expression in SH-SY5Y cells in a concentration-dependent manner. Puerarin (10 μM) slightly increased GPR30 expression (1.2-fold) after 24-h treatment, while it enhanced GPR30 expression to peak level (1.5-fold) at 50 μM. To verify the time-dependent effect of puerarin on GPR30 expression, we selected a concentration of 50 μM puerarin in the experiment. As shown in Fig. 2b, GPR30 expression was enhanced 3 h after 50-μM puerarin treatment, and the effects lasted for at least 24 h. By contrast, GPR30 messenger RNA (mRNA) level was not influenced by puerarin application (Fig. 2c, d). In addition, immunofluorescent staining was also used to detect GPR30 expression after puerarin treatment. As shown in Fig. 2e, GPR30 was extensively expressed in the cell membrane or cytoplasm but not in the nucleus. After exposure of the cells in puerarin, GPR30 was markedly up-regulated at both the 3- and 6-h time points.

Puerarin up-regulates GPR30 protein expression. a Puerarin increases the protein levels of GPR30 in a concentration-dependent manner (24-h treatment). b Puerarin (50 μM) increases GPR30 expression in a time-dependent manner. c Different concentrations of puerarin (1–100 μM) did not affect GPR30 expression detected by real-time PCR (24-h treatment). d Treatment with puerarin (50 μM) for different times (3–24 h) did not affect GPR30 expression detected by real-time PCR. e Immunofluorescence staining detecting GPR30 expression 3 and 6 h after puerarin treatment. Scale bar 10 μm. Data are presented as mean ± SEM with six repeats. *p < 0.05 compared with control
The signaling pathway involved in the puerarin effect was also accessed. As shown in Fig. 3a, b, p-mTor was significantly activated 3 h after puerarin treatment. The downstream effector of mTor, p70S6K, was also significantly phosphorylated. Rapamycin pre-treatment attenuated the phosphorylation of mTor and p70S6K. By contrast, puerarin or rapamycin alone did not affect the total mTor and p70S6 K levels.

Puerarin up-regulates GPR30 expression via activation of the mTor signaling pathway. a Puerarin treatment (50 μM) activates the p-mTor pathway. Representative blots of p-mTor, p-p70S6K, mTor, and p70S6K. b Quantification data of p-mTor/mTor. c Quantification data of p-p70S6K/p70S6K. d Puerarin treatment promotes newly synthesized proteins. e Puerarin treatment promotes newly synthesized GPR30. upper panel Representative blots and lower panel quantification data. Data are presented as mean ± SEM with six repeats. *p < 0.05 compared with control, # p < 0.05 compared with corresponding DMSO. Pue puerarin, Rap rapamycin, NL non-labeling
In order to examine whether puerarin promoted de novo GPR30 synthesis, we performed experiments in SH-SY5Y cells using a modified amino acid, L-azidohomoalanine, which is incorporated into newly synthesized proteins and can later be labeled with biotin. Total GPR30 was immunoprecipitated after metabolic labeling of nascent proteins. Newly synthesized (biotin labeled) and total GPR30 were then detected using infrared dye-conjugated streptavidin and anti-GPR30 antibody, respectively. Treatment of the cells with puerarin for 3 h significantly increased the levels of newly synthesized GPR30 as compared with controls (Fig. 3c). Rapamycin also blocked the newly synthesized GPR30 (Fig. 3d). These results further supported that puerarin incubation stimulated GPR30 protein expression, possibly through activating the mTor signaling pathway.
We also detected the effect of puerarin on GPR30 expression in MPP+-lesioned cells. As shown in Fig. 4a, MPP+ did not affect GPR30 expression, while puerarin remarkably increased GPR30 in MPP+-treated cells. As rapamycin blocked the new synthesis of GPR30, it should ideally also block the effect of puerarin on MPP+-induced cell death. As confirmed by MTT assay, puerarin protection was blocked by rapamycin pre-treatment (Fig. 4b). In addition, a protein synthesis inhibitor (cycloheximide, 1 μM) was also found to block the puerarin-mediated protection (Fig. 4c). These data suggested that the new protein synthesis of GPR30 was involved in puerarin-mediated protection of MPP+-induced cell death.

Puerarin prevents against MPP+-induced cell death via promoting GPR30 expression. a Puerarin promotes GPR30 expression in MPP+-induced cells. upper panel Representative blots and lower panel quantification data. b Rapamycin blocks puerarin-mediated protection of MPP+-induced cell death. c Cycloheximide blocks puerarin-mediated protection of MPP+-induced cell death. *p < 0.05 compared with MPP+, # p < 0.05 compared with puerarin + MPP+. Pue puerarin, Rap rapamycin, CXM cycloheximide
GPR30 Activation Prevented MPP+-Induced Decrease of GDNF
GDNF levels were detected after the indicated drug treatments in SH-SY5Y cells. We found that incubation with MPP+ decreased the GDNF level. Puerarin or GPR30 agonist activated GDNF level not only in normal but also in MPP+-induced SH-SY5Y cells (Fig. 5). Moreover, G15 blocked the effect of puerarin on GDNF expression in SH-SY5Y cells that underwent MPP+ treatment.

GPR30 activation by puerarin stimulates GDNF release. GPR30 agonist has similar effect as puerarin, while GPR30 antagonist blocks the effect of puerarin. G1 or puerarin alone does not affect GDNF in SH-SY5Y cells. *p < 0.05 compared with control, # p < 0.05 compared with MPP+, $ p < 0.05 compared with MPP+. Pue puerarin, G1 GPR30 agonist, G15 GPR30 antagonist
Discussion
In this study, we report the novel action of puerarin via GPR30 expression in MPP+-treated SH-SY5Y cells. GPR30 was up-regulated at the protein level, but not in the mRNA level, and this up-regulation of GPR30 played a critical role in preventing MPP+-induced cell death. We believe that GPR30 could be a potential target for PD therapy, consistent with previous studies (Bourque et al. 2013; Tang et al. 2014).
Similarly, consistent with our previous study, others have shown that puerarin prevented against neurotoxin-induced dopaminergic neuronal death (Zhu et al. 2010; Zhu et al. 2014; Zou et al. 2013). Firstly, puerarin regulates proteasome activity to facilitate the degradation of ubiquitinated proteins and α-synuclein (Cheng et al. 2009). Secondly, puerarin possesses anti-oxidative activity to antagonize the radical oxidative stress and reverses the impairment of mitochondrial membrane potential to prevent apoptosis (Cheng et al. 2011). Finally, we found that puerarin promoted Akt phosphorylation, and this process prohibited p53 protein nuclear translocation (Zhu et al. 2012). In this study, we showed that puerarin functions via facilitating GPR30 expression. As reported in previous studies, activation of GPR30 could stimulate Akt phosphorylation to exert its biological action (Ge et al. 2013; Yang et al. 2016). We previously also reported that puerarin activated the Akt signaling pathway to prohibit MPP+-induced cell death (Zhu et al. 2012). In this study, it is possible that puerarin activated Akt by facilitating GPR30, based on previous publication (Bourque et al. 2014). In addition, GPR30 also enhances melanogenesis via cAMP-protein kinase pathway (Sun et al. 2016).
Because puerarin has an estrogen-like structure, it could function by directly binding to GPR30 (Zhang et al. 2005; Zheng et al. 2002). Although we could not exclude whether puerarin acted as a direct agonist to GPR30 to exhibit its effect, its application increased GPR30 expression in both normal and MPP+-treated SH-SY5Y cells. Moreover, new synthesis of GPR30 contributed to puerarin-mediated protection, as both rapamycin and protein synthesis inhibitors blocked this action. Concurrently, the mTor pathway was activated upon puerarin treatment, and this signaling pathway regulated protein synthesis. Using a system to detect the newly synthesized proteins, we found that puerarin application increased protein synthesis, including that of GPR30. Moreover, new synthesis of GPR30 was blocked by an mTor inhibitor (Briz et al. 2015b). To the best of our knowledge, this is the first report implicating that phytoestrogens could activate protein synthesis similar to E2 (Tuscher et al. 2016; Xing et al. 2015). Most importantly, MPP+-induced cell death was mitigated by rapamycin application. Taken together, puerarin probably facilitates the Akt/mTor pathway to stimulate GPR30, and this process prevents MPP+-induced cell death (Bourque et al. 2014).
The question of how new synthesis of GPR30 was initiated is still debatable. It is possible that puerarin functions through binding to endogenous GPR30 to elicit the synthesis of GPR30 and membrane insertion. Thus, a feedback loop might enhance the activity of GPR30. It is also possible that puerarin activates the Akt/mTor pathway and subsequently stimulates GPR30 expression. We could thus conclude that the new synthesis of GPR30 performs critical roles in preventing MPP+-induced cell death. To clearly demonstrate these mechanisms, the endogenous GPR30 should be silenced to investigate the potential roles of endogenous GPR30.
GDNF is an important neurotropic factor. Several studies have shown its roles performed in regulating dopaminergic cell survival (d’Anglemont de Tassigny et al. 2015; Gao et al. 2016). Although several practical problems still exist regarding the viral vector delivery of neurotrophic factors for PD therapy (Kelly et al. 2015), adenovirus-, adeno-associated virus-, and lentivirus-mediated GDNF expressions were reportedly useful in PD models (Chen et al. 2014; Kells et al. 2010; Lu-Nguyen et al. 2014). In our previous study, we also found that GDNF expression was depleted in an 6-OHDA-induced PD model and in an MPTP-insulted model, and puerarin pre-treatment was able to reverse the decrease of GDNF in a PD model (Zhu et al. 2010). In this study, we further detected GDNF expression to observe the potential function of GPR30 in GNDF release. The up-regulation of GDNF is crucial for GPR30-mediated protection in PD (Bessa et al. 2015).
Conclusion
We showed a new puerarin-mediated protective mechanism in an MPP+-induced PD model. Puerarin stimulated GPR30 synthesis to exhibit its neuroprotection through activating the mTor pathway. Moreover, GDNF was activated following GPR30 activation. These data implicated that GPR30 served as a potential target for PD treatment.
References
Al Sweidi S, Sanchez MG, Bourque M, Morissette M, Dluzen D, Di Paolo T (2012) Oestrogen receptors and signalling pathways: implications for neuroprotective effects of sex steroids in Parkinson’s disease. J Neuroendocrinol 24:48–61
Bessa A, Campos FL, Videira RA, Mendes-Oliveira J, Bessa-Neto D, Baltazar G (2015) GPER: a new tool to protect dopaminergic neurons? Biochim Biophys Acta 1852:2035–2041
Bourque M, Morissette M, Cote M, Soulet D, Di Paolo T (2013) Implication of GPER1 in neuroprotection in a mouse model of Parkinson’s disease. Neurobiol Aging 34:887–901
Bourque M, Morissette M, Di Paolo T (2014) Raloxifene activates G protein-coupled estrogen receptor 1/Akt signaling to protect dopamine neurons in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice. Neurobiol Aging 35:2347–2356
Bourque M, Morissette M, Di Paolo T (2015) Neuroprotection in parkinsonian-treated mice via estrogen receptor alpha activation requires G protein-coupled estrogen receptor 1. Neuropharmacology 95:343–352
Briz V, Liu Y, Zhu G, Bi X, Baudry M (2015a) A novel form of synaptic plasticity in field CA3 of hippocampus requires GPER1 activation and BDNF release. J Cell Biol 210:1225–1237
Briz V, Zhu G, Wang Y, Liu Y, Avetisyan M, Bi X, Baudry M (2015b) Activity-dependent rapid local RhoA synthesis is required for hippocampal synaptic plasticity. J Neurosci 35:2269–2282
Chen SS, Yang C, Hao F, Li C, Lu T, Zhao LR, Duan WM (2014) Intrastriatal GDNF gene transfer by inducible lentivirus vectors protects dopaminergic neurons in a rat model of parkinsonism. Exp Neurol 261:87–96
Cheng Y, Zhu G, Guan Y, Liu Y, Hu Y, Li Q (2011) Protective effects of puerarin against 1-methyl-4-phenylpyridinium-induced mitochondrial apoptotic death in differentiated SH-SY5Y cells. Zhongguo Zhong Yao Za Zhi 36:1222–1226
Cheng YF, Zhu GQ, Wang M, Cheng H, Zhou A, Wang N, Fang N, Wang XC, Xiao XQ, Chen ZW, Li QL (2009) Involvement of ubiquitin proteasome system in protective mechanisms of puerarin to MPP(+)-elicited apoptosis. Neurosci Res 63:52–58
Cote M, Bourque M, Poirier AA, Aube B, Morissette M, Di Paolo T, Soulet D (2015) GPER1-mediated immunomodulation and neuroprotection in the myenteric plexus of a mouse model of Parkinson’s disease. Neurobiol Dis 82:99–113
d’Anglemont de Tassigny X, Pascual A, Lopez-Barneo J (2015) GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson’s disease Front Neuroanat 9:10
Gao J, Kang XY, Sun S, Li L, Zhang BL, Li YQ, Gao DS (2016) Transcription factor Six2 mediates the protection of GDNF on 6-OHDA lesioned dopaminergic neurons by regulating Smurf1 expression. Cell Death Dis 7:e2217
Ge X, Guo R, Qiao Y, Zhang Y, Lei J, Wang X, Li L, Hu D (2013) The G protein-coupled receptor GPR30 mediates the nontranscriptional effect of estrogen on the activation of PI3K/Akt pathway in endometrial cancer cells. Int J Gynecol Cancer 23:52–59
Jacenik D, Cygankiewicz AI, Krajewska WM (2016) The G protein-coupled estrogen receptor as a modulator of neoplastic transformation. Mol Cell Endocrinol 429:10–18
Javed H, Azimullah S, Abul Khair SB, Ojha S, Haque ME (2016) Neuroprotective effect of nerolidol against neuroinflammation and oxidative stress induced by rotenone. BMC Neurosci 17:58
Ji M, Liu Y, Yang S, Zhai D, Zhang D, Bai L, Wang Z, Yu J, Yu C, Cai Z (2013) Puerarin suppresses proliferation of endometriotic stromal cells in part via differential recruitment of nuclear receptor coregulators to estrogen receptor-alpha. J Steroid Biochem Mol Biol 138:421–426
Kells AP, Eberling J, Su X, Pivirotto P, Bringas J, Hadaczek P, Narrow WC, Bowers WJ, Federoff HJ, Forsayeth J, Bankiewicz KS (2010) Regeneration of the MPTP-lesioned dopaminergic system after convection-enhanced delivery of AAV2-GDNF. J Neurosci 30:9567–9577
Kelly MJ, O’Keeffe GW, Sullivan AM (2015) Viral vector delivery of neurotrophic factors for Parkinson’s disease therapy. Expert Rev Mol Med 17:e8
Kim J, Szinte JS, Boulware MI, Frick KM (2016) 17beta-estradiol and agonism of G-protein-coupled estrogen receptor enhance hippocampal memory via different cell-signaling mechanisms. J Neurosci 36:3309–3321
Lebesgue D, Chevaleyre V, Zukin RS, Etgen AM (2009) Estradiol rescues neurons from global ischemia-induced cell death: multiple cellular pathways of neuroprotection. Steroids 74:555–561
Lee TM, Lin SZ, Chang NC (2014) Both GPER and membrane oestrogen receptor-alpha activation protect ventricular remodelling in 17beta oestradiol-treated ovariectomized infarcted rats. J Cell Mol Med 18:2454–2465
Lu-Nguyen NB, Broadstock M, Schliesser MG, Bartholomae CC, von Kalle C, Schmidt M, Yanez-Munoz RJ (2014) Transgenic expression of human glial cell line-derived neurotrophic factor from integration-deficient lentiviral vectors is neuroprotective in a rodent model of Parkinson’s disease. Hum Gene Ther 25:631–641
Malkus KA, Tsika E, Ischiropoulos H (2009) Oxidative modifications, mitochondrial dysfunction, and impaired protein degradation in Parkinson’s disease: how neurons are lost in the Bermuda triangle. Mol Neurodegener 4:24
Ojha S, Javed H, Azimullah S, Haque ME (2016) Beta-caryophyllene, a phytocannabinoid attenuates oxidative stress, neuroinflammation, glial activation, and salvages dopaminergic neurons in a rat model of Parkinson disease. Mol Cell Biochem 418:59–70
Sun M, Xie HF, Tang Y, Lin SQ, Li JM, Sun SN, Hu XL, Huang YX, Shi W, Jian D ( 2016) G proteincoupled estrogen receptor enhances melanogenesis via cAMP-protein kinase (PKA) by upregulating microphthalmia-related transcription factor-tyrosinase in melanoma. J Steroid Biochem Mol Biol 2016 Jul 1. pii: S0960-0760(16)30195–9. doi:10.1016/j.jsbmb.2016.06.012
Surmeier DJ, Guzman JN, Sanchez-Padilla J, Goldberg JA (2010) What causes the death of dopaminergic neurons in Parkinson’s disease? Prog Brain Res 183:59–77
Tang H, Zhang Q, Yang L, Dong Y, Khan M, Yang F, Brann DW, Wang R (2014) GPR30 mediates estrogen rapid signaling and neuroprotection. Mol Cell Endocrinol 387:52–58
Tran QK, Firkins R, Giles J, Francis S, Matnishian V, Tran P, VerMeer M, Jasurda J, Burgard MA, Gebert- Oberle B (2016) Estrogen enhances linkage in the vascular endothelial calmodulin network via a feedforward mechanism at the G protein-coupled estrogen receptor 1. J Biol Chem 291(20):10805–23
Tuscher JJ, Luine V, Frankfurt M, Frick KM (2016) Estradiol-mediated spine changes in the dorsal hippocampus and medial prefrontal cortex of ovariectomized female mice depend on ERK and mTOR activation in the dorsal hippocampus. J Neurosci 36:1483–1489
Wang Y, Zhu G, Briz V, Hsu YT, Bi X, Baudry M (2014) A molecular brake controls the magnitude of long-term potentiation. Nat Commun 5:3051
Xing L, Esau C, Trudeau VL (2015) Direct regulation of aromatase B expression by 17beta-estradiol and dopamine D1 receptor agonist in adult radial glial cells. Front Neurosci 9:504
Yang WR, Zhu FW, Zhang JJ, Wang Y, Zhang JH, Lu C, Wang XZ (2016) PI3K/Akt activated by GPR30 and Src regulates 17beta-estradiol-induced cultured immature boar Sertoli cells proliferation. Reprod Sci 2016 May 24. pii: 1933719116649696. doi:10.1177/1933719116649696
Zhang Y, Chen J, Zhang C, Wu W, Liang X (2005) Analysis of the estrogenic components in kudzu root by bioassay and high performance liquid chromatography. J Steroid Biochem Mol Biol 94:375–381
Zhao Q, Ye J, Wei N, Fong C, Dong X (2016) Protection against MPP-induced neurotoxicity in SH-SY5Y cells by tormentic acid via the activation of PI3-K/Akt/GSK3beta pathway. Neurochem Int 97:117–123
Zheng G, Zhang X, Zheng J, Meng Q, Zheng D (2002) Estrogen-like effects of puerarin and total isoflavones from Pueraria lobata. Zhong Yao Cai 25:566–568
Zhu G, Wang X, Chen Y, Yang S, Cheng H, Wang N, Li Q (2010) Puerarin protects dopaminergic neurons against 6-hydroxydopamine neurotoxicity via inhibiting apoptosis and upregulating glial cell line-derived neurotrophic factor in a rat model of Parkinson’s disease. Planta Med 76:1820–1826
Zhu G, Wang X, Wu S, Li Q (2012) Involvement of activation of PI3K/Akt pathway in the protective effects of puerarin against MPP+-induced human neuroblastoma SH-SY5Y cell death. Neurochem Int 60:400–408
Zhu G, Wang X, Wu S, Li X, Li Q (2014) Neuroprotective effects of puerarin on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine induced Parkinson’s disease model in mice. Phytother Res 28:179–186
Zou Y, Hong B, Fan L, Zhou L, Liu Y, Wu Q, Zhang X, Dong M (2013) Protective effect of puerarin against beta-amyloid-induced oxidative stress in neuronal cultures from rat hippocampus: involvement of the GSK-3beta/Nrf2 signaling pathway. Free Radic Res 47:55–63
Acknowledgments
This work was supported by Natural Science Foundation of Hebei Province, China (Grant No. H2014209300), and Research Project of Administration of Traditional Chinese Medicine of Hebei Province, China (Grant No. 2013180), to YC. This work was also supported by the National Natural Science Foundation of China (81673716, 81601181) to GZ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Yue-Fa Cheng and Guoqi Zhu contributed equally to this work.
Rights and permissions
About this article

Cite this article
Cheng, YF., Zhu, G., Wu, QW. et al. GPR30 Activation Contributes to the Puerarin-Mediated Neuroprotection in MPP+-Induced SH-SY5Y Cell Death. J Mol Neurosci 61, 227–234 (2017). https://doi.org/10.1007/s12031-016-0856-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-016-0856-y