
Abstract
Neuropathic pain results in considerable trouble to people’s physical and mental health. The pathophysiological mechanisms underlying its occurrence and development remain unclear. A large number of experiments show that microRNAs (miRNAs) play a major role in the pathogenesis of neuropathic pain and neuroinflammation resulting from nerve injury. Among various miRNAs, microRNA-221 (miR-221) overexpression has been reported in a chronic constrictive injury (CCI)-induced rat model of neuropathic pain. However, the role of miR-221 in the regulation of neuropathic pain is unknown. In this study, we investigated the potential role and underlying mechanism of miR-221 in regulating neuropathic pain. Our findings show that miR-221 is overexpressed in the spinal cord and the isolated microglia of CCI rats. Intrathecal injection of a miR-221 inhibitor attenuated CCI-induced mechanical allodynia and thermal hyperalgesia, and reduced proinflammatory cytokine expression, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 in CCI rats. Using a dual-luciferase reporter assay, we show that suppressor of cytokine signaling 1 (SOCS1), an important regulator of inflammation, is a direct target of miR-221. Treatment with the miR-221 inhibitor significantly inhibited the expression of SOCS1. Furthermore, the miR-221 inhibitor markedly suppressed the activation of nuclear factor-kappa B (NF-κB) and the p38 mitogen-activated protein kinase (p38 MAPK) signaling pathway. Knockdown of SOCS1 in CCI rats abrogated the inhibitory effect of the miR-221 inhibitor on CCI-induced neuropathic pain and the NF-κB and p38 MAPK signaling pathways. Together, these results suggest that inhibition of miR-221 alleviates neuropathic pain and neuroinflammation through increasing SOCS1 and by inhibiting the NF-κB and p38 MAPK signaling pathways, indicating that miR-221 may be a promising molecular target for the treatment of neuropathic pain.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Neuropathic pain, which may result from neuronal tissue damage or a dysfunction in the nervous system, is characterized by spontaneous pain, allodynia, and hyperalgesia (Dalm et al. 2015) and affects the patients’ quality of life. The accumulating evidence suggests that neuroinflammation is associated with the development of neuropathic pain (Ellis and Bennett 2013). However, the pathophysiological mechanisms underlying its occurrence and development remain unclear, and therapies have also been inadequate, ineffective, or produce potentially severe adverse effects. Therefore, optimal management of neuropathic pain has become a major clinical challenge.
Neuroinflammation is a pathophysiological state associated with neuropathic pain. Following nerve injury, inflammatory cells, such as spinal microglia and astrocytes, are recruited to the site of nerve injury and are activated to release proinflammatory cytokines (e.g., tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6) that exacerbate pain (He et al. 2014). In addition, upregulation of these proinflammatory cytokines is associated with mechanical allodynia and hyperalgesia in a rat pain model (Sacerdote et al. 2013). Nuclear factor-kappa B (NF-κB), a critical transcriptional factor that regulates the inflammatory response, is activated during the development of neuropathic pain (Meunier et al. 2007). The p38 mitogen-activated protein kinase (p38 MAPK) also regulates the release of proinflammatory cytokines, which in turn activate p38 MAPK and contribute to the generation of neuropathic pain (Zhou et al. 2014). Therefore, inhibition of the inflammatory response could be a potential therapeutic strategy for treating neuropathic pain.
MicroRNAs (miRNAs) are small, non-coding RNAs with gene regulatory functions (Ha and Kim 2014). Emerging evidence indicates that miRNA expression in the nervous system is differentially regulated in the development of neuropathic pain (Andersen et al. 2014; Genda et al. 2013; Yu et al. 2011). MicroRNA-221 (miR-221) has been found to be upregulated in the dorsal horn of the spinal cord in chronic constrictive injury (CCI) model rats (Genda et al. 2013). miR-221 regulates NF-κB activity in colorectal cancer by targeting components of the NF-κB pathway (Liu et al. 2014b). In addition, miR-221 directly targets suppressor of cytokine signaling 1 (SOCS1), which is associated with inflammation in cells and animals (Xu et al. 2014). However, how the miR-221 contributes to neuroinflammation and neuropathic pain remains to be identified.
Given that SOCS1 in the spinal cord plays a critical role in neuroinflammation and neuropathic pain (Tan et al. 2015), and that miR-221 is a key regulator of the inflammatory response, we hypothesized that miR-221 may be involved in the pathogenesis of neuropathic pain via regulation of SOCS1 in the spinal cord. Our findings demonstrate that miR-221 not only inhibits the inflammatory response but also ameliorates CCI-induced neuropathic pain through regulating of the SOCS1 signaling pathway.
Materials and Methods
Animals
Sprague–Dawley rats (male, 250 ± 20 g) were provided by the animal center of Henan province (Zhengzhou, China). All animal experimental procedures were approved by the The Second Affiliated Hospital of Zhengzhou University Institutional Animal Care and Use Committee. Throughout the course of the study, rats were housed in cages under a controlled temperature, humidity, and 12-h light/dark cycle with free access to food and water.
Cell Culture
Rat microglia cells were purchased from Sciencell (Carlsbad, CA, USA). Primary microglia were isolated according to a recently reported method (Chen et al. 2014). In brief, rats were euthanized and sacrificed. Then, spinal cords from lumbar enlargements were minced, triturated, filtered, and resuspended in medium containing 20 % fetal bovine serum (FBS) in high glucose in Dulbecco’s modified Eagle’s medium (DMEM). Next, cells were plated into 75-cm2 flasks at 37 °C and 5 % CO2 in a humidified incubator, and the medium was changed once a week until the cells became confluent (approximately 14 days). The flasks were shaken at 180 rpm for 3 h, and the floating cells were collected and identified with Iba1 and CD11b+ staining. The isolated and purified microglia cells in the CD11b+ and Iba1 fraction were cultured in 10 % fetal bovine serum in high-glucose DMEM and were used for further analyses.
Intrathecal Catheter Insertion
Rats were anesthetized via an intraperitoneal injection of chloral hydrate (350 mg/kg). After their heads were fixed on a stereotaxic table under sterile conditions, the occipital muscles of the rats were separated, and the atlanto-occipital membrane was exposed. A small incision was then made on the midline of the membrane using the tip of a fresh 20-gauge Quincke point spinal needle (BD Medical Systems, Franklin Lakes, NJ, USA), leading to the release of clear cerebrospinal fluid. A PE-0402 catheter was slowly inserted 7.8 cm long into the subarachnoid space of the spinal cord to the lumbar enlargement (L4–L5) level according to the method of Yaksh and Rudy (1976). After placement, the catheter was cleared, fixed under the skin, and sealed. Motor deficiency and infection was monitored after the surgery, and rats with any deficits were excluded from further experimentation. A 10-μL dose of Xylocaine (2 %) was injected via the intrathecal catheter to determine catheter location. The rats were allowed to recover for 5 days and then used for the subsequent experiments.
Neuropathic Pain Model
The rat model of CCI was used for neuropathic pain according to the method of Bennett and Xie (1988). In brief, cannula-implanted rats were anesthetized with chloral hydrate (350 mg/kg). The left sciatic nerve was exposed at the mid-thigh level, and then four ligations with 5.0 chromic gut approximately 1 mm apart were tied loosely around the sciatic nerve. The same procedure was applied to the sham-operated group without nerve injury. The wound was sutured in the muscle and skin layers.
Intrathecal Injections
The miR-221 inhibitor, mimic, and its scrambled control were purchased from Genepharma (Shanghai, China). These oligonucleotide sequences (inhibitor: 5′-GAAACCCAGCAGACAAUGUAGCU-3′, mimics: 5′-AGCUACAUUGUCUGCUGGGUUUC/AACCCAGCAGACAAUGUAGCUUU-3′, and scrambled control: 5′-CAGUACUUUUGUGUAGUACAA-3′) were modified with 2′-methyl, glucosinolate skeleton and 3′-cholesterol. Intrathecal injection was used to deliver the miRNA oligonucleotide reagents (10 μL) once daily from days 0 to 5 after CCI using a microinjection syringe through an intrathecally implanted catheter. Throughout the experiment, the L4–5 lumbar segment of the spinal cord was removed for analysis.
Mechanical Withdrawal Threshold
The mechanical withdrawal threshold (MWT) in rats was determined to evaluate mechanical allodynia using an electronic von Frey Anesthesiometer (2390 series, IITC, CA, USA) according to a previously reported method (Mert et al. 2013) 1 day before surgery and on postoperative days 1, 3, 5, 7, and 14. All rats were placed individually inside wire mesh-bottomed Perspex boxes and habituated to the testing environment for 30 min prior to baseline tests. The plantar surface of each mid-hind paw was perpendicularly stimulated by von Frey filaments in a range of 0.1–60 g for 4–6 s with an increasing force (0.02–15 g, Stoelting) until the rat paw twitched. At the time of paw withdrawal, the maximum force was noted. Each rat was tested alternately eight times (interval 30 s). Based on Dixon’s up-down method (Chaplan et al. 1994), the 50 % probability of withdrawal threshold was calculated.
Thermal Withdrawal Latency
In order to evaluate thermal hyperalgesia with the Hargreaves method (Hargreaves et al. 1988), a BME-410C automatic heat pain stimulator (Biomedical Engineering Institute of Chinese Academy of Medical Sciences, Tianjin, China) was used to test the thermal withdrawal latency (TWL). In brief, the rats were individually placed into the Perspex box with a glass surface. After habituation for 30 min, a radiant heat stimulus was applied to the lower extremity plantar surface until the withdrawal response. The pain latency time was recorded. If the rat failed to withdraw its paw, the thermal source was automatically discontinued after 25 s to prevent any significant tissue damage. Each rat was tested six times (interval 6 min).
Dual-Luciferase Reporter Assay
The SOCS1-3′ untranslated region (UTR) containing the miR-221 binding side and the SOCS1-mutated-3′UTR fragment was amplified by PCR and then cloned into the pGL3 luciferase promoter vector (Promega, Madison, WI, USA) to develop the Luc-pGL3-SOCS1-3′UTR and Luc-pGL3-SOCS1-mut-3′UTR vectors. For the luciferase assay in rat microglial cells, cells seeded at the density of 1.0 × 106 cells per well in 24-well plates were co-transfected with Luc-pGL3-SOCS1-3′UTR or Luc-pGL3-SOCS1-mut-3′UTR vector and miR-221 mimic or scrambled miRNA using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Cells were also transfected with pRL-SV40 vector as an internal standard. After transfection for 48 h, cells were harvested and the luciferase activity was measured using a dual-luciferase reporter assay kit (Promega, Madison, WI, USA). The results are expressed as relative luciferase activity (firefly Luc/renilla Luc). All experiments were repeated three times in triplicate.
Western Blotting
Total protein of the L4–5 segments of the spinal cords and cultured cells was extracted and collected after treatment. The BCA protein assay (Pierce, Rockford, IL, USA) was used to determine protein concentrations. Protein samples were electrophoresed in SDS–PAGE gels and transferred onto PVDF membranes. After blocking with 5 % non-fat milk, the membranes were incubated overnight at 4 °C with specific primary antibodies: anti-SOCS1 (1:600; anti-mouse; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-NF-κB, anti-p38 MAPK, anti-phosphorylated NF-κB, and anti-phosphorylated p38 MAPK (1:600; anti-rabbit; Cell Signaling, Beverly, MA, USA), and anti-GAPDH (1:10,000; anti-mouse; Millipore, Billerica, MA, USA). These blots was then incubated with HRP-conjugated goat anti-rabbit or goat anti-mouse antibody (1:10,000 dilution; KPL, Gaithersburg, MA, USA) for 1 h at room temperature, developed in ECL solution, and detected using Pierce ECLWestern Blotting Substrate (Thermo Fisher Scientific, Waltham, MA, USA). The optical density of protein fragments was semi-quantitated using Quantity One software (Bio-Rad, Hercules, CA, USA).
Quantitative Real-Time PCR
Total RNA of the L4–5 segments of the spinal cords and cultured cells was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) and was reverse-transcribed to complementary DNA (cDNA) using M-MLV reverse transcriptase (Clontech, Palo Alto, CA, USA) and the PrimeScript® miRNA cDNA synthesis kit (Perfect Real Time; TaKaRa, Dalian, China) according to the manufacturers’ protocols. The primers shown in Table 1 and a SYBR RT-PCR kit (Takara, Shiga, Japan) were then used to amplify. The relative expression was normalized to an internal control (GAPDH and U6 SnRNA). All reactions were performed in triplicate. Data were analyzed using the 2−ΔΔCt method.
ELISA
Protein samples in the lumbar spinal cords were prepared in the same way as for Western blotting. The concentration of TNF-α, IL-1β, and IL-6 was determined with an ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Statistical Analysis
All data are expressed as the mean ± SD and were analyzed with SPSS 17.0 software. All variables measured in this study were normally distributed, and the groups were compared using the Student’s t test or one-way analysis of variance (ANOVA). Results were considered a significant difference at p < 0.05.
Results
Overexpression of miR-221 in CCI Rat Models
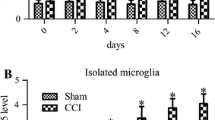
Recent reports (Genda et al. 2013; Yu et al. 2011; Zhu et al. 2011) demonstrated that miR-221 is associated with inflammation and neuropathic pain. However, the relationship between miR-221 and neuropathic pain and neuroinflammation is unknown. Here, we investigated the expression of miR-221 in a CCI-induced rat model of neuropathic pain. miR-221 was significantly upregulated in the spinal cord of the CCI rat model at different times (3, 7, or 14 days) compared with the sham group (Fig. 1a). To further evaluate the involvement of miR-221 in the regulation of neuroinflammation, we determined the expression of miR-221 in isolated microglia from the spinal cord. miR-221 expression was significantly upregulated in isolated microglia from CCI rats when compared with those from the sham group (Fig. 1b).

Overexpression of miR-221 in the CCI rat model. Expression of miR-221 in the spinal cord of the rat model was examined by QRT-PCR at 0, 3, 7, and 14 days after CCI (a). Expression of miR-221 in isolated microglia from the spinal cord was determined at the indicated times (b). a p < 0.05 vs. sham
Intrathecal Injection of a miR-221 Inhibitor Attenuates CCI-Induced Neuropathic Pain Development
To determine whether inhibition of miR-221 affects neuropathic pain development, we delivered a miR-221 inhibitor to CCI rats by intrathecal injection and measured its effect on mechanical allodynia and thermal hyperalgesia. The pain thresholds of the MWT and TWL following inhibition of miR-221 in CCI were significantly different from sham. Meanwhile, the miR-221 inhibitor markedly increased the MWT (Fig. 2a) and TWL (Fig. 2b) values in CCI rats compared with those of the scrambled miRNA group, which indicates that inhibition of miR-221 attenuates neuropathic pain in the CCI rat model.

Intrathecal injection of a miR-221 inhibitor produces an analgesic effect in the CCI rat model. The force of MWT (a) and the latency of TWL (b) were determined at 0, 1, 3, 5, 7, and 14 days post-CCI. CCI + scramble, CCI-operated rats administered with scrambled miRNA by intrathecal injection; CCI + miR-221 inhibitor, CCI-operated rats administered with miR-221 inhibitor by intrathecal injection. a p < 0.05 vs. sham; b p < 0.01 vs. CCI + scramble
Intrathecal Injection of a miR-221 Inhibitor Suppresses CCI-Induced Proinflammatory Cytokine Expression
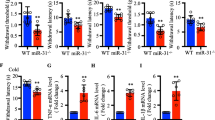
To further determine the relationship between miR-221 and neuropathic pain and neuroinflammation, we measured the expression of proinflammatory cytokines in the L4–5 lumbar segment of the spinal cord of CCI rats treated with the miR-221 inhibitor via quantitative real-time PCR (QRT-PCR) analysis and ELISA. The mRNA expression levels of TNF-α (Fig. 3a), IL-1β (Fig. 3b), and IL-6 (Fig. 3c) were significantly decreased in the miR-221 inhibitor group compared with the scrambled miRNA group. Similarly, miR-221 inhibitor also markedly reduced the protein levels of TNF-α (Fig. 3d), IL-1β (Fig. 3e), and IL-6 (Fig. 3f) in CCI rats.

The miR-221 inhibitor suppresses CCI-induced proinflammatory cytokine expression. The mRNA expression levels of TNF-α (a), IL-1β (b), and IL-6 (c) in the L4–5 lumbar segment of the spinal cord were tested by QRT-PCR at 7 days post-CCI. ELISA was used to detect the protein expression of TNF-α (d), IL-1β (e), and IL-6 (f). a p < 0.05 vs. sham; b p < 0.05 vs. CCI + scramble
SOCS1 Is a Direct Target of miR-221
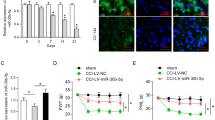
Recent reports showed that miR-221 could regulate the SOCS1 expression by directly targeting the 3′-UTR of the SOCS1 mRNA in Huh7.5.1 cells (Xu et al. 2014). To determine whether SOCS1 is a direct target of miR-221 in rat microglial cells, a dual-luciferase reporter assay was performed. The miR-221 mimics markedly decreased the luciferase activity of groups transfected with pGL3-SOCS1-3′-UTR compared with that of the scrambled, but the miR-221 inhibitor increased the luciferase activity up to the scramble group. In contrast, the miR-221 mimics and the inhibitor had no effect on the luciferase activity of the pGL3-Mut-SOCS1-3′-UTR transfected groups (Fig. 4a). Furthermore, the SOCS1 mRNA and protein expression in CCI were markedly reduced compared with that in the sham group, whereas intrathecal injection of the miR-221 inhibitor significantly reversed the decreased SOCS1 expression levels (Fig. 4b–d). These findings suggest that the SOCS1 gene is a direct target of miR-221 and that inhibition of miR-221 increased the expression of SOCS1.

SOCS1 is a direct target of miR-221. Validation of miR-221 binding to the SOCS1 3′-UTR using a dual-luciferase reporter assay (a). *p < 0.05 vs. scrambled. The SOCS1 mRNA (b) and protein (c) expression levels in the L4–5 lumbar segment of the spinal cord were tested by QRT-PCR and Western blotting at 0, 3, 7, and 14 days post-CCI. SOCS1 protein expression was quantified using Quantity One software after normalization with GAPDH (d). a p < 0.05 vs. sham; b p < 0.05 vs. CCI + scramble or scramble
Knockdown of SOCS1 Abrogates the Inhibitory Effect of miR-221 Inhibitor
To confirm the suppressive role of miR-221 inhibitor on neuropathic pain through direct targeting of SOCS1 in CCI rats, CCI rats were treated with the miR-221 inhibitor and LV-shSOCS1, which directly silences SOCS1 expression. LV-shSOCS1 treatment markedly suppressed SOCS1 expression in the spinal cord of CCI rats, which was significantly increased by miR-221 inhibitor (Fig. 5a, b). Knockdown of SOCS1 significantly reversed the promoting effect of the miR-221 inhibitor on SOCS1 expression in CCI rats (Fig. 5a, b). In addition, knockdown of SOCS1 markedly accentuated mechanical allodynia and thermal hyperalgesia in CCI rats and also blocked the inhibitory effect of the miR-221 inhibitor on neuropathic pain (Fig. 5c, d). Furthermore, knockdown of SOCS1 significantly increased the expression of proinflammatory cytokines, including TNF-α, IL-1β, and IL-6, and also markedly reversed the inhibitory effect of miR-221 inhibitor on the expression of proinflammatory cytokines (Fig. 5e–g). Collectively, these results show that knockdown of SOCS1 abrogated the inhibitory effect of the miR-221 inhibitor on neuropathic pain development and proinflammatory cytokine expression, indicating that the miR-221 inhibitor functions through SOCS1.

Knockdown of SOCS1 abrogates the effect of miR-221 inhibitor. The SOCS1 mRNA (a) and protein (b) expression levels in the L4–5 lumbar segment of the spinal cord were measured by QRT-PCR and Western blotting at 7 days post-CCI. The force of MWT (c) and the latency of TWL (d) were determined at 0, 1, 3, 5, 7, and 14 days after different treatments. The protein expression of TNF-α, IL-1β, and IL-6 (e–g) in the L4–5 lumbar segment of the spinal cord at 7 days post-CCI was tested by ELISA. a p < 0.05 vs. sham; b p < 0.05 vs. CCI + scramble; c p < 0.05 vs. CCI + miR-221 inhibitor
miR-221 Inhibitor Alleviates CCI-Induced Neuropathic Pain via Activation of NF-κB and p38 MAPK
Development of neuropathic pain and neuroinflammation involve the activation of NF-κB and p38 MAPK. To further investigate the underlying molecular mechanism of miR-221 in regulating neuropathic pain, the protein expression of NF-κB and p38 MAPK and the phosphorylation of NF-κB (p-NF-κB) and p38 MAPK (p-p38 MAPK) were measured. Suppression of miR-221 markedly inhibited NF-κB phosphorylation, which was significantly blocked by LV-shSOCS1 (Fig. 6a). In addition, the miR-221 inhibitor also reduced the phosphorylation of p38 MAPK, whereas knockdown of SOCS1 blocked the inhibitory effect of the miR-221 inhibitor on p38 MAPK phosphorylation (Fig. 6b).

The miR-221 inhibitor alleviated CCI-induced neuropathic pain via activation of NF-κB and p38 MAPK. The protein expression levels of NF-κB (a) and p38 MAPK (b) in the L4–5 lumbar segment of the spinal cord were tested by Western blotting at 7 days post-CCI. The relative protein expression of total NF-κB and p38 MAPK and their phosphorylated forms was quantified using Quantity One software after normalization with GAPDH. a p < 0.05 vs. sham; b p < 0.05 vs. CCI + scramble (CCI); c p < 0.05 vs. CCI + miR-221 inhibitor
Discussion
In this study, we show that miR-221 is involved in the regulation of neuropathic pain and neuroinflammation in a CCI rat model. The miR-221 expression levels were markedly increased in the L4–5 lumbar segment of the spinal cord and microglia. Moreover, intrathecal injection of a miR-221 inhibitor significantly attenuated mechanical allodynia and thermal hyperalgesia, reduced proinflammatory cytokine (TNF-α, IL-1β, and IL-6) expression, and suppressed the activation of NF-κB and p38 MAPK signaling pathways in CCI rats. These effects of miR-221 inhibitor were markedly abrogated by knockdown of SOCS1, suggesting that miR-221 regulates neuropathic pain and neuroinflammation through targeting SOCS1.
Alterations in the expression of a large number of miRNAs play an important role in health and disease (Kusuda et al. 2011). Recently, numerous examples of dysregulation in microRNA expression have been reported in patients suffering from painful disorders, such as complex regional pain syndrome, cystitis-induced chronic pain, and irritable bowel disorder (Andersen et al. 2014). Moreover, microRNA expression levels in dorsal root ganglion neurons and the spinal dorsal horn are dysregulated in animal models of inflammatory and neuropathic pain (Genda et al. 2013; Norcini et al. 2014). Here, our findings indicate that miR-221 expression is increased in the L4–5 lumbar segment of the spinal cord in the CCI-induced rat model. These data are consistent with the results obtained using a microarray analysis, which showed that the expression of miR-221 was increased in a neuropathic pain model involving ligation of the sciatic nerve (Genda et al. 2013). Furthermore, ligated sciatic nerve injury led to significant mechanical allodynia and thermal hyperalgesia in a rat model, similar to previous studies (Zhang et al. 2014). Interestingly, pain was markedly attenuated by intrathecal administration of a miR-221 inhibitor, suggesting that inhibition of miR-221 upregulation attenuated neuropathic pain in the CCI rat model.
The inflammatory response within the nervous system plays a major role in the development of neuropathic pain (Liu et al. 2014a; Scholz and Woolf 2007). It was also reported that miR-221 provides an important target for regulating neuroinflammation in microglial cells (Brown and Yin 2013). According to these findings, we isolated microglia from the spinal cord in CCI and assessed the miR-221 expression by QRT-PCR and showed that mRNA expression levels were markedly upregulated. Inhibition of miR-221 upregulation decreased the mRNA and protein expression of proinflammatory cytokines, including TNF-α, IL-1β, and IL-6, suggesting that miR-221 takes part in the inflammatory response in CCI-induced neuropathic pain.
Neuropathic pain-relevant miRNAs affect translation of multiple proteins by binding to their mRNAs and then contribute to the initiation and maintenance of neuropathic pain and neuroinflammation following various types of nerve injury (Andersen et al. 2014; Genda et al. 2013). SOCS-1 is a member of the SOCS family, which includes seven other proteins that share a small C-terminal region of homology with SOCS-1, as well as a central Src homology 2 domain (Losman et al. 1999). SOCS1 is involved in inflammatory reactions by regulating Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathways in the innate immune cells and non-immune cells (Hashimoto et al. 2011; Sachithanandan et al. 2011; Torisu et al. 2008; Yoshimura et al. 2012). The blockade of the JAK/STAT transduction pathway in spinal cord microglial cells and astrocytes during peripheral nerve injury attenuates local inflammation and pain hypersensitivity (Molet et al. 2016; Tsuda et al. 2011). In line with these findings, our results showed that miR-221 directly targeted SOCS1 and then regulated neuropathic pain and neural inflammation during CCI. Knockdown of SOCS1 abrogated the inhibitory effect of the miR-221 inhibitor. In addition, the NF-κB/p38 MAPK pathway is central to the regulation of inflammation by targeting SOCS1 (Baig et al. 2015; Guimaraes et al. 2013), and mimics of miR-221 activated NF-κB in colorectal cancer cells (Liu et al. 2014b). According to these findings, we examined the phosphorylation of NF-κB and p38 MAPK in the spinal cord of the CCI rat model and found that the upregulation of SOCS1 by intrathecal administration of a miR-221 inhibitor diminished NF-κB and p38 MAPK phosphorylation.
Other miRNAs have also been reported to be involved in regulating neuropathic pain and neural inflammation. Upregulation of miR-195, which was examined in the rat model of L5 spinal nerve ligation (PNI), contributed to expression of proinflammatory cytokines in cultured microglia and promoted mechanical and cold hypersensitivity by regulating autophagy after PNI (Shi et al. 2013). miR-124 increased the transition from acute to persistent IL-1β-induced hyperalgesia in LysM-GRK2+/− mice and aggravated the mechanical allodynia in mice with spinal nerve injury (Willemen et al. 2012). A miR-146a-5p mimic attenuated neuropathic pain partly through inhibition of tumor necrosis factor receptor-associated factor 6 and its downstream JNK/CCL2 signaling pathway (Lu et al. 2015).
Here, for the first time, we demonstrate that miR-221 regulates neuropathic pain through targeting SOCS1. Interestingly, a recent study reported that miR-19a regulated the progression of neuropathic pain through targeting SOCS1 in bilateral CCI rats (Wang et al. 2015). Similarly, Tan et al. revealed that inhibition of miR-155 decreased neuropathic pain through upregulation of SOCS1 in the CCI rats (Tan et al. 2015). These studies, as well as our findings, suggest that targeting SOCS1 by using specific miRNAs is a promising method for the treatment of neuropathic pain.
In conclusion, our findings show that inhibition of miR-221 attenuated neuropathic pain and neuroinflammation through increasing SOCS1 and by inhibiting the NF-κB and p38 MAPK signaling pathways. Our study provides evidence that miR-221 may be a promising target for the development of novel therapeutics for the treatment of neuropathic pain. However, the precise mechanism by which miR-221 regulates neuropathic pain and neural inflammation warrants further investigation.
Abbreviations
- miR-221:
-
MicroRNA-221
- SOCS1:
-
Suppressor of cytokine signaling 1
- CCI:
-
Chronic constrictive injury
- UTR:
-
Untranslated region
- NF-κB:
-
Nuclear factor-kappa B
- p38 MAPK:
-
p38 mitogen-activated protein kinase
- TWL:
-
Thermal withdrawal latency
- MWT:
-
Mechanical withdrawal threshold
References
Andersen HH, Duroux M, Gazerani P (2014) MicroRNAs as modulators and biomarkers of inflammatory and neuropathic pain conditions. Neurobiol Dis 71:159–168
Baig MS, Zaichick SV, Mao M, de Abreu AL, Bakhshi FR, Hart PC, Saqib U, Deng J, Chatterjee S, Block ML, et al. (2015) NOS1-derived nitric oxide promotes NF-kappaB transcriptional activity through inhibition of suppressor of cytokine signaling-1. J Exp Med 212:1725–1738
Bennett GJ, Xie YK (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33:87–107
Brown PN, Yin H (2013) PNA-based microRNA inhibitors elicit anti-inflammatory effects in microglia cells. Chem Commun 49:4415–4417
Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63
Chen X, Zhang T, Li CH, Wang W, Luo X, Yu ZY (2014) Primary culture and purification of microglia from rat spinal cord. Neural injury and functional reconstruction 9:455–457
Dalm BD, Reddy CG, Howard MA, Kang S, Brennan TJ (2015) Conditioned place preference and spontaneous dorsal horn neuron activity in chronic constriction injury model in rats. Pain 156:2562–2571
Ellis A, Bennett DL (2013) Neuroinflammation and the generation of neuropathic pain. Br J Anaesth 111:26–37
Genda Y, Arai M, Ishikawa M, Tanaka S, Okabe T, Sakamoto A (2013) MicroRNA changes in the dorsal horn of the spinal cord of rats with chronic constriction injury: A TaqMan(R) Low Density Array study. Int J Mol Med 31:129–137
Guimaraes MR, Leite FR, Spolidorio LC, Kirkwood KL, Rossa C Jr (2013) Curcumin abrogates LPS-induced pro-inflammatory cytokines in RAW 264.7 macrophages. evidence for novel mechanisms involving SOCS-1, -3 and p38 MAPK. Arch Oral Biol 58:1309–1317
Ha M, Kim VN (2014) Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15:509–524
Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32:77–88
Hashimoto M, Hiwatashi K, Ichiyama K, Morita R, Sekiya T, Kimura A, Sugiyama Y, Sibata T, Kuroda K, Takahashi R, et al. (2011) SOCS1 regulates type I/type II NKT cell balance by regulating IFNgamma signaling. Int Immunol 23:165–176
He ZW, Wei W, Li SP, Ling Q, Liao KJ, Wang X (2014) Anti-allodynic effects of obtusifolin and gluco-obtusifolin against inflammatory and neuropathic pain. Biol Pharm Bull 37:1606–1616
Kusuda R, Cadetti F, Ravanelli MI, Sousa TA, Zanon S, De Lucca FL, Lucas G (2011) Differential expression of microRNAs in mouse pain models. Mol Pain 7:17
Liu M, Liao K, Yu C, Li X, Liu S, Yang S (2014a) Puerarin alleviates neuropathic pain by inhibiting neuroinflammation in spinal cord. Mediat Inflamm 2014:485927
Liu S, Sun X, Wang M, Hou Y, Zhan Y, Jiang Y, Liu Z, Cao X, Chen P, Liu Z, et al. (2014b) A microRNA 221- and 222-mediated feedback loop maintains constitutive activation of NFkappaB and STAT3 in colorectal cancer cells. Gastroenterology 147(847–859):e811
Losman JA, Chen XP, Hilton D, Rothman P (1999) Cutting edge: SOCS-1 is a potent inhibitor of IL-4 signal transduction. J Immunol 162:3770–3774
Lu Y, Cao DL, Jiang BC, Yang T, Gao YJ (2015) MicroRNA-146a-5p attenuates neuropathic pain via suppressing TRAF6 signaling in the spinal cord. Brain Behav Immun 49:119–129
Mert T, Gunes Y, Gunay I (2013) Comparison of actions of systemically and locally administrated local anaesthetics in diabetic rats with painful neuropathy. Fundam Clin Pharmacol 27:161–168
Meunier A, Latremoliere A, Dominguez E, Mauborgne A, Philippe S, Hamon M, Mallet J, Benoliel JJ, Pohl M (2007) Lentiviral-mediated targeted NF-kappaB blockade in dorsal spinal cord glia attenuates sciatic nerve injury-induced neuropathic pain in the rat. Mol Ther 15:687–697
Molet J, Mauborgne A, Diallo M, Armand V, Geny D, Villanueva L, Boucher Y, Pohl M (2016) Microglial Janus kinase/signal transduction and activator of transcription 3 pathway activity directly impacts astrocyte and spinal neuron characteristics. J Neurochem 136:133–147
Norcini M, Sideris A, Martin Hernandez LA, Zhang J, Blanck TJ, Recio-Pinto E (2014) An approach to identify microRNAs involved in neuropathic pain following a peripheral nerve injury. Front Neurosci 8:266
Sacerdote P, Franchi S, Moretti S, Castelli M, Procacci P, Magnaghi V, Panerai AE (2013) Cytokine modulation is necessary for efficacious treatment of experimental neuropathic pain. J NeuroImmune Pharmacol 8:202–211
Sachithanandan N, Graham KL, Galic S, Honeyman JE, Fynch SL, Hewitt KA, Steinberg GR, Kay TW (2011) Macrophage deletion of SOCS1 increases sensitivity to LPS and palmitic acid and results in systemic inflammation and hepatic insulin resistance. Diabetes 60:2023–2031
Scholz J, Woolf CJ (2007) The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 10:1361–1368
Shi G, Shi J, Liu K, Liu N, Wang Y, Fu Z, Ding J, Jia L, Yuan W (2013) Increased miR-195 aggravates neuropathic pain by inhibiting autophagy following peripheral nerve injury. Glia 61:504–512
Tan Y, Yang J, Xiang K, Tan Q, Guo Q (2015) Suppression of microRNA-155 attenuates neuropathic pain by regulating SOCS1 signalling pathway. Neurochem Res 40:550–560
Torisu T, Nakaya M, Watanabe S, Hashimoto M, Yoshida H, Chinen T, Yoshida R, Okamoto F, Hanada T, Torisu K, et al. (2008) Suppressor of cytokine signaling 1 protects mice against concanavalin A-induced hepatitis by inhibiting apoptosis. Hepatology 47:1644–1654
Tsuda M, Kohro Y, Yano T, Tsujikawa T, Kitano J, Tozaki-Saitoh H, Koyanagi S, Ohdo S, Ji RR, Salter MW, et al. (2011) JAK-STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain J Neurol 134:1127–1139
Wang C, Jiang Q, Wang M, Li D (2015) MiR-19a targets suppressor of cytokine signaling 1 to modulate the progression of neuropathic pain. Int J Clin Exp Pathol 8:10901–10907
Willemen HL, Huo XJ, Mao-Ying QL, Zijlstra J, Heijnen CJ, Kavelaars A (2012) MicroRNA-124 as a novel treatment for persistent hyperalgesia. J Neuroinflammation 9:143
Xu G, Yang F, Ding CL, Wang J, Zhao P, Wang W, Ren H (2014) MiR-221 accentuates IFNs anti-HCV effect by downregulating SOCS1 and SOCS3. Virology 462-463:343–350
Yaksh TL, Rudy TA (1976) Chronic catheterization of the spinal subarachnoid space. Physiol Behav 17:1031–1036
Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H (2012) SOCS, inflammation, and autoimmunity. Front Immunol 3:20
Yu B, Zhou S, Qian T, Wang Y, Ding F, Gu X (2011) Altered microRNA expression following sciatic nerve resection in dorsal root ganglia of rats. Acta Biochim Biophys Sin 43:909–915
Zhang K, Ramamurthy S, Prihoda TJ, Eckmann MS (2014) Effect of delayed intrathecal administration of capsaicin on neuropathic pain induced by chronic constriction injury of the sciatic nerve in rats. J Pain Res 7:547–554
Zhou C, Shi X, Huang H, Zhu Y, Wu Y (2014) Montelukast attenuates neuropathic pain through inhibiting p38 mitogen-activated protein kinase and nuclear factor-kappa B in a rat model of chronic constriction injury. Anesth Analg 118:1090–1096
Zhu N, Zhang D, Chen S, Liu X, Lin L, Huang X, Guo Z, Liu J, Wang Y, Yuan W, et al. (2011) Endothelial enriched microRNAs regulate angiotensin II-induced endothelial inflammation and migration. Atherosclerosis 215:286–293
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All animal experimental procedures were approved by The Second Affiliated Hospital of Zhengzhou University Institutional Animal Care and Use Committee.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Xia, L., Zhang, Y. & Dong, T. Inhibition of MicroRNA-221 Alleviates Neuropathic Pain Through Targeting Suppressor of Cytokine Signaling 1. J Mol Neurosci 59, 411–420 (2016). https://doi.org/10.1007/s12031-016-0748-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-016-0748-1