
Abstract
Spinal and bulbar muscular atrophy (SBMA) or Kennedy’s disease is an X-linked disease associated with the expansion of the CAG triplet repeat present in exon 1 of the androgen receptor (AR) gene. This results in the production of a mutant AR containing an elongated polyglutamine tract (polyQ) in its N-terminus. Interestingly, the ARpolyQ becomes toxic only after its activation by the natural androgenic ligands, possibly because of aberrant androgen-induced conformational changes of the ARpolyQ, which generate misfolded species. These misfolded ARpolyQ species must be cleared from motoneurons and muscle cells, and this process is mediated by the protein quality control (PQC) system. Experimental evidence suggested that failure of the PQC pathways occurs in disease, leading to ARpolyQ accumulation and toxicity in the target cells. In this review, we summarized the overall impact of mutant and misfolded ARpolyQ on the PQC system and described how molecular chaperones and the degradative pathways (ubiquitin-proteasome system (UPS), the autophagy-lysosome pathway (ALP), and the unfolded protein response (UPR), which activates the endoplasmic reticulum-associated degradation (ERAD)) are differentially affected in SBMA. We also extensively and critically reviewed several molecular and pharmacological approaches proposed to restore a global intracellular activity of the PQC system. Collectively, these data suggest that the fine and delicate equilibrium existing among the different players of the PQC system could be restored in a therapeutic perspective by the synergic/additive activities of compounds designed to tackle sequential or alternative steps of the intracellular defense mechanisms triggered against proteotoxic misfolded species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Almost all inherited and sporadic neurodegenerative diseases are characterized by the presence of proteins that, either because of their natural structure and/or as a consequence of genetic mutation(s), have a tendency to aberrantly fold (misfold), generating uncommon conformations. When this happens, these proteins become prone to oligomerize and/or aggregate. These misfolded proteins, either in their monomeric, oligomeric, or aggregated form, are recognized by the protein quality control (PQC) system, which surveys proteostasis, by facilitating the removal of potentially toxic species and/or attenuating their translation (Carra et al. 2013; Seguin et al. 2014). However, when not properly disposed, these aberrant misfolded species can exert toxic effects, thereby contributing to cell vulnerability and death. In the presence of aberrantly folded species, the PQC system activates the so-called proteotoxic stress response, which involves (a) the chaperone proteins, including members of the heat shock protein (HSP) family, and the two major intracellular degradative systems, comprising the (b) ubiquitin-proteasome system (UPS) and the (c) autophagy-lysosome pathway (ALP). Molecular chaperones recognize the misfolded species and assist their refolding; when this fails, as in the case of misfolded proteins, chaperones assist their clearance by cooperating with the degradative systems. ALP can be subdivided into at least three different pathways, the macroautophagy (herein referred to autophagy), microautophagy, and chaperone-mediated autophagy (CMA). In addition, even if more indirectly, the unfolded protein response (UPR), which involves the endoplasmic reticulum-associated degradation (ERAD), is part of this protective mechanism which aims to reduce proteotoxic stress to neurons. The UPR system works commonly in conjunction with molecular chaperones to determine the fate of misfolded proteins, leading to either refolding of the protein or UPS/ALP-mediated degradation. Imbalance in the PQC system has been described in a number of neurodegenerative diseases (Carra et al. 2012, 2013; Ciechanover and Kwon, 2015; Kakkar et al. 2014; Kampinga and Craig 2010; Klionsky et al. 2012; Senft and Ronai 2015; Xilouri and Stefanis 2015). Here, we will focus on the role of the PQC in the pathogenesis of spinal and bulbar muscular atrophy (SBMA). In addition, we will discuss why and how boosting specific molecular chaperones and/or enhancing the activity of the degradative systems may have beneficial and therapeutic implications for SBMA.
SBMA Is a Protein Misfolding Disease
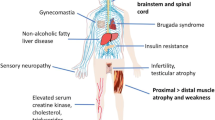
SBMA, also known as Kennedy’s disease, is an inherited X-linked motor neuron disease (MND) mainly characterized by motor neuron loss in the brain stem and in the anterior horns of the spinal cord. However, other cell types are also directly affected, such as the dorsal root ganglia (DRG) sensory neurons and some cells involved in male reproductive functions (Atsuta et al. 2006; Fischbeck 1997; Kennedy et al. 1968; La Spada et al. 1991; Malena et al. 2013; Soraru et al. 2008). In recent years, a significant contribution of muscle has emerged in SBMA as in other MNDs (Boyer et al. 2013a, b). Indeed, recent data suggest that muscle pathology maybe a primary player in the pathogenesis of SBMA, underscoring the importance of exploring therapeutic approaches targeting muscle as well as motor neurons (Cortes et al. 2014a; Lieberman et al. 2014; Rinaldi et al. 2014). Most of the cells that are affected in SBMA are postmitotic (Campisi and d’Adda di Fagagna 2007; Grunseich et al. 2014b), and their loss gives rise to a variety of symptoms. The most debilitating of these symptoms are caused by atrophy of bulbar, facial, and limb muscles (Fratta et al. 2014b; Kennedy et al. 1968; Sobue et al. 1989), alterations in sensory function (Adachi et al. 2005; Atsuta et al. 2006; Polo et al. 1996; Suzuki et al. 2008), as well as specific endocrine alterations (Fischbeck 1997, 2012). The molecular basis of the disease is associated with an expansion of a CAG triplet repeat sequence that codes for the amino acid glutamine (Q), located in exon 1 of the androgen receptor (AR) gene (La Spada et al. 1991). In the normal population, this CAG repeat is comprised between 9 and 37 repeats (average = 22), but in SBMA patients, expansions of more than 38 (up to 68) repeats (Fratta et al. 2014a; Grunseich et al. 2014a) result in disease. The CAG repeat codes for a polyglutamine (polyQ) tract, which is located in the N-terminal transactivation domain of the AR (ARpolyQ) protein. The elongated polyQ confers toxicity to the ARpolyQ, but in a ligand-dependent manner (Fig. 1) (Katsuno et al. 2002, 2003). This toxicity has been linked to the acquisition of polyQ-induced aberrant protein conformations (misfolding), which are prone to aggregation. The excess of misfolded ARpolyQ may perturb the PQC system, specifically in cells expressing high levels of ARpolyQ, which include both neuronal and nonneuronal cells including lower spinal cord motor neurons, DRG neurons, muscle cells, and Sertoli and Leydig cells.

Proteostasis regulation in spinal and bulbar muscular atrophy (SBMA). Inactive ARpolyQ is located in the cytoplasm associated with HSP proteins, in a “non-toxic” conformation. Testosterone (T) binding induces the HSP dissociation unmasking the polyQ tract, which is prone to misfold. Misfolded ARpolyQ aggregates in the cytoplasm and/or nuclei, and an excess of misfolded ARpolyQ may alter the intracellular proteostasis. This leads to activation of the proteotoxic response mediated by the protein quality control (PQC) system. The PQC comprises (a) the chaperone proteins (including the heat shock proteins (HSPs)), the degradative pathways (b) ubiquitin-proteasome system (UPS), (c) autophagy-lysosome pathway (ALP), and (d) the unfolded protein response (UPR), which activates the endoplasmic reticulum-associated degradation (ERAD). Misfolded ARpolyQ is recognized by the HSPs which may assist in its refolding or, alternatively, when folding cannot be reached, direct it to degradation. The UPR comprises three parallel signaling branches—PERK, IRE1α, and ATF6—and works with chaperones to determine the fate of misfolded proteins. ARpolyQ is mainly refolded by the ATP-dependent HSP70 and HSP40 and, when this fails, it is ubiquitinated (by ubiquitin ligases E1, E2, E3) and degraded via UPS and/or ALP. HSP70 and HSP40 allow E1-E2-E3 ubiquitination of misfolded ARpolyQ degraded by UPS. ARpolyQ ubiquitination is also mediated by the E3-ligase C-terminus of HSC70-interacting protein (CHIP) complexed to HSC70. The fate of this complex is determined by BAG family members acting as HSC70 co-chaperones. BAG1 links HSC70 to UPS through its ubiquitin-like (UBL) domain. BAG3 and its partner HSPB8 promote ALP degradation of HSC70 substrates. BAG3 interacts with the dynein complex moving misfolded ARpolyQ to the microtubule organization center (MTOC) where aggresomes are formed. Alternatively, the histone deacetylase 6 (HDAC6) may also interact with the dynein complex, but apparently after CHIP-mediated ubiquitination of substrates escaping UPS degradation. Polyubiquitinated proteins bound to the HSC70-BAG3 are recognized by p62/SQSTM1, which interacts with the autophagy marker LC3 to allow insertion of substrates into the autophagosome for degradation
ARpolyQ nuclear inclusions were first identified back in 1998 by Li and colleagues in several autopsy tissues from SBMA patients (Li et al. 1998a). These inclusions are heavily ubiquitinated, a clear index of alteration of UPS-mediated degradation. They are also particularly reactive with antibodies recognizing the N-terminus of the AR, suggesting that they originate from proteolytic cleavage of the AR. The nuclear inclusions were found in spinal and brainstem motor neurons, but not in unaffected neural tissues, although they were also present in several different nonneural tissues (Li et al. 1998b). Subsequent studies further characterized their intracellular distribution in affected neurons and showed that ARpolyQ inclusions are present in the nuclei of spinal cord motor neurons and the cytoplasm of DRG sensory neurons (Adachi et al. 2005; Suzuki et al. 2008). In muscle, inclusions were mainly found in the nuclei (Li et al. 1998b). Subsequently, we and others were able to show that in reconstituted motor neuronal systems, these inclusions are formed exclusively after ARpolyQ activation either by its endogenous ligand testosterone (or the testosterone derivative, dihydrotestosterone, DHT) (Simeoni et al. 2000; Stenoien et al. 1999), or by some (but not all, i.e., bicalutamide or casodex) specific AR selective modulators (SARMs), such as cyproterone acetate (Rusmini et al. 2007; Whitaker et al. 2004). However, it is still not clear why ARpolyQ toxicity strictly depends on androgens. Several data suggest that testosterone triggers ARpolyQ toxicity by inducing a switch in its structure from “nontoxic” to “toxic” conformations, which generates ARpolyQ aggregates (Simeoni et al. 2000). Thus, both the process of misfolding and the consequent aggregation possibly occur during the androgen-induced AR maturation to its active form (Poletti 2004; Poletti et al. 2005). The AR maturation/activation process requires the ligand-induced dissociation from HSP proteins (Fig. 1). This step allows the unmasking of the polyQ tract of the AR, which may then act either intramolecularly (intrinsic propensity to misfold), preventing proper folding because of its elongated size, or intermolecularly (extrinsic propensity to oligomerize and possibly aggregate), by its interaction with other polyQs in different ARpolyQ molecules. Moreover, ARpolyQ could interact with nonpolyQ proteins able to exacerbate ARpolyQ misfolding (see Poletti (2004) for review) and possibly ARpolyQ cleavage, with the consequent release of a potentially “supertoxic” N-terminal fragment containing the polyQ repeat. This process has long been considered one of the major steps required for the acquisition of ARpolyQ toxicity. However, recent data have raised the possibility that caspase-mediated release of ARpolyQ fragments may not be the cause of cell death. In fact, while the “toxic fragment” may directly induce neuronal dysfunction and death in a soluble form, its toxicity could be buffered by its sequestration into amyloid-like inclusions. The fragment containing the polyQ may also contribute to (or initiate) the seeding process required to generate aggregates in many misfolded protein diseases. However, the ARpolyQ proteolysis is a late event in SBMA mice, and most of the ARpolyQ aggregates that originate in the early stage of the disease contain the full-length protein. Finally, the ARpolyQ undergoes cleavage to a smaller fragment via the UPS and only after its incorporation into intranuclear inclusions (Heine et al. 2015), further supporting the possibility that ARpolyQ fragments may not be the “culprit.”
An alternative but not exclusive mechanism proposed to explain the ligand-induced toxicity to ARpolyQ is based on the fact that testosterone induces nuclear translocation of ARpolyQ. Several lines of evidence have demonstrated that the nucleus is the site where ARpolyQ exerts most of its toxic effects (Montie et al. 2009, 2011; Nedelsky et al. 2010). A specific interdomain interaction, which is required for the nuclear activities of AR, is also crucial for ARpolyQ aggregation and toxicity. This is the amino-terminal FXXLF motif capable of interacting in a ligand-dependent manner with the carboxyl-terminal AF-2 domain (N/C interaction). Genetic mutation of the FXXLF motif can prevent ARpolyQ aggregation and toxicity (Orr et al. 2010). Apart from the molecular mechanisms of aggregation, during this phenomenon, the aggregating ARpolyQ also sequesters several other proteins which are fundamental for cell functions. These include proteins that can associate to the AR (e.g., CREB and the steroid receptor coactivator 1, SRC-1) or proteins recruited by misfolded species during their processing (e.g., proteins involved in the PQC, like NEDD8, HSP70, HSP90, and HDJ2/HSDJ or also components of the PA700 proteasome caps, but not 20S core particles) (Stenoien et al. 1999).
While the original interpretation suggested that aggregates/inclusions of ARpolyQ were responsible for motor neuronal death, other data revealed that this process may be more complex than expected. A recent study performed with atomic force microscopy (AFM) has focused attention to inclusions normally generated by the wild-type (wt) AR (a protein particularly prone to aggregate even in its normal amino acid sequence) in aged tissue and compared these inclusions with those produced by the ARpolyQ in SBMA (Jochum et al. 2012). The results evidenced the existence of annular oligomers (120–180 nm in diameter) for the wtAR and of small oligomeric fibrils (up to 300–600 nm in length) for the ARpolyQ, similar to those described by us and others in SBMA (Li et al. 2007; Palazzolo et al. 2008) and also similar to inclusions described in other related CAG/polyQ neurodegenerative diseases. Thus, the elongated polyQ seems to exacerbate the natural propensity of wtAR to aggregate, but with a distinct aggregate maturation.
Whether these steps are associated with the intrinsic properties of the ARpolyQ, to a deleterious activity of the ARpolyQ on the degradative system, or rather due to its insufficient handling and clearance by an overwhelmed PQC system is still a matter of discussion. It is conceivable that all of these mechanisms are involved, and the accumulation of ARpolyQ depends on a reduced capability of aged cells to counteract the intrinsic tendency of ARpolyQ to generate immature inclusions. In general, it is accepted that the aggregation process occurs through several successive steps that require misfolded protein oligomerization into soluble deposits, with generation of different peculiar structures, followed by maturation into insoluble fibrils. In SBMA mice, ARpolyQ oligomers appear several weeks prior to symptom onset and are rather soluble, since they disappear after testosterone removal by castration. Thus, these aggregates are structurally distinct from intranuclear inclusions that appear at later stages (Li et al. 2007). With these subsequent steps, the formation of ARpolyQ aggregates may initially serve to protect cells from the aberrant toxic conformation of ARpolyQ. In fact, during aggregation, these toxic species are sequestered into a physically defined subcellular compartment (the aggregate), waiting for their possible intracellular degradation (see below) (Arrasate et al. 2004; Klement et al. 1998; Rusmini et al. 2007; Saudou et al. 1998; Simeoni et al. 2000). However, if not removed, at later stages and possibly because of reduced PQC activity in aged cells, ARpolyQ aggregates may then have an impact on several other intracellular mechanisms, thus becoming toxic to cells (Piccioni et al. 2001, 2002; Poletti 2004), by affecting several different intracellular functions. Therefore, the toxic role of aggregates is rather controversial, although the search for the “toxic specie(s)” of ARpolyQ (and of other polyQ containing proteins) is still in progress.
The Protein Quality Control System in SBMA
Besides the search for the “toxic specie(s)”, the presence of insoluble ARpolyQ in affected SBMA cells is the index of defective activity of the PQC system.
The Chaperones
The first line of defense of the PQC system is represented by several families of HSPs. HSPs act as molecular chaperones capable of both recognizing and stabilizing misfolded proteins, including ARpolyQ (Fig. 1). The effect of a variety of chaperones has been tested on ARpolyQ clearance, accumulation, and aggregation in different models of SBMA. The heat shock factor-1 (HSF-1), which is a transcriptional regulator of HSPs, strongly reduces ARpolyQ aggregation by enhancing the levels of different HSPs, thus with a specific mechanism associated with the pan-activation of the heat shock response (Kondo et al. 2013). The mediators of the anti-aggregation chaperone activity have been identified mainly in the combined activities of HSP70 and HSP40 (Fig. 1) and of HDJ2/HSDJ (Adachi et al. 2003; Bailey et al. 2002; Howarth et al. 2007; Kobayashi et al. 2000; Stenoien et al. 1999). Other chaperones active against ARpolyQ aggregation include HSP90, HSP105 (Adachi et al. 2009; Ishihara et al. 2003; Katsuno et al. 2005) and the DnaJ-like-1 (HSJ1) proteins (which contain DnaJ and ubiquitin-interacting motifs) (Howarth et al. 2007). As mentioned above, these chaperones are often sequestered into ARpolyQ inclusions, and/or their levels in affected cells may be significantly reduced. In general, their overexpression correlates with a reduction in the total amount of insoluble ARpolyQ accumulated in neuronal cells (Adachi et al. 2009). The complex interplay between the chaperones and the ARpolyQ has been extensively reviewed in Pratt et al. (2014), in which the authors proposed that the misfolded ARpolyQ accumulates as a consequence of being a “client” of HSP90, which is then recognizable by the other HSPs. The same concept is applied to other misfolded polyQ containing proteins (e.g., Huntingtin (HD)) or mutant proteins associated with other neurodegenerative diseases (e.g., tau (AD), α-synuclein (PD)). In these models, HSP90 exerts its action by acting on a specific protein-folding cleft, which can be used to drive the clearance of the client protein (Pratt et al. 2014). In addition, the overall power of molecular chaperones against ARpolyQ aggregation may depend both on the type of chaperones and on their specific combinations (e.g., synergic activity of HSP70 with its co-chaperone HSP40) (Kobayashi et al. 2000; Stenoien et al. 1999). For example, HSP70, HSP40, HSJ1a, and HSJ1b have all been shown to reduce ARpolyQ inclusions; HSP70 and HSP40 increase chaperone-mediated refolding, while HSJ1 proteins mainly avoid ARpolyQ aggregation and assist in its ubiquitination and UPS-mediated degradation, without promoting its refolding (Howarth et al. 2007). Another HSP70 interactor, the C-terminus of HSC70 (heat shock cognate protein 70)-interacting protein (CHIP), which has a U-box type E3 ubiquitin ligase activity, has been shown to be a potent modulator of ARpolyQ accumulation. Following interaction with HSP70, CHIP can ubiquitinate the ARpolyQ entrapped by the chaperone machinery, allowing its subsequent degradation by the PQC degradative pathway (Fig. 1) (Adachi et al. 2007). Indeed, in studies performed on neuronal models of SBMA, CHIP greatly reduced ARpolyQ accumulation, while CHIP overexpression in a SBMA transgenic mouse model resulted in a clear amelioration of the motor behavior of affected mice, accompanied with a reduced neuronal nuclear accumulation of ARpolyQ (Adachi et al. 2007).
It is not only the classical ATP-dependent HSPs that can exert anti-aggregation activity on ARpolyQ, but also small ATP-independent HSPs, such as HSPB8 (Carra et al. 2005; Rusmini et al. 2013) which are potent inhibitors of ARpolyQ aggregation. In vitro, HSPB8 displays chaperone activity (Carra 2009; Carra et al. 2008a, b, 2012, 2013; Rusmini et al. 2013). In cells, HSPB8 forms a complex with BAG3, a co-chaperone of HSP70, and promotes the degradation of misfolded proteins resistant to HSP-dependent refolding (Fig. 1). Degradation of the bound substrate (ARpolyQ and several other disease-associated misfolded proteins) occurs via autophagy rather than via the UPS and in different cell types, including motor neurons, HeLa cells, COS cells, and HEK293T cells (Carra et al. 2012; Crippa et al. 2010b; Gamerdinger et al. 2011; Rusmini et al. 2013).
The Proteasome
When the first PQC defense line is activated, but refolding fails, specific chaperones assure cell protection against proteotoxicity by routing misfolded proteins to the degradative systems (UPS and autophagy) (Fig. 1). With regard to proteasome, misfolded proteins are recognized by several different chaperones (mainly the HSP70/HSP40 complex), which allow the selective ubiquitination of the target misfolded substrates (mediated by the complex system of the ubiquitin ligases) to be directed to the 20S core of the proteasome for degradation (Fig. 1). Both the wtAR and the ARpolyQ are efficiently processed by the proteasome, and inhibition of the UPS results in accumulation of large amounts of AR (either wt or mutant) in neuronal and nonneuronal cells (Dossena et al. 2014; Rusmini et al. 2007, 2010, 2011, 2013). It is unclear whether neuronal cells have a different UPS vs. autophagic power compared to other cell types (Crippa et al. 2013b; Galbiati et al. 2014; Onesto et al. 2011), but possible modifications to the equilibrium between these two pathways may be responsible for selective modification of ARpolyQ clearance in different cells.
It has been shown that the propensity of ARpolyQ to form insoluble species is accentuated in neuronal cells, possibly because of a less efficient PQC system compared to other cell types. In fact, using SBMA patient-derived induced pluripotent stem cells (iPSCs), Nihei and colleagues showed that ligand-induced ARpolyQ aggregation can be detected in filter retardation assays (FRA) only in cells differentiated to a motor neuronal phenotype and not in undifferentiated iPSCs or fibroblasts from SBMA patients (Nihei et al. 2013). However, in nonneuronal cells, such as adipose mesenchymal cells derived from SBMA patients, inhibition of the PQC (in this particular case, the inhibition of proteasome but not of autophagy) induces ARpolyQ accumulation into ubiquitinated and HSP70-positive nuclear aggregates (Dossena et al. 2014). Because of its structure, the wtAR is already prone to aggregate (Jochum et al. 2012), and the AR utilizes the same route of degradation, which is normally based on the proteasome. However, the presence of the elongated polyQ makes this protein more resistant to UPS handling and degradation, and the long polyQ may impair and/or overwhelm the proteasome capabilities; as a consequence, this phenomenon will require the involvement of the autophagic pathway, which is an alternative pathway of degradation characterized by lower selectivity, but higher capacity than the UPS (see Rusmini et al. (2010) for review). The aggregates therefore serve to temporarily sequester those fractions of ARpolyQ misfolded species that may have a direct impact on the UPS. Indeed, by analyzing UPS functions during ligand-induced ARpolyQ cytoplasmic aggregation, we clearly noted that the proteasome reporter, YFPu, accumulates in the absence of ARpolyQ aggregates (untreated ARpolyQ is cleared by the UPS, thus impairing the pathway), but testosterone-induced ARpolyQ aggregation correlates with a normal YFPu clearance (Rusmini et al. 2007). Thus, aggregation contributes to proteasome desaturation (but this effect is not related to AR nuclear translocation), protecting the proteasome from an excess of misfolded protein to be processed, and the forming aggregates then have to be processed by the autophagic pathway.
Autophagy
Autophagy may take place in different cytoplasmic regions and the insoluble forms of ubiquitinated ARpolyQ are recognized by specific proteins, including p62/SQSTM1. p62/SQSTM1 interacts with the autophagosome-associated lipidated form of LC3 (LC3-II) and is engulfed into nascent autophagosomes; later, these autophagosomes fuse to lysosomes, where misfolded proteins and aggregates are degraded (Fig. 1). Misfolded species can also be directed to the microtubule organization center (MTOC) in a microtubule- and dynein-dependent manner (Fig. 1) (Johnston et al. 2002; Webb et al. 2004). Here, misfolded species can form the “aggresomes” to facilitate engulfment into the nascent autophagosomes (Fujinaga et al. 2009; Iwata et al. 2005; Johnston et al. 1998, 2002; Kopito 2000; Kopito and Ron 2000; Taylor et al. 2003). The MTOC region is particularly enriched of autophagosomal components (Johnston et al. 2002). Depletion of the essential autophagic component p62/SQSTM1 results in an earlier disease onset and a worsening of motor deficits in a transgenic SBMA mouse model (Doi et al. 2013), which correlate with increased levels of insoluble ARpolyQ in affected cells. In addition, in an autophagy-related manner, p62/SQSTM1 silencing increased, while p62/SQSTM1 overexpression reduced the total amounts of monomeric soluble and oligo/heteromeric insoluble complexes of ARpolyQ in cultured cells and in transgenic SBMA mice (Doi et al. 2013). If the proteasome is responsible for wt and ARpolyQ degradation in basal conditions, and autophagy takes over only when the UPS is impaired or overwhelmed, the accumulation of ARpolyQ (in particular in nuclear inclusions) may be linked to a defect in the secondary activation of the protective cytoplasmic autophagic pathway, rather than to an initial failure in proteasome function. A delicate equilibrium exists between the UPS and autophagy, which are both essential for the correct functioning of the PQC system (Carra et al. 2012, 2013; Rusmini et al. 2010, 2013). The ARpolyQ rerouting from the proteasome to autophagy may be of relevance for its clearance, and it requires specific chaperones that are able to sense proteasome inhibition, such as HSPB8 (Crippa et al. 2010a, b) and the co-chaperones BAG3 (for autophagic clearance) and BAG1 (for UPS clearance) (Fig. 1) (Gamerdinger et al. 2011). The complex HSPB8 and BAG3 (in a 2:1 ratio) interacts both with misfolded proteins and with HSC70/CHIP for substrate ubiquitination (Fig. 1) (Arndt et al. 2010; Crippa et al. 2010a, b). It is of note that conditions that lead to proteasome overwhelming and/or impairment result in a selective “de novo” transcriptional activation of the HSPB8 promoter (Crippa et al. 2010b). The raised levels of HSPB8 permit an increase in the total amount of the 2×(HSPB8)/BAG3 complex, with an enhancement of its overall activity against misfolded species (Crippa et al. 2010a). Interestingly, we found that HSPB8, by interacting with BAG3, decreases ARpolyQ aggregation, by increasing its solubility and clearance (Rusmini et al. 2013). In motor neuronal cells, HSPB8 does not modify the expression of p62/SQSTM1 and LC3 (the two key autophagic molecules). However, HSPB8 prevents p62 body formation, restoring a normal autophagic flux, which is known to be impaired by testosterone-activated ARpolyQ (Rusmini et al. 2013). As a consequence of the restored autophagic flux, HSPB8 facilitates the ARpolyQ autophagic removal, thereby limiting its aggregation and toxicity in motor neuronal cells (Crippa et al. 2010a, b, 2013; Rusmini et al. 2013). Upregulation of the HSPB8-BAG3 complex may thus play a key role in the defense against ARpolyQ and in the maintenance of motor neuron viability. Notably, HSPB8 is mutated (at K141 with E or N) in some forms of motor neuron diseases (Charcot-Marie-Tooth type 2L disease and in hereditary distal motor neuropathy type II (dHMNII); Fontaine et al. 2006; Irobi et al. 2010), and the chaperone activity of HSPB8 is lost in the mutant dHMNII-HSPB8 (Kwok et al. 2011). This supports the possibility that HSPB8 is crucial for motor neuron function and viability. Moreover, in ALS mice, both anterior horn spinal cord motor neurons and skeletal muscle cells respond to proteotoxicity by activating a robust HSPB8-mediated PQC system response (Crippa et al. 2010b, 2013a, b). Furthermore, the motor neurons that survive in the spinal cord at disease end stage have particularly enhanced levels of HSPB8 protein in association with soluble mutant SOD1 (Crippa et al. 2010b). Similar data have been reported in autopsy specimens of ALS patients’ spinal cord (Anagnostou et al. 2010). It must be noted that in mouse spinal cord, HSPB8 expression level declines with age (Crippa et al. 2010b), suggesting that motor neuronal cells may become more vulnerable to misfolded protein toxicity during aging, as a result of low levels of this pro-autophagic protein. HSPB8 also exerts a physiological role in the PQC response in muscle (Arndt et al. 2010), another target of ARpolyQ toxicity. However, although the expression levels of HSPB8 dramatically increase with disease progression in muscle of ALS mice (Carra et al. 2013; Crippa et al. 2013a, b), nothing is known at present about the possible involvement of the HSPB8 and BAG3 machinery in SBMA skeletal muscle. Our preliminary results suggest that in symptomatic SBMA male mice, the expression level of HSPB8 in muscle is increased, but not at levels comparable to those found in the corresponding muscles of symptomatic ALS mice.
As stated above, many of the genes involved in autophagy have been found altered in response to ARpolyQ expression or activation by ligands. However, one of the most relevant defects is associated with the fact that ARpolyQ is able to reduce the long-term turnover of proteins and to block the cytoplasmic autophagic flux (Carra et al. 2012; Cortes et al. 2014b; Giorgetti et al. 2014; Rusmini et al. 2007, 2010, 2011, 2013). As will be discussed in the next sections, the pharmacological restoration of a normal autophagic flux, for example by treatment with trehalose, a transcription factor EB (TFEB) activator (Dehay et al. 2010), greatly increases the clearance of the mutant misfolded ARpolyQ (Giorgetti et al. 2014; Rusmini et al. 2013). Defects in autophagy flux have been clearly identified in SBMA mice (Cortes et al. 2014b), and the importance of autophagy in SBMA is confirmed by several recent studies (Cortes et al. 2014b; Settembre and Ballabio 2011). The ARpolyQ processing may differ considerably in neuronal and muscle cells, both targets of ARpolyQ toxicity, and the identification of specific autophagy-related molecular markers of skeletal muscle degeneration in SBMA might represent a valuable diagnostic tool for monitoring disease progression. In SBMA muscle, the toxicity of activated ARpolyQ is rather complex and controversial, and autophagy may exert either a protective or a detrimental effect. Autophagy is dysregulated in muscle of AR113Q knock-in SBMA mice, and autophagic genes appear to be regulated by a physiological antagonism between TFEB and ZKSCAN3 (Chua et al. 2014), which surprisingly, are both greatly increased. In muscle of SBMA mice, TFEB levels are increased, and consequently most of the TFEB target genes are upregulated, including LC3, Vps11, Vps18, and Lamp1 (Chua et al. 2014). Inhibition of Beclin-1 induced autophagy-diminished skeletal muscle atrophy in AR113Q knock-in SBMA mice, improved motor behavior, and extended lifespan (Yu et al. 2011). On the other hand, overactivation of autophagy worsened the motor phenotype (Yu et al. 2011). Therefore, restoration of both a normal flux and a dysregulated autophagy may help in the ARpolyQ clearance in muscle of SBMA mice.
Endoplasmic Reticulum-Associated Degradation in Stress Conditions and the Unfolded Protein Response
Alteration of the proteasomal and/or autophagic pathways may involve a proteotoxic response to cell stress that induces the UPR at the level of the endoplasmic reticulum (ER). The UPR is normally activated in the ER lumen to restore normal cell function via transient translational blockage, activation of degradative pathways, and induction of molecular chaperones. When protein misfolding occurs, hydrophobic amino acids are usually exposed to the surface of the protein, which can be recognized by proteins such as BiP/Grp78, a member of the HSP70 family. These proteins can be driven to the ERAD to initiate the signaling of the UPR, involving factors including PKR-like endoplasmic reticulum kinase (PERK) and inositol requiring enzyme 1 (IRE1) (Fig. 1). Translation attenuation is mediated by the PERK receptor soon after UPR initiation and induced by PERK oligomerization and autophosphorylation, and involves phosphorylation of the eIF2 messenger RNA (mRNA) translation machinery. At the same time, the mRNA coding for the transcription factor XBP1 is activated by alternative splicing and the resulting protein product upregulates the transcription of UPR stress genes. In addition, the activating transcription factor 6 (ATF6) is activated by protein cleavage and migrates from the ER to the nucleus, where it controls other UPR genes. These simultaneous activities lead to the restoration of normal ER functions. It has been shown, using a disease-associated fragment of the ARpolyQ, that this is capable of activating an ER stress-inducible promoter. When the contribution of the three proximal sensors of ER stress, ATF6, IRE1, and PERK, to the UPR was analyzed in detail, it was found that ARpolyQ toxicity increased when ATF6 was blocked, but decreased by overexpression of a constitutively active mutant of ATF6. Conversely, modification of the IRE1 arm of UPR did not modify ARpolyQ toxicity, while PERK activation resulted in increased ARpolyQ toxicity (Thomas et al. 2005). Thus, the UPR is activated by ARpolyQ and UPR activation may reduce its toxicity. ARpolyQ-induced toxicity in skeletal muscle also induces the UPR. In knock-in SBMA mice, characterized by muscle atrophy induced by Beclin-1-mediated autophagy, deletion of the transcription factor C/EBP homologous protein (CHOP), which is normally induced following ER stress, significantly increased muscle atrophy, proving the existence of a cross talk between the UPR and macroautophagy in SBMA (Yu et al. 2011).
In mouse embryonic stem cells (ESCs), ARpolyQ is able to induce a testosterone-dependent cell stress phenotype during cell differentiation, characterized by the formation of ARpolyQ inclusions positive for E3 ubiquitin ligase, CHIP and BiP/GRP78, as well as by caspase-3 activation. ARpolyQ aggregation was accompanied by ER stress and increased susceptibility to apoptosis. These modifications were paralleled by a robust increase in the levels of ER chaperones (GRP78/BiP and GRP94) and ER stress markers (ATF6, phosphorylated PERK, GADD153/CHOP, and spliced XBP-1). ARpolyQ activation was also responsible for the dissociation of BiP/GRP78 from ATF6. Interestingly, exogenous overexpression of GRP78/BiP reduced ARpolyQ ubiquitination and aggregation, while GRP78/BiP silencing of GRP78/BiP correlated with increased ARpolyQ aggregation, caspase-3 activity, and cell apoptosis (Yang et al. 2013).
The ER may be regarded as a quality control system for newly synthesized proteins in cells. The ER is associated not only with protein folding but also the control of Ca2+ homeostasis and is the main intracellular Ca2+ reservoir (Hetz and Mollereau 2014). Therefore, accumulation of misfolded proteins within the ER and/or depletion of ER Ca2+ may compromise normal cellular function and subsequently trigger ER stress. The mechanisms that attempt to restore ER homeostasis include store-operated Ca2+ influx, which replenishes Ca2+ levels, as well as activation of the UPR (Szegezdi et al. 2006). Sustained activation of ER stress diminishes the ability of the UPR to restore ER homeostasis and results in ER stress-induced apoptosis, mediated by caspase-12 (Yoneda et al. 2001). The role of ER stress and Ca2+ homeostasis was recently examined in the AR100 mouse model of SBMA. AR100 mice develop a late onset, slowly progressive neuromuscular phenotype, accompanied by motor neuron degeneration and muscle atrophy (Malik et al. 2011; Montague et al. 2014; Sopher et al. 2004). Dysregulation of Ca2+ homeostasis in embryonic motor neurons of AR100 mice results in ER stress, which enhances the vulnerability of AR100 motor neurons to ER-stress-induced apoptosis (Montague et al. 2014). These findings suggest that in cultured motor neurons of AR100 SBMA mice, ligand-dependent DHT activation of ARpolyQ triggers a depletion of ER Ca2+ levels and a reduction of store-operated Ca2+ influx. As a consequence, ER stress is more likely to occur in AR100 embryonic motor neurons, as the normal function of the ER is dependent on Ca2+ levels (Hetz and Mollereau 2014; Tadic et al. 2014). The reduction in ER Ca2+ may in part be explained by the finding that the expression of the SERCA2b pump was significantly lower in AR100 motor neurons than in wild-type motor neurons. SERCA2b belongs to the sarcoendoplasmic reticulum Ca2+ ATPases (SERCAs) which enable ER Ca2+ reuptake (Foradori and Handa 2008). Furthermore, the levels of the ER stress markers BiP, ATF4, and CHOP were all also significantly elevated in DHT-treated AR100 embryonic motor neurons in vitro. As a result, ER stress-mediated apoptosis was activated in AR100 motor neurons in vitro, leading to an increase in the expression of activated caspase-12. Similarly, there was elevation of ER stress markers in vivo, in spinal cord motor neurons of AR100 mice (Montague et al. 2014). Importantly, this ER stress was detected in presymptomatic mice, long before the onset of any pathology and development of disease, suggesting that ER stress may be an early event in the pathogenesis of SBMA which possibly plays a causal role in the development of the disease. Furthermore, preliminary findings suggest that there is a difference in activation of the ER stress in muscle compared to spinal cord in AR100 mice, indicating that there may be an alternative mechanism responsible for muscle dysfunction in SBMA (Malik and Greensmith, unpublished observation).
Approaches Aimed to Enhance the Activity of the Protein Quality Control System to Counteract ARpolyQ Toxicity in SBMA
The findings discussed so far indicate that the PQC system exerts protective effects against ARpolyQ toxicity. On the other hand, ARpolyQ may impair the PQC system, generating a vicious circle in which the reduced activity of the PQC leads to less protection against cell stress induced by the ARpolyQ, thereby enhancing its toxicity. As a result, several investigators have developed different approaches aimed to improve the overall activity of the PQC system in neurons or muscle cells. Indeed, the simple overexpression of chaperones and/or the stimulation of autophagic pathway has been shown to positively modulate ARpolyQ deposition and to reduce its toxicity.
Enhancement of the Chaperone Response
One of the first attempts to increase the chaperone response in models of SBMA involved the use of geranylgeranylacetone (GGA), a nontoxic anti-ulcer drug, which has been shown to stimulate HSP70, HSP90, and HSP105 expression (Katsuno, et al., 2005). In transgenic SBMA mice, oral GGA treatment induced a significant enhancement of the chaperone activity mediated by these HSPs, resulting in an amelioration of neuromuscular deficits and an increase in survival. This protective effect of GGA was mediated by a significant reduction in ARpolyQ accumulation in the affected areas of the central nervous system (Adachi et al. 2009; Katsuno et al. 2005). Unfortunately, although oral GGA was well tolerated in mice, the effective dose was very high, so that translation to human patients would be likely to cause significant side effects. Further studies by other groups have shown that specific lysine (Lys) residues of the wtAR (acetylated in a ligand-dependent manner) may significantly affect AR dynamics, inducing a behavior (in terms of trafficking, misfolding, and aggregation) similar to that caused by ARpolyQ. When the Lys 630 or 632 and 633 are converted to alanine (K/A conversion), wtAR nuclear translocation is greatly delayed, accompanied by the formation of inclusions that entrap HSP40, HSP70, and the ubiquitin-protein isopeptide ligase (E3) CHIP. This aberrant activity can be blocked with radicicol, an inhibitor of HSP90, suggesting that the ligand-dependent K/A ARpolyQ intracellular distribution, folding, and aggregation are linked to HSP90 functions (Thomas et al. 2004). Whether or not the effect of radicicol is extended to ARpolyQ and whether this approach may have relevance for SBMA remain to be tested.
Following these two initial proofs of principle, other molecules targeting the chaperone response have been tested in SBMA models. For example, geldanamycin and other related HSP90 inhibitors can activate HSF-1, which in turn results in an enhanced expression of HSP70 and HSP40. Surprisingly, treatment with geldanamycin completely prevented ARpolyQ aggregate formation in HSF-1(−/−) mouse embryonic fibroblasts, in which HSP70 and HSP40 cannot be induced by HSF-1 (Thomas et al. 2006). Since geldanamycin also inhibits HSP90-mediated trafficking in cells, it may also act via HSP90-dependent trafficking that involves the immunophilins (IMM). As a proof of concept, overexpression of p23, a co-chaperone of HSP90, was able to inhibit both ARpolyQ trafficking and aggregation (Thomas et al. 2006). Thus, different druggable sites can be identified in the chaperone system that plays a role in response to proteotoxicity.
A study using Drosophila models of SBMA has demonstrated that some small molecules may act by mimicking the activity of HSP70 interacting protein (Hip) (Wang et al. 2013). Hip is a co-chaperone that can enhance the binding of HSP70 to the target misfolded substrates. Together with HSP70, Hip promotes the ubiquitination of the client ARpolyQ, facilitating its degradation. In this study, a series of small molecules was designed based on the rhodocyanine MKT-077, a known interactor of HSP70, and capable of binding the nucleotide-binding domain of ADP, but not ATP-bound HSP70. A derivative of MKT-077, named YM-1, was able to promote HSP70-dependent steps in nNOS maturation, thus possibly favoring the tight-binding form of HSP70 (Wang et al. 2013). YM-1 potently stimulated the binding of HSP70 to unfolded substrates and converted HSP70 to its tight-affinity conformation. Hip blocks binding of YM-1 to HSP70, indicating that this synthetic co-chaperone uses a mechanism similar to Hip to interact with HSP70. Using this mechanism, YM-1 was able to increase ubiquitination and degradation of client ARpolyQ (Wang et al. 2013). YM-1 reduced the accumulation of RIPA-insoluble ARpolyQ species and the number of intranuclear inclusions induced by testosterone, but had no effect on soluble ARpolyQ, suggesting that YM-1 acts primarily on unfolded AR species. Treatment with YM-1 was also able to rescue toxicity in Drosophila expressing ARpolyQ. In fact, the DHT-dependent pupal toxicity of the ARpolyQ was significantly rescued by YM-1 (Wang et al. 2013). Thus, the Hip mimetics act allosterically, promoting the binding of HSP70 to unfolded substrates, like ARpolyQ, and enhance its ubiquitination and degradation (Wang et al. 2013).
An alternative approach to modulate chaperone function is based on the use of 17-allylamino-17-demethoxygeldanamycin (17-AAG), which is a potent HSP90 inhibitor, and an anticancer agent which is currently in phase II clinical trials for a variety of cancers. HSP90 is a fundamental protein involved in the assembly of the multichaperone complex responsible for the folding, activation, and assembly of the AR. HSP90 also has an important role in the response to misfolded proteins like ARpolyQ (Poletti 2004). The ARpolyQ has a higher affinity for the HSP90-p23 complex than for wtAR (Waza et al. 2005). Treatment of transgenic SBMA mice with 17-AAG markedly ameliorates ARpolyQ-induced motor impairments (Waza et al. 2005, 2006b). This effect was attributed to the ability of 17-AAG to reduce both the monomeric and aggregated forms of ARpolyQ, an effect which is highly selective for ARpolyQ compared to wtAR (Waza et al. 2005). The effect of 17-AAG does not simply involved HSP90, but is associated with a mild increase in HSP70 and HSP40 (Waza et al. 2006a). Interestingly, 17-AAG acts on ARpolyQ by enhancing the overall prodegradative activity in motor neuronal cells, but without impacting on proteasome function. 17-AAG clears the misfolded fraction of ARpolyQ and the resulting ARpolyQ aggregates by activating the autophagic system (Rusmini et al. 2011). The effect of 17-AAG on the aggregation and clearance of ARpolyQ has been confirmed in iPSCs derived from SBMA patients and induced to differentiate to motor neurons (Nihei et al. 2013). Unfortunately, 17-AAG is not well tolerated in humans and cannot be administered orally. A series of compounds has therefore been developed to reduce these adverse effects. One of these is 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), an oral HSP90 inhibitor, that can ameliorate motor deficits in SBMA mice, by reducing the levels of monomeric and nuclear-accumulated ARpolyQ, and which is far less toxic than 17-AAG (Tokui et al. 2009). Similar to 17-AAG, 17-DMAG also increases HSP70 and HSP40 expression, thus contributing to the enhancement of the chaperone response in motor neuronal cells.
Recently, arimoclomol, a novel, well-tolerated, and orally administrable co-inducer of the heat shock response (HSR) and chaperone system, was tested in SBMA mice (Malik et al. 2013). Arimoclomol is a hydroxylamine derivative of bimoclomol and has been shown to upregulate the expression of HSPs via co-activation of HSF-1 (Hargitai et al. 2003). Unlike other drugs which target chaperone expression and the HSR, arimoclomol acts as a “smart drug” and enhances the HSR only in cells already under stress in which HSF-1 is already activated, thereby co-inducing the HSR rather than actually activating this stress response (Kalmar et al. 2008; Vigh et al. 1997). This precise targeting of only cells exhibiting a stress response reduces the possibility of nonspecific effects in otherwise unstressed cells, which occurs following the use of other compounds that upregulate HSP expression by direct global activation of the HSR which may reflect a more generalized stress response that actually results in cell toxicity and death (Kalmar et al. 2008). In the AR100 mouse model of SBMA, treatment with arimoclomol increased HSP expression only in affected tissue (spinal cord and muscle) and not in unaffected regions (e.g., cortex and liver). Moreover, oral administration of AR100 mice with arimoclomol significantly delayed disease progression by preventing motor neuron degeneration, consequently improving the survival of functional motor units, reducing muscle atrophy, and alleviating the significant deterioration in body weight. Importantly, arimoclomol also improved hindlimb muscle force and contractile characteristics. Since arimoclomol upregulates HSP expression in muscles and the spinal cord of AR100 mice, it is likely that its beneficial effects in AR100 mice are a result of its effects both in the periphery in muscles under stress, as well as within the CNS in motor neurons. In addition, arimoclomol also upregulated the expression of vascular endothelial growth factor (VEGF), both in the spinal cord and hindlimb muscles of diseased AR100 mice. VEGF is known to have neurotrophic effects in the survival of motor neurons (Azzouz et al. 2004; Cvetanovic et al. 2011; Jin et al. 2002; Lambrechts et al. 2003; Sopher et al. 2004; Storkebaum et al. 2005), as well as protect against muscle degeneration in neuromuscular diseases (Lambrechts et al. 2003; Storkebaum et al. 2005). VEGF is regulated by the HIF1A transcription factor, and as HIF1A has been shown to interact with HSPs (Huang et al. 2009; Isaacs et al. 2002), this may provide an explanation for the upregulation induced by arimoclomol. Arimoclomol has been previously shown to be effective in delaying disease progression in the SOD1G93A mouse model of ALS (Kalmar et al. 2008; Kieran et al. 2004) and to protect against retinal degeneration in rhodopsin retinitis pigmentosa (Parfitt et al. 2014). It is therefore an attractive therapeutic candidate for SBMA, as it has already been shown to be safe and well tolerated in healthy volunteers and patients with ALS (Cudkowicz et al. 2008) and is currently undergoing a phase II/III trial in the USA in SOD1-ALS (www.ClinicalTrials.gov), as well as in patients with the inflammatory myopathy inclusion body myositis (IBM) (Machado et al. 2013).
Activation of Autophagy
Several attempts have been made to enhance the overall cellular autophagy power to promote the clearance of toxic ARpolyQ species from SBMA-affected cells. The advantage of this strategy is that autophagy is thought to be selectively regulated in cells which are specifically sensitive to its activation (i.e., hepatocytes, muscle cells, and neurons), and therefore, it may be possible to identify small molecules capable of selectively intervening in neuronal autophagy. The biological relevance of autophagy in these cells is related to their energy requirements as well as to the need for protection against different proteotoxic or organelle-mediated stresses. Thus, the goal for SBMA is to identify compounds that increase autophagic clearance of misfolded ARpolyQ selectively in SBMA-affected cells, motor neurons, and/or muscle cells. Using primary cultures of motor neurons derived from SBMA mice, Montie and Merry have shown that both the autophagy activator mTOR-dependent phenoxazine (or AKTi, a potent Akt inhibitor or rapamicyn) and the mTOR-independent trehalose were able to rescue motor neurons from ARpolyQ-mediated ligand-dependent death (Montie et al. 2009; Montie and Merry 2009). In motor neurons, both AKTi (Montie et al. 2009) and trehalose (Giorgetti et al. 2015; Montie et al. 2009; Rusmini et al. 2013) induced the classical cytoplasmic punctate distribution of the lipidated form of LC3 (LC3-II) which is associated with nascent autophagosomes, a clear index of autophagy activation, and this correlated with the full removal of aggregated, insoluble ARpolyQ (Giorgetti et al. 2014; Montie et al. 2009; Montie and Merry 2009; Rusmini et al. 2013). Other small molecules structurally related to trehalose may have the potential to revert ARpolyQ accumulation and aggregation. Trehalose is considered a natural osmolyte and/or “chemical chaperone” (Kumar 2012). Its advantage as a small molecule, which is shared by other osmolytes, is that it can cross the blood-brain barrier with high efficiency. In addition, its activity can be enhanced by the synergic effects of other compounds that target ARpolyQ. Indeed, since testosterone activates ARpolyQ toxicity by inducing its nuclear translocation, the prevention of ARpolyQ localization in the nucleus, combined with an increased cytoplasmic clearance, may be protective. Notably, cytoplasmic retention of ARpolyQ obtained using the SARM Casodex not only prevented ARpolyQ aggregation and toxicity, enhancing motor neuronal survival (Orr et al. 2010; Rusmini et al. 2007), but also potentiated the pro-autophagic activity of trehalose (Giorgetti et al. 2014). This synergistic effect may be due to an increased time window for the recognition of misfolded ARpolyQ species in the cytoplasm, soon after AR dissociation from HSPs, and their autophagic engulfment prior to migration into the nucleus. This, together with improved ARpolyQ autophagic clearance in the cytoplasm, leads to an indirect reduction of ARpolyQ nuclear accumulation, preventing its nuclear toxicity by maintaining normal motor neuron proteostasis and viability.
Although the toxic effects of ARpolyQ are thought to be exerted at the nuclear level, cytoplasmic autophagic degradation of ARpolyQ greatly reduces its toxic effects. Interestingly, the autophagic activation that protects SBMA motor neurons has only mild effects on nuclear ARpolyQ aggregation, but significant effects on cytoplasmic ARpolyQ aggregation. This pharmacological data has been corroborated by evidence obtained in SBMA mice genetically manipulated to produce an AR lacking the nuclear localization signal (NLS) (Montie et al. 2009). In these ARpolyQΔNLS mice, the AR is poorly translocated into the nuclei upon androgenic activation, and this correlates with a substantial improvement in motor function compared with classical SBMA mice (Montie et al. 2009). Furthermore, using a Drosophila model of SBMA, it has also been shown that the retention of an ARpolyQ fragment in the cytoplasm ameliorates the disease (Takeyama et al. 2002). Moreover, in cultured motor neurons of ARpolyQΔNLS mice, the autophagic pathway was capable of fully degrading the cytoplasmically retained ARpolyQ, activated by androgens, suggesting that authopagy may even protect against nuclear ARpolyQ toxicity. As mentioned above, trehalose not only is a potent autophagic inducer but also enhances the expression of HSPB8 (Rusmini et al. 2013), known to actively participate in the protection against cell stress by assisting the autophagic removal of misfolded ARpolyQ. HSPB8 is able to restore a normal autophagic flux. We recently screened for a large library of FDA-approved drugs for their ability to stimulate the HSPB8 promoter in a reconstituted transcriptional assay. Several estrogenic compounds (and selective estrogen receptor modulators, SERMs) were able to enhance the expression of HSPB8, suggesting that these steroids may have a protective activity against proteotoxicity induced by misfolded proteins. It will be of interest to evaluate whether SERMs, by modulating HSPB8, may exert pharmacological protection against misfolded ARpolyQ and related proteins (Crippa et al., unpublished results). In the same screening, we also confirmed that proteasome inhibitors are potent inducers of HSPB8 expression, leading us to hypothesize that when the proteasome is overwhelmed and consequently impaired, transcriptional activation of the HSPB8 gene may be triggered in order to facilitate the removal of proteins via autophagy (Carra et al. 2013; Crippa et al. 2010b; Giorgetti et al. 2014; Rusmini et al. 2013). While the use of proteasome inhibitors is not appropriate for disorders such as SBMA, their effects on HSPB8 can be taken as a proof of principle that proteins that accumulate when the proteasome is blocked may be molecularly rerouted by HSPB8 (in complex with BAG3) to the autophagy pathway for clearance.
Activation of the UPR Response
ER stress has been shown to be an early pathological feature of motor neuron disorders, in both the SOD1G93A mouse model of ALS and the AR100 model of SBMA (Montague et al. 2014; Saxena et al. 2009). The effect of pharmacological targeting of ER stress was examined by treatment of AR100 SBMA embryonic motor neuron cultures with the ER inhibitor, salubrinal, in order to establish whether motor neuron survival was related to the ER stress observed in AR100 motor neurons (Montague et al. 2014). Salubrinal is a selective inhibitor of eIF2α dephosphorylation and consequently diminishes ER stress-mediated apoptosis (Boyce et al. 2005; Kessel 2006). Treatment with salubrinal improved the survival of cultured motor neurons, in particular AR100 motor neurons. The expression of activated caspase-12 was significantly higher in AR100 motor neurons, but treatment with salubrinal resulted in a marked decrease in expression in AR100 motor neurons. Sustained ER stress is known to diminish the ability of the UPR to regulate ER homeostasis, resulting in apoptosis. Caspase-12 plays a key role in this ER stress-induced apoptosis in mice (Yoneda et al. 2001). These results therefore show that treatment with an ER stress inhibitor prevents activation of the ER-associated cell death pathway in AR100 motor neurons. Importantly, inhibition of ER stress in AR100 motor neurons in vitro, by treatment with salubrinal, significantly reduces activation of ER stress-associated apoptosis, by suppressing activation of caspase-12. Treatment of mutant SOD1 transgenic mice with salubrinal has been shown to delay disease progression and attenuate the disease phenotype (Saxena et al. 2009). ER stress has also been implicated in motor neuron degeneration in Caenorhabditis elegans and zebrafish models of TDP-43 ALS, which is reduced by treatment with salubrinal, resulting in protection against TDP-43 neuronal toxicity in vivo (Vaccaro et al. 2013). In addition, treatment of α-synuclein transgenic mice with salubrinal has also been shown to be neuroprotective in mouse model of Parkinson’s disease (Colla et al. 2012a, b). Thus, pharmacological targeting of the ER stress pathway, one of the earliest pathological mechanisms observed in SBMA motor neurons, may be a potential therapeutic strategy for SBMA.
Targeting Other Components of the Proteostasis Network
The proteostasis network is an integrated system of pathways that regulate protein synthesis, trafficking, and degradation, to ensure proper protein folding and to eliminate misfolded proteins when folding repair mechanisms are unsuccessful (Balch et al. 2008). The overall purpose of this network is to maintain the integrity of the proteome in response to environmental or genetic stressors. The accumulation of aberrant protein species in SBMA, as in other diseases caused by protein misfolding, challenges the proteostasis machinery and, at the same time, may accelerate aging by interfering with protein folding and clearance and other key cellular processes (Hipp et al. 2014). Along with the HSR, other critical components of this network are mediated by nuclear factor (erythroid-derived 2)-like 1 (NFE2L1 or Nrf1) and nuclear factor (erythroid-derived 2)-like 2 (NFE2L2 or Nrf2). Nrf2 controls the antioxidant response by activating the expression of antioxidant and detoxification enzymes. Oxidative stress plays a major role in the pathogenesis of SBMA (Iida et al. 2015; Ranganathan and Fischbeck 2010) and neurodegeneration in general (Lin and Beal 2006). Nrf2 overexpression decreases neuronal loss in a mouse model of amyotrophic lateral sclerosis (Vargas et al. 2008). The Nrf1 pathway is strongly linked to the antioxidant stress response, which has been shown to enhance proteasome activity, thus representing a promising target for therapeutic intervention in neurodegenerative diseases (Radhakrishnan et al. 2010). To date, attempts to improve UPS activity to counteract ARpolyQ toxicity in SBMA have been unsuccessful. The major challenge is to find compounds that are safe but capable of inducing expression of several components of the proteasome, with the aim of enhancing its activity in cells. Paeoniflorin, a major component of Paeonia plants, has been shown to be effective in vivo in SBMA mice, where it mitigates the behavioral and pathological impairments typical of these model mice and induces ARpolyQ clearance via the UPS and autophagy (Tohnai et al. 2014). The molecular mechanism of paeoniflorin involves upregulation of nuclear factor-YA (NF-YA), a potent inducer of the PQC system. The effect of NF-YA is mediated by several molecular chaperones, CHIP and TFEB, thus possibly activating a general response to proteotoxic stress, which is able to activate both the UPS and autophagy, rather than to promote a focused and selective activity of the UPS.
In the context of the regulation of UPS, HDAC6 modulators have also been shown to exert a beneficial role. Their activity is mainly related to the tight functional relationship that exists between the UPS and autophagy. Indeed, in a Drosophila model of SBMA, HDAC6 overexpression was able to rescue the eye degeneration induced by ARpolyQ as a consequence of a malfunctioning UPS (see Beitel et al. (2013) for review). The action of HDAC6 was initially related to its potential to activate a compensatory activation of the autophagic pathway (see also below) in response to UPS impairment (Pandey et al. 2007), since HDAC6 also controls autophagosome maturation (Lee et al. 2010b). In addition, an enhanced chaperone response may also be involved (Hageman et al. 2010). Importantly, several selective modulators of HDAC6 are now available (d’Ydewalle et al. 2012; Dallavalle et al. 2012) and may have therapeutic potential in SBMA. Efforts in targeting the proteostatic networks and the UPS with small molecules are starting to show some promise: using a high-throughput screening, a selective small molecule inhibitor of the deubiquitinating activity of human USP14 has been recently identified and shown to enhance degradation of several proteasome substrates implicated in neurodegenerative diseases (Lee et al. 2012; Lee et al. 2010a). Therefore, modulation of small molecules of the proteostasis network represents a promising strategy for the treatment of SBMA and other diseases caused by the accumulation of misfolded proteins, as it offers the opportunity to both decrease the levels of the toxic protein and alleviate its pathogenic effects.
Conclusions
Based on the data reported above, SBMA onset and progression may depend on several alterations of the pathways composing the intracellular response to proteotoxic stress, known as the PQC system. These alterations are associated with the formation of testosterone-inducible misfolded species of the ARpolyQ responsible for SBMA, that, by escaping the PQC system, trigger a series of downstream cytotoxic events and accumulate in motoneuronal and muscle cells. In the last decades, the role of proteostasis in SBMA has been widely studied, not only for the understanding of the disease but also with the aim to find druggable targets to ameliorate disease course. All major components of the PQC system are deregulated in SBMA and their functions could be potentially rescued with appropriated molecular and pharmacological approaches, already tested both on SBMA cell and/or animal models. In particular, the first intracellular response, represented by molecular chaperones localized in the cytoplasm or associated with intracellular organelles (including ER), can be boosted with commercially available drugs that may be promising for future therapeutic approaches. It is possibly the failure or a downregulation of the chaperone system that, in SBMA, leads initially both to the ARpolyQ intracellular accumulation and to the activation of the degradative pathways, the UPR, the UPS, and the ALP. Since these three systems work synergically and in concert for counteracting the ARpolyQ toxicity, it may be more difficult to act selectively on one of these aiming for restoration of the global equilibrium on the PQC system. However, interesting approaches to enhance the functions of the degradative systems have been proposed (in particular of the autophagic system) as it has been described in this review. In the perspective of future therapeutic approaches, it will be important to deepen the temporal and spatial cross talk between UPS, UPR, and ALP for identifying other key molecules involved in the clearance of ARpolyQ.
Although several different therapeutic approaches have already been tested, SBMA is still without a cure. Fortunately, among the various effective compounds here reviewed, most of them able to modulate different players and able to enhance the PQC system activity in SBMA, it might be worth to test the possibility of their combined additive/synergic action. This might allow to tackle different steps of the pathways damaged by the ARpolyQ in target cells. The combined use of PQC modulators might represent a novel therapeutic perspective. Moreover, these compounds could be used even with several active drugs targeting other intracellular processes altered in SBMA.
References
Adachi H, Katsuno M, Minamiyama M, et al. (2003) Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci 23:2203–2211
Adachi H, Katsuno M, Minamiyama M, et al. (2005) Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain Res 128:659–670
Adachi H, Katsuno M, Waza M, Minamiyama M, Tanaka F, Sobue G (2009) Heat shock proteins in neurodegenerative diseases: pathogenic roles and therapeutic implications. Int J Hyperther 25(8):647–654. doi:10.3109/02656730903315823
Adachi H, Waza M, Tokui K, et al. (2007) CHIP overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. J Neurosci 27(19):5115–5126. doi:10.1523/jneurosci.1242-07.2007
Anagnostou, G., Akbar, M.T., Paul, P., Angelinetta, C., Steiner, T.J., de Belleroche, J. 2010. Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol Aging 31(969–985). doi:10.1016/j.neurobiolaging.2008.07.005.
Arndt V, Dick N, Tawo R, et al. (2010) Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol 20(2):143–148. doi:10.1016/j.cub.2009.11.022
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431(7010):805–810
Atsuta N, Watanabe H, Ito M, et al. (2006) Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain 129(Pt 6):1446–1455. doi:10.1093/brain/awl096
Azzouz M, Ralph GS, Storkebaum E, et al. (2004) VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 429(6990):413–417. doi:10.1038/nature02544
Bailey CK, Andriola IF, Kampinga HH, Merry DE (2002) Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum Mol Genet 11:515–523
Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319(5865):916–919. doi:10.1126/science.1141448
Beitel LK, Alvarado C, Mokhtar S, Paliouras M, Trifiro M (2013) Mechanisms mediating spinal and bulbar muscular atrophy: investigations into polyglutamine-expanded androgen receptor function and dysfunction. Front Neurol 4:53. doi:10.3389/fneur.2013.00053
Boyce M, Bryant KF, Jousse C, et al. (2005) A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307(5711):935–939. doi:10.1126/science.1101902
Boyer JG, Ferrier A, Kothary R (2013a) More than a bystander: the contributions of intrinsic skeletal muscle defects in motor neuron diseases. Front Physiol 4:356. doi:10.3389/fphys.2013.00356
Boyer JG, Murray LM, Scott K, De Repentigny Y, Renaud JM, Kothary R (2013b) Early onset muscle weakness and disruption of muscle proteins in mouse models of spinal muscular atrophy. Skelet Muscle 3(1):24. doi:10.1186/2044-5040-3-24
Campisi J, d’Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat rev Mol Cell Biol 8(9):729–740. doi:10.1038/nrm2233
Carra S (2009) The stress-inducible HspB8-Bag3 complex induces the eIF2alpha kinase pathway: implications for protein quality control and viral factory degradation? Autophagy 5(3):428–429
Carra S, Crippa V, Rusmini P, et al. (2012) Alteration of protein folding and degradation in motor neuron diseases: implications and protective functions of small heat shock proteins. Prog Neurobiol 97(2):83–100. doi:10.1016/j.pneurobio.2011.09.009
Carra S, Rusmini P, Crippa V, et al. (2013) Different anti-aggregation and pro-degradative functions of the members of the mammalian sHSP family in neurological disorders. Philos Trans R Soc Lond Ser B Biol Sci 368(1617):20110409. doi:10.1098/rstb.2011.0409
Carra S, Seguin SJ, Lambert H, Landry J (2008a) HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem 283(3):1437–1444. doi:10.1074/jbc.M706304200
Carra S, Seguin SJ, Landry J (2008b) HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 4(2):237–239
Carra S, Sivilotti M, Chavez Zobel AT, Lambert H, Landry J (2005) HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum Mol Genet 14(12):1659–1669
Chua JP, Reddy SL, Merry DE, et al. (2014) Transcriptional activation of TFEB/ZKSCAN3 target genes underlies enhanced autophagy in spinobulbar muscular atrophy. Hum Mol Genet 23(5):1376–1386. doi:10.1093/hmg/ddt527
Ciechanover A, Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47:e147. doi:10.1038/emm.2014.117
Colla E, Coune P, Liu Y, et al. (2012a) Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J Neurosci 32(10):3306–3320. doi:10.1523/JNEUROSCI.5367-11.2012
Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK (2012b) Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J Neurosci 32(10):3301–3305. doi:10.1523/JNEUROSCI.5368-11.2012
Cortes CJ, Ling SC, Guo LT, et al. (2014a) Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron 82(2):295–307. doi:10.1016/j.neuron.2014.03.001
Cortes CJ, Miranda HC, Frankowski H, et al. (2014b) Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat Neurosci 17(9):1180–1189. doi:10.1038/nn.3787
Crippa V, Boncoraglio A, Galbiati M, et al. (2013a) Differential autophagy power in the spinal cord and muscle of transgenic ALS mice. Front Cell Neurosci 7:234. doi:10.3389/fncel.2013.00234
Crippa V, Carra S, Rusmini P, et al. (2010a) A role of small heat shock protein B8 (HspB8) in the autophagic removal of misfolded proteins responsible for neurodegenerative diseases. Autophagy 6(7):958–960. doi:10.4161/auto.6.7.13042
Crippa V, Galbiati M, Boncoraglio A, et al. (2013b) Motoneuronal and muscle-selective removal of ALS-related misfolded proteins. Biochem Soc T 41(6):1598–1604. doi:10.1042/BST20130118
Crippa V, Sau D, Rusmini P, et al. (2010b) The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet 19(17):3440–3456. doi:10.1093/hmg/ddq257
Cudkowicz ME, Shefner JM, Simpson E, et al. (2008) Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve 38(1):837–844. doi:10.1002/mus.21059
Cvetanovic M, Patel JM, Marti HH, Kini AR, Opal P (2011) Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1. Nature Med 17(11):1445–1447. doi:10.1038/nm.2494
D’Ydewalle C, Bogaert E, Van Den Bosch L (2012) HDAC6 at the intersection of neuroprotection and neurodegeneration. Traffic 13(6):771–779. doi:10.1111/j.1600-0854.2012.01347.x
Dallavalle S, Pisano C, Zunino F (2012) Development and therapeutic impact of HDAC6-selective inhibitors. Biochem Pharmacol 84(6):756–765. doi:10.1016/j.bcp.2012.06.014
Dehay B, Bove J, Rodriguez-Muela N, et al. (2010) Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci 30(37):12535–12544. doi:10.1523/jneurosci.1920-10.2010
Doi H, Adachi H, Katsuno M, et al. (2013) p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J Neurosci 33(18):7710–7727. doi:10.1523/JNEUROSCI.3021-12.2013
Dossena M, Bedini G, Rusmini P, et al. (2014) Human adipose-derived mesenchymal stem cells as a new model of spinal and bulbar muscular atrophy. PLoS One 9(11):e112746. doi:10.1371/journal.pone.0112746
Fischbeck KH (1997) Kennedy disease. J Inher Metab Dis 20(2):152–158
Fischbeck KH (2012) Developing treatment for spinal and bulbar muscular atrophy. Prog Neurobiol 99(3):257–261. doi:10.1016/j.pneurobio.2012.05.012
Fontaine JM, Sun X, Hoppe AD, et al. (2006) Abnormal small heat shock protein interactions involving neuropathy-associated HSP22 (HSPB8) mutants. FASEB J 20(12):2168–2170
Foradori CD, Handa RJ (2008) Living or dying in three quarter time: neonatal orchestration of hippocampal cell death pathways by androgens and excitatory GABA. Exp Neurol 213(1):1–6. doi:10.1016/j.expneurol.2008.04.035
Fratta P, Collins T, Pemble S, et al. (2014a) Sequencing analysis of the spinal bulbar muscular atrophy CAG expansion reveals absence of repeat interruptions. Neurobiol Aging 35(2):443 e1-3. doi:10.1016/j.neurobiolaging.2013.07.015.
Fratta P, Nirmalananthan N, Masset L, et al. (2014b) Correlation of clinical and molecular features in spinal bulbar muscular atrophy. Neurology 82(23):2077–2084. doi:10.1212/WNL.0000000000000507
Fujinaga R, Takeshita Y, Uozumi K, et al. (2009) Microtubule-dependent formation of the stigmoid body as a cytoplasmic inclusion distinct from pathological aggresomes. Histochem Cell Biol 132(3):305–318. doi:10.1007/s00418-009-0618-9
Galbiati M, Crippa V, Rusmini P, et al. (2014) ALS-related misfolded protein management in motor neurons and muscle cells. Neurochem Int 79:70–78. doi:10.1016/j.neuint.2014.10.007
Gamerdinger M, Carra S, Behl C (2011) Emerging roles of molecular chaperones and co-chaperones in selective autophagy: focus on BAG proteins. J Mol Med 89(12):1175–1182. doi:10.1007/s00109-011-0795-6
Giorgetti E, Rusmini P, Crippa V, et al. (2015) Synergic prodegradative activity of bicalutamide and trehalose on the mutant androgen receptor responsible for spinal and bulbar muscular atrophy. Hum Mol Genet. 24(1):64–75. doi:10.1093/hmg/ddu419
Grunseich C, Kats IR, Bott LC, et al. (2014a) Early onset and novel features in a spinal and bulbar muscular atrophy patient with a 68 CAG repeat. Neuromuscular Disord 24(11):978–981. doi:10.1016/j.nmd.2014.06.441
Grunseich C, Rinaldi C, Fischbeck KH (2014b) Spinal and bulbar muscular atrophy: pathogenesis and clinical management. Oral Dis 20(1):6–9. doi:10.1111/odi.12121
Hageman J, Rujano MA, van Waarde MA, et al. (2010) A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol Cell 37(3):355–369. doi:10.1016/j.molcel.2010.01.001
Hargitai J, Lewis H, Boros I, et al. (2003) Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem Biophys Res Com 307(3):689–695
Heine, E.M., Berger, T.R., Pluciennik, A., Orr, C.R., Merry, D.E. 2015. Proteasome-mediated proteolysis of the polyglutamine-expanded androgen receptor is a late event in SBMA pathogenesis. J Biol Chem. doi:10.1074/jbc.M114.617894.
Hetz C, Mollereau B (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 15(4):233–249. doi:10.1038/nrn3689
Hipp MS, Park SH, Hartl FU (2014) Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol 24(9):506–514. doi:10.1016/j.tcb.2014.05.003
Howarth JL, Kelly S, Keasey MP, et al. (2007) Hsp40 molecules that target to the ubiquitin-proteasome system decrease inclusion formation in models of polyglutamine disease. Mol Ther 15(6):1100–1105. doi:10.1038/sj.mt.6300163
Huang X, Ding L, Bennewith KL, et al. (2009) Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell 35(6):856–867. doi:10.1016/j.molcel.2009.09.006
Iida M, Katsuno M, Nakatsuji H, et al. (2015) Pioglitazone suppresses neuronal and muscular degeneration caused by polyglutamine-expanded androgen receptors. Hum Mol Genet 24(2):314–329. doi:10.1093/hmg/ddu445
Irobi J, Almeida-Souza L, Asselbergh B, et al. (2010) Mutant HSPB8 causes motor neuron-specific neurite degeneration. Hum Mol Genet 19(16):3254–3265. doi:10.1093/hmg/ddq234
Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM (2002) Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem 277(33):29936–29944. doi:10.1074/jbc.M204733200
Ishihara K, Yamagishi N, Saito Y, et al. (2003) Hsp105alpha suppresses the aggregation of truncated androgen receptor with expanded CAG repeats and cell toxicity. J Biol Chem 278(27):25143–25150. doi:10.1074/jbc.M302975200
Iwata A, Christianson JC, Bucci M, et al. (2005) Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci USA 102:13135–13140
Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA (2002) Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci USA 99(18):11946–11950. doi:10.1073/pnas.182296499
Jochum T, Ritz ME, Schuster C, et al. (2012) Toxic and non-toxic aggregates from the SBMA and normal forms of androgen receptor have distinct oligomeric structures. Biochim Biophys Acta 1822(6):1070–1078. doi:10.1016/j.bbadis.2012.02.006
Johnston JA, Illing ME, Kopito RR (2002) Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil Cytoskeleton 53(1):26–38. doi:10.1002/cm.10057
Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143(7):1883–1898
Kakkar V, Meister-Broekema M, Minoia M, Carra S, Kampinga HH (2014) Barcoding heat shock proteins to human diseases: looking beyond the heat shock response. Dis Model Mech 7(4):421–434. doi:10.1242/dmm.014563
Kalmar B, Novoselov S, Gray A, Cheetham ME, Margulis B, Greensmith L (2008) Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J Neurochem 107(2):339–350. doi:10.1111/j.1471-4159.2008.05595.x
Kampinga HH, Craig EA (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nature Rev Mol Cell Biol 11(8):579–592. doi:10.1038/nrm2941
Katsuno M, Adachi H, Doyu M, et al. (2003) Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nature Med 9(6):768–773. doi:10.1038/nm878
Katsuno M, Adachi H, Kume A, et al. (2002) Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35(5):843–854
Katsuno M, Sang C, Adachi H, et al. (2005) Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. P Natl Acad Sci USA 102(46):16801–16806
Kennedy WR, Alter M, Sung JH (1968) Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-Linked recessive Trait Neurology 18:671–680
Kessel D (2006) Protection of Bcl-2 by salubrinal. Bioche Biophys Res Com 346(4):1320–1323. doi:10.1016/j.bbrc.2006.06.056
Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L (2004) Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med 10(4):402–405. doi:10.1038/nm1021
Klement IA, Skinner PJ, Kaytor MD, et al. (1998) Ataxin-1 nuclear localization and aggregation—role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95(1):41–53
Klionsky DJ, Abdalla FC, Abeliovich H, et al. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8(4):445–544
Kobayashi Y, Kume A, Li M, et al. (2000) Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem 275(12):8772–8778
Kondo N, Katsuno M, Adachi H, et al. (2013) Heat shock factor-1 influences pathological lesion distribution of polyglutamine-induced neurodegeneration. Nat Com 4:1405. doi:10.1038/ncomms2417
Kopito RR (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10(12):524–530
Kopito RR, Ron D (2000) Conformational disease. Nat Cell Biol 2(11):E207–E209. doi:10.1038/35041139
Kumar R (2012) Role of androgen receptor polyQ chain elongation in Kennedy’s disease and use of natural osmolytes as potential therapeutic targets. IUBMB Life 64(11):879–884. doi:10.1002/iub.1088
Kwok AS, Phadwal K, Turner BJ, et al. (2011) HspB8 mutation causing hereditary distal motor neuropathy impairs lysosomal delivery of autophagosomes. J Neurochem 119(6):1155–1161. doi:10.1111/j.1471-4159.2011.07521.x
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352(6330):77–79
Lambrechts D, Storkebaum E, Morimoto M, et al. (2003) VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet 34(4):383–394
Lee BH, Finley D, King RW (2012) A high-throughput screening method for identification of inhibitors of the deubiquitinating enzyme USP14. Curr Protoc Chem Biol 4(4):311–330. doi:10.1002/9780470559277.ch120078
Lee BH, Lee MJ, Park S, et al. (2010a) Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467(7312):179–184. doi:10.1038/nature09299
Lee JY, Koga H, Kawaguchi Y, et al. (2010b) HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 29(5):969–980. doi:10.1038/emboj.2009.405
Li M, Chevalier-Larsen ES, Merry DE, Diamond MI (2007) Soluble androgen receptor oligomers underlie pathology in a mouse model of SBMA. J Biol Chem 282:3157–3164
Li M, Miwa S, Kobayashi Y, et al. (1998a) Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol 44(2):249–254
Li M, Nakagomi Y, Kobayashi Y, et al. (1998b) Nonneural nuclear inclusions of androgen receptor protein in spinal and bulbar muscular atrophy. Am J Pathol 153(3):695–701
Lieberman AP, Yu Z, Murray S, et al. (2014) Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep 7(3):774–784. doi:10.1016/j.celrep.2014.02.008
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443(7113):787–795. doi:10.1038/nature05292
Machado P, Brady S, Hanna MG (2013) Update in inclusion body myositis. Curr Opin Rheumatol 25(6):763–771. doi:10.1097/01.bor.0000434671.77891.9a
Malena A, Pennuto M, Tezze C, et al. (2013) Androgen-dependent impairment of myogenesis in spinal and bulbar muscular atrophy. Acta Neuropathol 126(1):109–121. doi:10.1007/s00401-013-1122-9
Malik B, Nirmalananthan N, Bilsland LG, et al. (2011) Absence of disturbed axonal transport in spinal and bulbar muscular atrophy. Hum Mol Genet 20(9):1776–1786. doi:10.1093/hmg/ddr061
Malik B, Nirmalananthan N, Gray AL, La Spada AR, Hanna MG, Greensmith L (2013) Co-induction of the heat shock response ameliorates disease progression in a mouse model of human spinal and bulbar muscular atrophy: implications for therapy. Brain 136(Pt 3):926–943. doi:10.1093/brain/aws343
Montague K, Malik B, Gray AL, et al. (2014) Endoplasmic reticulum stress in spinal and bulbar muscular atrophy: a potential target for therapy. Brain 137(Pt 7):1894–1906. doi:10.1093/brain/awu114
Montie HL, Cho MS, Holder L, et al. (2009) Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 18(11):1937–1950. doi:10.1093/hmg/ddp115
Montie HL, Merry DE (2009) Autophagy and access: understanding the role of androgen receptor subcellular localization in SBMA. Autophagy 5(8):1194–1197
Montie HL, Pestell RG, Merry DE (2011) SIRT1 modulates aggregation and toxicity through deacetylation of the androgen receptor in cell models of SBMA. J Neurosci 31(48):17425–17436. doi:10.1523/JNEUROSCI.3958-11.2011
Nedelsky NB, Pennuto M, Smith RB, Palazzolo I, Moore J, Nie Z, Neale G, Taylor JP (2010) Native functions of the androgen receptor are essential to pathogenesis in a drosophila model of spinobulbar muscular atrophy. Neuron 67(6):936–952. doi:10.1016/j.neuron.2010.08.034
Nihei Y, Ito D, Okada Y, et al. (2013) Enhanced aggregation of androgen receptor in induced pluripotent stem cell-derived neurons from spinal and bulbar muscular atrophy. J Biol Chem. doi:10.1074/jbc.M112.408211
Onesto E, Rusmini P, Crippa V, et al. (2011) Muscle cells and motoneurons differentially remove mutant SOD1 causing familial amyotrophic lateral sclerosis. J Neurochem 118(2):266–280. doi:10.1111/j.1471-4159.2011.07298.x
Orr CR, Montie HL, Liu Y, et al. (2010) An interdomain interaction of the androgen receptor is required for its aggregation and toxicity in spinal and bulbar muscular atrophy. J Biol Chem 285(46):35567–35577. doi:10.1074/jbc.M110.146845
Palazzolo I, Gliozzi A, Rusmini P, et al. (2008) The role of the polyglutamine tract in androgen receptor. J Steroid Biochem Mol Biol 108(3–5):245–253. doi:10.1016/j.jsbmb.2007.09.016
Pandey UB, Nie Z, Batlevi Y, et al. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447(7146):859–863
Parfitt DA, Aguila M, McCulley CH, et al. (2014) The heat-shock response co-inducer arimoclomol protects against retinal degeneration in rhodopsin retinitis pigmentosa. Cell Death Dis 5:e1236. doi:10.1038/cddis.2014.214
Piccioni F, Pinton P, Simeoni S, et al. (2002) Androgen receptor with elongated polyglutamine tract forms aggregates that alter axonal trafficking and mitochondrial distribution in motor neuronal processes. FASEB J 16(11):1418–1420. doi:10.1096/fj.01-1035fje
Piccioni F, Simeoni S, Andriola I, et al. (2001) Polyglutamine tract expansion of the androgen receptor in a motoneuronal model of spinal and bulbar muscular atrophy. Brain Res Bull 56(3–4):215–220
Poletti A (2004) The polyglutamine tract of androgen receptor: from functions to dysfunctions in motor neurons. Front Neuroendocrinol 25(1):1–26. doi:10.1016/j.yfrne.2004.03.001
Poletti A, Negri-Cesi P, Martini L (2005) Reflections on the diseases linked to mutations of the androgen receptor. Endocrine 28(3):243–262. doi:10.1385/ENDO:28:3:243
Polo A, Teatini F, D’Anna S, et al. (1996) Sensory involvement in X-linked spino-bulbar muscular atrophy (Kennedy’s syndrome): an electrophysiological study. J Neurol 243(5):388–392
Pratt WB, Morishima Y, Gestwicki JE, Lieberman AP, Osawa Y (2014) A model in which heat shock protein 90 targets protein-folding clefts: rationale for a new approach to neuroprotective treatment of protein folding diseases. Exp Biol Med 239(11):1405–1413. doi:10.1177/1535370214539444
Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ (2010) Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell 38(1):17–28. doi:10.1016/j.molcel.2010.02.029
Ranganathan S, Fischbeck KH (2010) Therapeutic approaches to spinal and bulbar muscular atrophy. Trends Pharmacol Sci 31(11):523–527. doi:10.1016/j.tips.2010.08.005
Rinaldi C, Bott LC, Fischbeck KH (2014) Muscle matters in Kennedy’s disease. Neuron 82(2):251–253. doi:10.1016/j.neuron.2014.04.005
Rusmini P, Bolzoni E, Crippa V, et al. (2010) Proteasomal and autophagic degradative activities in spinal and bulbar muscular atrophy. Neurobiol Dis 40(2):361–369. doi:10.1016/j.nbd.2010.06.016
Rusmini P, Crippa V, Giorgetti E, et al. (2013) Clearance of the mutant androgen receptor in motoneuronal models of spinal and bulbar muscular atrophy. Neurobiol Aging 34(11):2585–2603. doi:10.1016/j.neurobiolaging.2013.05.026
Rusmini P, Sau D, Crippa V, et al. (2007) Aggregation and proteasome: the case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol Aging 28(7):1099–1111. doi:10.1016/j.neurobiolaging.2006.05.015
Rusmini P, Simonini F, Crippa V, et al. (2011) 17-AAG increases autophagic removal of mutant androgen receptor in spinal and bulbar muscular atrophy. Neurobiol Dis 41(1):83–95. doi:10.1016/j.nbd.2010.08.023
Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95(1):55–66
Saxena S, Cabuy E, Caroni P (2009) A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci 12(5):627–636. doi:10.1038/nn.2297
Seguin SJ, Morelli FF, Vinet J, et al. (2014) Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell Death Diff. doi:10.1038/cdd.2014.103
Senft D, Ronai ZA (2015) UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 40(3):141–148. doi:10.1016/j.tibs.2015.01.002
Settembre C, Ballabio A (2011) TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy 7(11):1379–1381
Simeoni S, Mancini MA, Stenoien DL, et al. (2000) Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum Mol Genet 9(1):133–144
Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A (1989) X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain 112:209–232
Sopher BL, Thomas Jr PS, LaFevre-Bernt MA, et al. (2004) Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron 41(5):687–699
Soraru G, D’Ascenzo C, Polo A, et al. (2008) Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J Neurol Sci 264(1–2):100–105. doi:10.1016/j.jns.2007.08.012
Stenoien DL, Cummings CJ, Adams HP, et al. (1999) Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet 8(5):731–741
Storkebaum E, Lambrechts D, Dewerchin M, et al. (2005) Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci 8(1):85–92
Suzuki K, Katsuno M, Banno H, et al. (2008) CAG repeat size correlates to electrophysiological motor and sensory phenotypes in SBMA. Brain 131(1):229–239. doi:10.1093/brain/awm289
Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7(9):880–885. doi:10.1038/sj.embor.7400779
Tadic V, Prell T, Lautenschlaeger J, Grosskreutz J (2014) The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front Cell Neurosci 8:147. doi:10.3389/fncel.2014.00147
Takeyama K, Ito S, Yamamoto A, et al. (2002) Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron 35(5):855–864
Taylor JP, Tanaka F, Robitschek J, et al. (2003) Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet 12:749–757
Thomas M, Dadgar N, Aphale A, et al. (2004) Androgen receptor acetylation site mutations cause trafficking defects, misfolding, and aggregation similar to expanded glutamine tracts. J Biol Chem 279(9):8389–8395
Thomas M, Harrell JM, Morishima Y, Peng HM, Pratt WB, Lieberman AP (2006) Pharmacologic and genetic inhibition of hsp90-dependent trafficking reduces aggregation and promotes degradation of the expanded glutamine androgen receptor without stress protein induction. Hum Mol Genet 15(11):1876–1883. doi:10.1093/hmg/ddl110
Thomas M, Yu Z, Dadgar N, et al. (2005) The unfolded protein response modulates toxicity of the expanded glutamine androgen receptor. J Biol Chem 280(22):21264–21271
Tohnai G, Adachi H, Katsuno M, et al. (2014) Paeoniflorin eliminates a mutant AR via NF-YA-dependent proteolysis in spinal and bulbar muscular atrophy. Hum Mol Genet 23(13):3552–3565. doi:10.1093/hmg/ddu066
Tokui K, Adachi H, Waza M, et al. (2009) 17-DMAG ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in an SBMA model mouse. Hum Mol Genet 18(5):898–910. doi:10.1093/hmg/ddn419
Vaccaro A, Patten SA, Aggad D (2013) Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol Dis 55:64–75. doi:10.1016/j.nbd.2013.03.015
Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA (2008) Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci 28(50):13574–13581. doi:10.1523/JNEUROSCI.4099-08.2008
Vigh L, Literati PN, Horvath I, et al. (1997) Bimoclomol: a nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nat Med 3(10):1150–1154
Wang AM, Miyata Y, Klinedinst S, et al. (2013) Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol 9(2):112–118. doi:10.1038/nchembio.1140
Waza M, Adachi H, Katsuno M, et al. (2005) 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med 11(10):1088–1095
Waza M, Adachi H, Katsuno M, et al. (2006a) Modulation of Hsp90 function in neurodegenerative disorders: a molecular-targeted therapy against disease-causing protein. J Mol Med 84(8):635–646
Waza M, Adachi H, Katsuno M, Minamiyama M, Tanaka F, Sobue G (2006b) Alleviating neurodegeneration by an anticancer agent: an Hsp90 inhibitor (17-AAG). Ann N Y Acad Sci 1086:21–34. doi:10.1196/annals.1377.012
Webb JL, Ravikumar B, Rubinsztein DC (2004) Microtubule disruption inhibits autophagosome-lysosome fusion: implications for studying the roles of aggresomes in polyglutamine diseases. Int J Biochem Cell Biol 36(12):2541–2550. doi:10.1016/j.biocel.2004.02.003
Whitaker HC, Hanrahan S, Totty N, et al. (2004) Androgen receptor is targeted to distinct subcellular compartments in response to different therapeutic antiandrogens. Clin Cancer Res 10(21):7392–7401
Xilouri M, Stefanis L (2015) Chaperone mediated autophagy to the rescue: a new-fangled target for the treatment of neurodegenerative diseases. Mol Cell Neurosci. doi:10.1016/j.mcn.2015.01.003
Yang YC, Fu HC, Hsiao BL, et al. (2013) Androgen receptor inclusions acquire GRP78/BiP to ameliorate androgen-induced protein misfolding stress in embryonic stem cells. Cell Death Dis 4:e607. doi:10.1038/cddis.2013.122
Yoneda T, Imaizumi K, Oono K, et al. (2001) Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem 276(17):13935–13940. doi:10.1074/jbc.M010677200
Yu Z, Wang AM, Adachi H, et al. (2011) Macroautophagy is regulated by the UPR-mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet 7(10):e1002321. doi:10.1371/journal.pgen.1002321
Acknowledgments
The following grants are gratefully acknowledged: Fondazione Telethon, Italy (n. GGP14039); Fondazione Cariplo, Italy (n. 2014–0686); Fondazione AriSLA, Italy (n. ALS_HSPB8 and ALS_Granulopathy); Association Française contre les Myopathies (AFM Telethon), France (n. 16,406); Regione Lombardia; Università degli Studi di Milano e piano di sviluppo UNIMI - linea B; and Italian Ministry of Health (n. GR-2011-02347198); EU Joint Programme - Neurodegenerative Disease Research (JPND).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rusmini, P., Crippa, V., Cristofani, R. et al. The Role of the Protein Quality Control System in SBMA. J Mol Neurosci 58, 348–364 (2016). https://doi.org/10.1007/s12031-015-0675-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-015-0675-6
Keywords
- Androgen receptor
- Polyglutamine
- CAG triplet repeat
- Neurodegeneration
- Motoneuron diseases
- Muscle
- SBMA
- Testosterone
- Autophagy
- Protein quality control system
- Proteotoxicity
- Proteasome
- UPR
- ERAD
- HSPB8
- Chaperones
- Misfolded protein
- Aggregation
- BAG3
- Rusmini Paola, Crippa Valeria, Greensmith Linda, and Poletti Angelo equally contributed to this study.