
Abstract
Parkinson's disease (PD) is the second most common neurodegenerative disease in humans. The effect of Krüppel-like factor (KLF) 4 in PD is unknown. In this study, KLF4 was found to be increased in both a time-dependent manner and a dose-dependent manner in response to the incubation with 1-methyl-4-phenylpyridinium (MPP+) in human dopamine neuroblastoma M17 cells, suggesting a potential role in MPP + −induced neurotoxicity. Following experiments showed that overexpression of KLF4 in M17 cells promoted MPP + −induced oxidative stress, embodied by exacerbated reactive oxygen species, 4-hydroxy-2-nonenal, and protein carbonyls. Furthermore, overexpression of KLF4 slowed cell proliferation and promoted lactate dehydrogenase release. Conversely, inhibition of KLF4 in M17 cells attenuated MPP + −induced neurotoxicity. The expression of superoxide dismutase (SOD) 1 in both mRNA and protein levels was found to be decreased by overexpressing KLF4, while increased by knockdown of KLF4. Moreover, promoter luciferase experiments showed that transcriptional activity on SOD1 was inhibited by KLF4. All the results indicated that KLF4 promoted the neurotoxicity of MPP + via inhibiting the transcription of SOD1, suggesting a potential mechanism of increased oxidative stress and cell death in Parkinson’s disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson's disease (also called Parkinson disease, PD) is a progressive degenerative central nervous system disorder, which is characterized by the loss of dopaminergic neurons and muscular rigidity (Cookson 2009). PD is the second most common neurodegenerative disease after Alzheimer’s disease of the elderly in the world. It attracted more and more attentions from both academic scientists and clinical physicians. The causes and mechanisms of PD are complex and still unknown. And it is considered as a multifactorial disease, resulting from interaction between one or more genes and the environment (Goedert et al. 2012).
1-Methyl-4-phenylpyridinium (MPP+), a major product of the oxidation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), has been extensively used in a variety of in vivo and in vitro systems to model PD. It has been reported that MPP + is actively transported into DAergic neurons through the plasma membrane in a dopamine transporter fashion (Kitao et al. 2007). Neurotoxicity of MPTP/MPP + is complicated. It involves the NO production (Barc et al. 2001), hydroxyl radical generation (Obata. 2006), and apoptosis. Moreover, MPP + was reported to be a potent inhibitor of complex I and thereby induce mitochondrial dysfunction (Dauer and Przedborski 2003). However, the cellular mechanisms of MPP-induced neurotoxicity underlying the degenerative process are not yet fully understood. Further understanding the mechanisms by which MPTP/MPP + leads to dopaminergic neuronal death is helpful to provide insights into therapeutic targets for PD.
Krüppel-like factor 4 (KLF4) is a member of the family of proteins KLF (Shields et al. 1996), which are transcription factors that exhibit homology to Krüppel from Drosophila (Ghaleb et al. 2005). KLF4 were reported to regulate multiple biological functions, including proliferation and differentiation. KLF4 also acts either as a tumor suppressor gene or as an oncogene in different kinds of cell types and cell contexts (Ghaleb et al. 2005; Rowland et al. 2005). Apoptosis has also been implicated in bladder cancer cells by KLF4 (Ohnishi et al. 2003) Previous studies have demonstrated that KLF4 was expressed in neural stem cells and acted as a transcriptional repressor of axonal growth in regenerating retinal ganglion cells (Moore et al. 2009). But whether KLF4 is involved in neurodegenerative disease, especially Parkinson’s disease, is still unknown.
In the present study, the effect of KLF4 in MPP + −induced neurotoxicity was investigated, and we demonstrated that KLF4-mediated transcriptional inactivation of superoxide dismutase 1 (SOD1) is critical in MPP + −induced oxidative stress and neurotoxicity.
Materials and Methods
Cell Culture, Treatment, and Transfection
The M17 neuroblastoma cell line was used in this study. Briefly, cells were maintained in Opti-MEM medium (Invitrogen, Eugene, OR, USA), supplemented with 5 % fetal bovine serum and 1 % penicillin–streptomycin, in a humid incubator with 5 % CO2 at 37 °C. Human KLF4 lentiviral vectors were purchased from Qiagen, USA. Briefly, HEK 293T cells were used to generate lentiviruses, and virus-containing supernatants were titrated on M17 cells to determine the titers needed to transduce > 95 % of the cells (Dekker et al. 2005). Loss-of-function experiments were performed with M17 transfected with nonspecific or KLF4-specific siRNA (50 nM, Dharmacon) using Lipofectamine RNAiMAX (Invitrogen). MPP + was used to treat M17 cells for various periods of time with various doses as follows: to detect the alternations of KLF4 expression in response to MPP + in a dose-dependent manner, cells were treated with MPP + for 48 h at the concentrations of 5, 10, 25, and 50 μM, respectively; to detect the alternations of KLF4 expression in response to MPP + in a time-dependent manner, cells were treated with 25 μM MPP + for 12, 24, and 48 h, respectively. For MTT and lactate dehydrogenase (LDH) assay, cells were treated with 50 μM MPP + for 48 or 72 h; for reactive oxygen species (ROS), 4-hydroxy-2-nonenal (4HNE), protein carbonyl, and SOD activity experiments, cells were treated with 50 μM MPP + for 48 h. A 100-μM H2O2 was used to treat M17 cells for 48 h as a positive control.
Measurement of Intracellular Reactive Oxygen Species
The intracellular ROS generation of cells was investigated using the 2′,7′-dichlorofluorescein-diacetate (DCFH-DA). Briefly, M17 cells were plated at a density of 1 × 105 cells/ml. After being incubated with the indicated concentrations of MPP+, cells were rinsed with Krebs’ ringer solution (100 mM NaCl, 2.6 mM KCl, 25 mM NaHCO3, 1.2 mM MgSO4, 1.2 mM KH2PO4, and 11 mM glucose), and 10 μM DCFH-DA was loaded. Then cells were incubated at 37 °C for 2 h and were washed five times with the same buffer. Fluorescence signals were examined under a fluorescence microscope. The average fluorescent density of intracellular areas was measured to index the ROS level by Image-Pro Plus.
Protein Carbonyl Assay
After diluted in phosphate-buffered saline (PBS), protein samples from cells were adsorbed to wells of an ELISA plate and then reacted with dinitrophenylhydrazine (DNPH), and the hydrazone adducts were probed by an anti-DNPH antibody, followed by quantification with a second antibody conjugated with horseradish peroxidase. The method was calibrated using oxidized BSA, prepared following the instructions described previously (Sheng et al. 2009a).
Determination of Superoxide Dismutase Activity
To determine the SOD activity in M17 cells after the indicated transfection and treatment, the SOD Assay Kit-WST (S311-08; Dojindo Laboratories, Kumamoto, Japan) was used in this study. Optical density (OD) values were recorded and normalized based on protein concentrations to reflect SOD activity according to the manual instruction.
4-Hydroxy-2-Nonenal Immunofluorescence
4-HNE is a major lipid peroxidation product. After the indicated transfection and incubation, M17 cells were fixed in 4 % paraformaldehyde for 10 min at RT, followed by permeabilization with 0.4 % Triton X-100 and blocked with 5 % BSA and 2.5 % FBS in PBST. Then cells were incubated with anti-4HNE (Cell Signaling, USA) for 2 h at RT and followed by Alexa-594 conjugated secondary antibodies (Invitrogen, USA) for 1 h at RT.
Lactate Dehydrogenase Determination
The release of LDH into medium is a marker of cell membrane integrity. Cells were plated in 24-well plates. After being treated with the indicated concentrations of MPP + for various periods of time, LDH activity measurements were performed using a commercially available assay (Roche Applied Science). Absorbance at 490 nm was read according to the manufacturer's protocol of LDH assay kit. The ratio of LDH activity in the supernatant to the total LDH activity was taken as the percentage of cell death.
Cell Viability
MTT reduction assay was used to determine cell viability. Under different conditions, M17 cells were cultured with the indicated concentrations of MPP + for various periods of time in 24-well plates. Briefly, MTT was added to each well with a final concentration of 1.0 mg/ml and incubated for 4 h at 37 °C. After removing MTT solution and washing for three times with PBS, the formed formazane crystal was dissolved in DMSO. The OD value was measured at 570 nm using a microplate reader (He et al. 2006).
Luciferase Reporter Gene Assay
An upstream fragment (approximately 2,200 bp) of SOD1 proximal promoters was generated by polymerase chain reaction (PCR) and subsequently cloned into pGL3 firefly luciferase reporter (Promega) using human genomic DNA as template in accordance with the manufacturer’s instructions. The following primers were used: 5′-AGGCTCGAGAGAATCACTTGAACCCAGCA-3′ and 5′-CGTAAGCTTCGCCATAACTCGCTAGGCCACGC-3′. Transfections and luciferase experiments were performed as described previously (Niemann et al. 2007). All transfections from at least three independent experiments were performed in triplicate.
Real-time Polymerase Chain Reaction
After various treatments, total RNA was isolated from cells using Trizol reagent (Invitrogen) according to the manufacturer’s protocol. After extraction, 2 μg of total RNA was then used as template for reverse transcription PCR to synthesize cDNA. After being diluted for ten times, the cDNA was used in quantitative real-time PCR analysis using SYBR Green qPCR Master Mix (Roche). The following primer pairs of human origin were used: KLF4, 5′-CAAGTCCCGCCGCTCCATTACCAA-3′ (forward) and 5′-CCACAGCCGTCCCAGTCACAGTGG-3′ (reverse). The following primers were used: catalase, 5′-CGTGCTGAATGAGGAACAGA-3′ (forward) and 5′-AGTCAGGGTGGACCTCAGTG-3′ (reverse); SOD1, 5′-GGCAAAGGTGGAAATGAAGA-3′ (forward) and 5′-GGGCCTCAGACTACATCCAA-3′ (reverse).
Western Blot Analysis
Whole-cell proteins lysate were seperated on 10 % SDS-PAGE and then transferred onto Immobilon-P membrane (Millipore, Billerica, MA, USA). After being blocked for 2 h at RT in TBS containing 10 % nonfat dry milk and 0.5 % Tween-20, membranes were sequentially incubated with primary antibodies for 3 h and horseradish peroxidase-conjugated secondary antibodies for 2 h. Blots were developed with Immobilon Western Chemiluminescent HRP Substrate (Millipore) or the Immunocruz (Santa Cruz Biotechnology) (Sheng et al. 2012).
Results
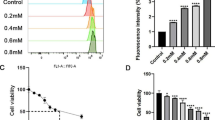
Firstly, we determined KLF4 expression in M17 cells incubated with MPP + for various periods of time and various doses. MPP + treatment at the concentrations of 5, 10, 25, and 50 μM on M17 cells for 48 h led to a sustainable increase in messenger RNA (mRNA) levels of KLF4 (Fig. 1a) and in protein levels (Fig. 1b). Moreover, M17 cells were incubated with 25 μM MPP + for various time. And the result showed a sustainable increase at both mRNA levels (Fig. 1c) and protein levels of KLF4 in a time-dependent manner from 12 to 48 h (Fig. 1d). To support this finding, the expression levels of KLF4 in H2O2-treated M17 cells were investigated as a positive control, and the results showed that the expression of KLF4 increased at both mRNA and proteins levels after treatment with 100 μM H2O2 for 48 h (Fig. 1e, f). The increased levels of KLF4 induced by MPP + suggested a potential role in MPP + neurotoxicity.

Expression of KLF4 in M17 cells after MPP + treatment. M17 cells were stimulated with MPP + at various concentrations for 48 h, and a mRNA levels of KLF4 at various concentrations were determined by real-time PCR (the number sign indicates P < 0.01 vs nontreated control, n = 4). b Protein levels of KLF4 at various concentrations were determined by western blot analysis (the number sign indicates P < 0.01 vs nontreated control, n = 4). M17 cells were stimulated with MPP + at indicated doses (25 μM) for varying periods of time. c mRNA levels of KLF4 at varying time periods were determined by real-time PCR. The relative values of all results were determined and expressed as the mean ± SEM of three experiments performed in duplicate (the number sign indicates P < 0.01 vs nontreated control, n = 4). d Protein levels of KLF4 at varying time periods were determined by western blot analysis. e mRNA levels of KLF4 were significantly increased by incubation with 100 μM H2O2 for 48 h. f Protein levels of KLF4 were significantly increased by incubation with 100 μM H2O2 for 48 h (the number sign indicates P < 0.01 vs nontreated control, n = 4)
We overexpressed KLF4 in M17 cells using lentiviral KLF4 construct, with the null as control. Cell viability was determined by MTT reduction assay (Sheng et al. 2009b). As shown in Fig. 2a, b, MTT assay results showed that overexpression of KLF4 exacerbated the impaired cell viability induced by 50 μM MPP + treatment for 48 and 72 h. To confirm that overexpression of KLF4 increased cell vulnerability to MPP+, the levels of cellular toxicity were determined using a LDH assay. As expected, overexpressing KLF4 in M17 cells exacerbated MPP + −induced LDH release after 48 and 72 h of incubation (Fig. 2c, d).

Cell viability and LDH release in KLF4-overexpressed M17 cells after incubation with MPP+. Neo, null control group; KLF4, KLF4 overexpression group. a After incubation with MPP + for 48 h, MTT assay results revealed that KLF4 overexpressing cells were more vulnerable to MPP + −induced neurotoxicity (the asterisk indicates P < 0.01 vs Neo group; the number sign indicates P < 0.01 vs Neo + (MPP+) group). b After incubation with MPP + for 72 h, MTT assay results revealed that KLF4 overexpressing cells were more vulnerable to MPP + −induced neurotoxicity (the asterisk indicates P < 0.01 vs Neo group; the number sign indicates P < 0.01 vs Neo + (MPP+) group). c After incubation with MPP + for 48 h, LDH assay results revealed that KLF4 overexpressing cells showed recued resistibility to MPP + −induced neurotoxicity (the asterisk indicates P < 0.01 vs Neo group; the number sign indicates P < 0.01 vs Neo + (MPP+) group). d After incubation with MPP + for 72 h, LDH assay results revealed that KLF4 overexpressing cells showed recued resistibility to MPP + −induced neurotoxicity (the asterisk indicates P < 0.01 vs Neo group; the number sign indicates P < 0.01 vs Neo + (MPP+) group)
The oxidative stress is the main cause of MPP + −induced neurotoxicity. Thus, the oxidative stress patterns were investigated. The levels of ROS, 4HNE, and protein carbonyls are three essential parameters of oxidative stress. To study the possible roles of KLF4 in regulating oxidative stress, we measured the intracellular ROS, 4HNE levels, and protein carbonyls in M17 cells after MPP + incubation and KLF4 transfection. The results indicated that overexpression of KLF4 promoted the increased levels of ROS (Fig. 3a), 4HNE (Fig. 3b), and protein carbonyls (Fig. 3c) induced by MPP + treatment. Increased susceptibility to oxidative conditions upon KLF4 overexpression is in agreement with the studies showing that transient overexpression of KLF4 in human myeloid leukemia cells increased hydrogen peroxide-induced apoptosis (Li et al. 2010). Furthermore, MPP + significantly decreased SOD activity, which was exacerbated by overexpression of KLF4 (Fig. 3d).

Overexpression of KLF 4 promoted MPP + −induced oxidative stress. Neo, null control group; KLF4, KLF4 overexpression group. a After incubation with MPP+, KLF4 overexpressing cells showed more intracellular ROS accumulation than neo control (the asterisk indicates P < 0.01 vs Neo group; the number sign indicates P < 0.01 vs Neo + (MPP+) group). b After incubation with MPP+, KLF4 overexpressing M17 cells exhibited a significant increase of 4HNE (the asterisk indicates P < 0.01 vs Neo group; the number sign indicates P < 0.01 vs Neo + (MPP+) group). c After incubation with MPP+, KLF4 overexpressing M17 cells displayed a significant increase of protein carbonyls (the asterisk indicates P < 0.01 vs Neo group; the number sign indicates P < 0.01 vs Neo + (MPP+) group). d Overexpressing KLF4 in M17 cells exacerbated reduced SOD activity induced by MPP + (the asterisk indicates P < 0.01 vs Neo group; the number sign indicates P < 0.01 vs Neo + (MPP+) group)
On further study, we investigated the effects of KLF4 in MPP + neurotoxicity with inhibition of KLF4 using KLF4 small RNA interference in M17 cells. As shown in Fig. 4a, b, MTT assay results showed that inhibition of KLF4 could rescue the impaired cell viability induced by MPP + for 48 and 72 h. The integrity of cell membrane was determined by the release of LDH after 48 and 72 h of exposure to MPP+, and results displayed that inhibition of KLF4 protected M17 cells from MPP + −induced LDH release (Fig. 4c, d).

Cell viability and LDH release in KLF4 knockdown M17 cells after incubation with MPP+. siKLF4, KLF4 siRNA group; NS, nonspecific siRNA group. a After incubation with MPP + for 48 h, MTT assay results revealed that KLF4 knockdown cells were more vulnerable to MPP + −induced neurotoxicity (the asterisk indicates P < 0.01 vs NS group; the number sign indicates P < 0.01 vs NS + (MPP+) group). b After incubation with MPP + for 72 h, MTT assay results revealed that KLF4 knockdown cells were more vulnerable to MPP + −induced neurotoxicity (the asterisk indicates P < 0.01 vs NS group; the number sign indicates P < 0.01 vs NS + (MPP+) group). c After incubation with MPP + for 48 h, LDH assay results revealed that KLF4 knockdown cells showed recued resistibility to MPP + −induced neurotoxicity (the asterisk indicates P < 0.01 vs NS group; the number sign indicates P < 0.01 vs NS + (MPP+) group). d After incubation with MPP + for 72 h, LDH assay results revealed that KLF4 knockdown cells showed recued resistibility to MPP + −induced neurotoxicity (the asterisk indicates P < 0.01 vs NS group; the number sign indicates P < 0.01 vs NS + (MPP+) group)
Oxidative stress was observed in PD brains. We further measured the intracellular ROS, 4HNE levels, and protein carbonyls in M17 cells after MPP + incubation and KLF4 inhibition. The results indicated that inhibition of KLF4 attenuated the increased levels of ROS (Fig. 5a), 4HNE (Fig. 5b), and protein carbonyls (Fig. 5c) induced by MPP + treatment. Moreover, MPP + significantly decreased SOD activity, which could be protected by knockdown of KLF4 (Fig. 5d).

Knockdown of KLF4 protected MPP + −induced oxidative stress. siKLF4, KLF4 siRNA group; NS, nonspecific siRNA group. a After MPP + treatment, KLF4 knockdown cells showed less intracellular ROS accumulation than nonspecific controls (the asterisk indicates P < 0.01 vs NS group; the number sign indicates P < 0.01 vs NS + (MPP+) group). b After incubation with MPP+, KLF4 knockdown M17 cells exhibited a significant decrease of 4HNE compared with nonspecific controls (the asterisk indicates P < 0.01 vs NS group; the number sign indicates P < 0.01 vs NS + (MPP+) group). c After incubation with MPP+, KLF4 knockdown M17 cells exhibited a significant reduction of protein carbonyls (the asterisk indicates P < 0.01 vs NS group; the number sign indicates P < 0.01 vs NS + (MPP+) group). d Overexpressing KLF4 in M17 cells reversed reduced SOD activity induced by MPP + (the asterisk indicates P < 0.01 vs NS group; the number sign indicates P < 0.01 vs NS + (MPP+) group)
In order to explore the mechanisms of which the reduced resistance to the oxidant stress challenge induced by KLF4, we measured the expression patterns of catalase and SOD 1, two important cellular oxidative defense mechanisms. The level of catalase was not influenced by KLF4 (Fig. 6a for mRNA and Fig. 6b for protein), while the level of SOD1 was significantly reduced by overexpressing KLF4 at both mRNA (Fig. 6a) and protein levels (Fig. 6b). Conversely, the inhibition of KLF4 by siRNA was detected by real-time PCR and Western blot analysis, resulting in induction of the expressions of SOD1 at both mRNA levels (Fig. 6c) and protein levels (Fig. 6d), without affecting the expression levels of catalase. These results suggested a potentially negative effect of KLF4 on SOD1. In order to understand how KLF4 regulates SOD1, we detected its effects on their promoter activity of SOD1. A strong transinactivation effect on the SOD1 promoter is shown in Fig. 6e after transfection with KLF4.

KLF4 inhibited SOD1 expression at transcriptional level in M17 cell. Neo, null control group; KLF4, KLF4 overexpression group; siKLF4, KLF4 siRNA group; NS, nonspecific siRNA group. a Overexpression of KLF4 decreased mRNA levels of SOD1 but not catalase. b Overexpression of KLF4 decreased protein levels of SOD1 but not catalase. c Knockdown of KLF4 increased mRNA levels of SOD1 but not catalase. d Knockdown of KLF4 increased protein levels of SOD1 but not catalase. e KLF4 inhibited SOD1 promoter activity. Activity of SOD1 promoter was measured in M17 cells after overexpression of lentiviral KLF4; normalized (firefly/protein concentration) promoter activity is expressed relative to vector control (the number sign indicates P < 0.01; n = 4, Student’s t test)
Discussion
PD is a progressive neurodegenerative disease caused by the death of dopaminergic neurons in the substantia nigra. The pathogenesis of PD still needs to be elucidated. Previous studies have reported that multiple pathways were involved in PD. These pathways included oxidative and nitrosative stress, defects in the ubiquitin–proteasome system, protein aggregation, mitochondrial damage, and apoptosis (Dawson and Dawson 2003). But the activation mechanism of these pathways remains unknown. Dopaminergic human neuroblastoma M17 cell lines, which were reported to possess dopamine synthetic, metabolistic, and transporting enzymes, have been widely used to study MPP + −induced neurotoxicity in PD (Marco et al. 2010). The present study demonstrates that regulation of SOD1 mediated by KLF4 is crucial for oxidative stress and cell death caused by MPP+. In this investigation, we first detected the expression of KLF4 in M17 cells and found that KLF4 levels were increased by MPP + in both a time-dependent manner and a dose-dependent manner. Overexpression of KLF4 promoted the neurotoxicity of MPP + in the forms of increasing cell vulnerability and oxidative stress, while knockdown of KLF4 attenuated the neurotoxicity of MPP+. These data suggest a potential role for KLF4 in MPP + neurotoxicity.
Oxidative damage is responsible for most of the neurodegenerative diseases including PD. Antioxidant status is contributive in playing a critical role in protecting MPP + −induced dopaminergic neuron loss. SOD is the most important enzyme in all kinds of tissues and protects against oxidative stress. It also can help to prevent neuronal cells from apoptosis. Reduced SOD1 expression in M17 cells is associated with alternation of KLF4. KLF transcription factors was reported to bind to “CACCC” elements or “CT box”, and GC-rich sequences that also serve as binding sites for Sp1/3 transcription factors (Bieker 2001; Wu and Lingrel 2004). The presented data demonstrated that MPP + −induced KLF4 expression leads to reduced SOD1 expression in M17 cells. Reduced SOD activity induced by MPP + is in consistence with the finding that MPP + significantly decreased GSH level and SOD activity in the rat primary neurons (Lo et al. 2012).The result was verified by the data that overexpression of KLF4 could reduce the level of SOD1, while knockdown of KLF4 could increase the level of SOD1. A previous study demonstrated that the SOD1 promoter contained multiple transcription factor binding motifs including Sp1 and Egr-1 binding sites (Minc et al. 1999). Thus, it may be speculated that KLF4 binds to the SOD1 promoter and thereby inhibits transcriptional activity of SOD1.
KLF4 has been implicated with slowing cell proliferation and apoptosis in a lot of studies. Previous studies indicated that KLF4 blocked the transition from the G (1) to S phase in the cell cycle to slow cell growth through repressing several positive cell-cycle regulatory gene promoters, such as cyclin D1 and ornithine decarboxylase (Wassmann et al. 2007; Shie et al. 2000; Chen et al. 2000). KLF4 was reported to induce apoptosis in leukemia cells involving the bcl-2/bax pathway during H2O2 stimulation through transactivating the promoter activity of bax and transinactivating the promoter activity of bcl-2 (Li et al. 2010). A recent study reported that the expression of KLF4 induced by H2O2 could be decreased by quercetin in human neuroblastoma SH-SY5Y cells, suggesting the mediated role of KLF4 for the protective effect of quercetin on cell damage induced by H2O2 (Xi et al. 2012). Moreover, overexpression of KLF4 was demonstrated to participate in heat stress-induced apoptosis in murine macrophages (Liu et al. 2007). There are some evidences showed that KLF4 could act as a tumor suppressor for both gastric and colorectal cancers through promoting apoptosis (Li et al. 2002). Although these evidences did not occur in the central neural system, they still supported our findings that KLF4 could slow cell proliferation and induce apoptosis. Microglial inflammation is observed in PD. It was reported that overexpression of KLF4 resulted in the increased production of iNOS and Cox-2 in BV-2 cells associated with a decrease of anti-inflammatory activity of honokiol (HNK), suggesting KLF4 as a potential target for therapeutic activities of HNK (Kaushik et al. 2012).
In summary, our study demonstrated the increasing expression of KLF4 in M17 cells after MPP + incubation, and it was identified that KLF4 reduced the expression of SOD1, potentially resulting in the neurotoxicity on M17 cells induced by MPP+. Our findings suggest an implication that KLF4 is a potential therapeutic target for PD treatment. Further research will provide us with a more complete picture of the underlying mechanisms in PD and other neurodegenerative diseases.
References
Barc S, Page G, Barrier L, Piriou A, Fauconneau B (2001) Impairment of the neuronal dopamine transporter activity in MPP + −treated rat was not prevented by treatments with nitric oxide synthase or poly (ADP-ribose) polymerase inhibitors. Neurosci Lett 314:82–86
Bieker JJ (2001) Kruppel-like factors: three fingers in many pies. J Biol Chem 276:34355–34358
Chen ZY, Shie J, Tseng C (2000) Up-regulation of gut-enriched Krüppel-like factor by interferon-gamma in human colon carcinoma cells. FEBS Lett 477:67–72
Cookson MR (2009) Alpha-synuclein and neuronal cell death. Mol Neurodegener 4:9
Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39:889–909
Dawson TM, Dawson VL (2003) Molecular pathways of neurodegeneration in Parkinson’s disease. Science 302:819–822
Dekker RJ, van Thienen JV, Rohlena J et al (2005) Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone-regulating genes. Am J Pathol 167(2):609–618
Ghaleb AM, Nandan MO, Chanchevalap S, Dalton WB, Hisamuddin IM, Yang VW (2005) Kruppel-like factors 4 and 5: the yin and yang regulators of cellular proliferation. Cell Res 15(2):92–96
Goedert M, Spillantini MG, Del Tredici K, Braak H (2012) 100 years of Lewy pathology. Nat Rev Neurol 10:242
He Y, Zhou H, Tang H, Luo Y (2006) Deficiency of disulfide bonds facilitating fibrillogenesis of endostatin. J Biol Chem 281(2):1048–1057
Kaushik DK, Mukhopadhyay R, Kumawat KL, Gupta M, Basu A (2012) Therapeutic targeting of Krüppel-like factor 4 abrogates microglial activation. J Neuroinflammation 9:57
Kitao Y, Matsuyama T, Takano K et al (2007) Does ORP150/HSP12A protect dopaminergic neurons against MPTP/MPP(+)-induced neurotoxicity? Antioxid Redox Signal 9:589–595
Li QL, Ito K, Sakakura C et al (2002) Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 109:113–124
Li Z, Zhao J, Li Q et al (2010) KLF4 promotes hydrogen-peroxide-induced apoptosis of chronic myeloid leukemia cells involving the bcl-2/bax pathway. Cell Stress Chaperones 15(6):905–912
Liu MD, Liu Y, Liu JW, Zhang HL, Xiao XZ (2007) Effect of Kruppel-like factor 4 overexpression on heat stress-induced apoptosis of Raw264.7 macrophages. Zhong Nan Da Xue Xue Bao Yi Xue Ban 32:1002–1006
Lo YC, Shih YT, Tseng YT, Hsu HT (2012) Neuroprotective effects of San-Huang-Xie-Xin-Tang in the MPP(+)/MPTP models of Parkinson's disease in vitro and in vivo. Evid Based Complement Alternat Med 2012:501032
Marco B, Elisa G, Dragan M et al (2010) Alpha-synuclein overexpression increases dopamine toxicity in BE2-M17 cells. BMC Neurosci 11:41
Minc E, de Coppet P, Masson P et al (1999) The human copper-zinc superoxide dismutase gene (SOD1) proximal promoter is regulated by Sp1, Egr-1, and WT1 via non-canonical binding sites. J Biol Chem 274(1):503–509
Moore DL, Blackmore MG, Hu Y et al (2009) KLF family members regulate intrinsic axon regeneration ability. Science 326:298–301
Niemann HH, Jäger V, Butler PJ et al (2007) Structure of the human receptor tyrosine kinase Met in complex with the Listeria invasion protein lnlB. Cell 130(2):235–246
Obata T (2006) Nitric oxide and MPP + −induced hydroxyl radical generation. J Neural Transm 113(9):1131–1144
Ohnishi S, Ohnami S, Laub F et al (2003) Downregulation and growth inhibitory effect of epithelial-type Kruppel-like transcription factor KLF4, but not KLF5, in bladder cancer. Biochem Biophys Res Commun 308(2):251–256
Rowland BD, Bernards R, Peeper DS (2005) The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol 7:1074–1082
Sheng B, Gong K, Niu Y et al (2009a) Inhibition of gamma-secretase activity reduces Abeta production, reduces oxidative stress, increases mitochondrial activity and leads to reduced vulnerability to apoptosis: implications for the treatment of Alzheimer's disease. Free Radic Biol Med 46(10):1362–1375
Sheng B, Song B, Zheng Z et al (2009b) Abnormal cleavage of APP impairs its functions in cell adhesion and migration. Neurosci Lett 450(3):327–331
Sheng B, Wang X, Su B et al (2012) Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer's disease. J Neurochem 120(3):419–429
Shie JL, Chen ZY, Fu M, Pestell RG, Tseng CC (2000) Gut-enriched Krüppel-like factor represses cyclin D1 promoter activity through Sp1 motif. Nucleic Acids Res 28:2969–2976
Shields JM, Christy RJ, Yang VW (1996) Identification and characterization of a gene encoding a gut-enriched Kruppel-like factor expressed during growth arrest. J Biol Chem 271:20009–200017
Wassmann S, Wassmann K, Jung A et al (2007) Induction of p53 by GKLF is essential for inhibition of proliferation of vascular smooth muscle cells. J Mol Cell Cardiol 43:301–307
Wu J, Lingrel JB (2004) KLF2 inhibits Jurkat T leukemia cell growth via upregulation of cyclin-dependent kinase inhibitor p21WAF1/CIP1. Oncogene 23:8088–8096
Xi J, Zhang B, Luo F, Liu J, Yang T (2012) Quercetin protects neuroblastoma SH-SY5Y cells against oxidative stress by inhibiting expression of Krüppel-like factor 4. Neurosci Lett 527(2):115–120
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chen, J., Wang, X., Yi, X. et al. Induction of KLF4 Contributes to the Neurotoxicity of MPP + in M17 Cells: A New Implication in Parkinson’s Disease. J Mol Neurosci 51, 109–117 (2013). https://doi.org/10.1007/s12031-013-9961-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-013-9961-3