
Abstract
Alpha-synuclein gene (SNCA) polymorphisms have been associated with the common sporadic form of Parkinson’s disease (PD). We searched for DNA variants at the SNCA 3′ UTR through single strand conformation analysis and direct sequencing in a cohort of Spanish PD patients and controls. We have genotyped the rs356165 SNCA 3′ UTR polymorphism in a total of 1,135 PD patients and 772 healthy controls from two Spanish cohorts (Asturias and Navarre). We identified six SNCA 3′ UTR variants. Single nucleotide polymorphism (SNP) rs356165 was significantly associated with PD risk in the Spanish cohort (p = 0.0001; odd ratio = 1.37, 95%CI = 1.19–1.58). This SNP was also significantly associated with early age at onset of PD. Our work highlights rs356165 as an important determinant of the risk of developing PD and early age at onset and encourages future research to identify a functional effect on SNCA expression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) (OMIM *168600) is the second most common neurodegenerative disorder affecting 1–2% of people older than 60 years (de Lau and Breteler 2006). Mutations at several genes have been linked to rare familial forms of PD. Most of the PD cases are sporadic, although a family history of the disease is also a well-recognized risk factor for PD (Dickson et al. 2009). Sporadic PD has a multifactorial origin owing to the interaction of genetic/inherited and environmental/acquired risk factors (Warner and Schapira 2003). Rare mutations and genomic rearrangements in the alpha-synuclein gene (SNCA) account for approximately 2% of the autosomal dominant familial forms of early-onset PD (Polymeropoulos et al. 1997). SNCA encodes a pre-synaptic protein that is the main component of the Lewy bodies, the pathological hallmark of sporadic and many familial PD cases (Spillantini et al. 1997).
SNCA polymorphisms have been associated with sporadic PD through several case–control studies (Mizuta et al. 2006; Mueller et al. 2005; Pals et al. 2004; Yu et al. 2010). Recent genome-wide association studies confirmed the association between some of these single nucleotide polymorphisms (SNPs) with PD, with odd ratios (OR) in the range 1.20–1.70 (Edwards et al. 2010; Satake et al. 2009; Simon-Sanchez et al. 2009). Some of them mapped downstream of the SNCA gene and a direct functional effect was thus unlikely. For 3′UTR variants, post-transcriptional regulation of SNCA expression through binding of micro-RNAs (miRNAs) to this region could explain the association to PD (Junn et al. 2009; Doxakis 2010). In this context, the association of some SNPs/haplotypes with PD could be explained by Linkage Disequilibrium (LD) with some functional variants in the 3′UTR. To address this issue, we characterized the SNCA 3′UTR variation in a large cohort of PD patients and healthy controls. We found several variants and a significant effect of the SNP rs356165 on PD risk and disease age at onset.
Methods
Patients and Controls
PD patients and healthy controls were Caucasians from two different Spanish regions (Asturias and Navarre). The Asturias cohort consisted of 752 PD patients (mean age at diagnosis 59 ± 13 years; 47% male) and 480 healthy controls (mean age at enrollment 62 ± 18 years; 55% male), and the Navarre cohort consisted of 417 PD patients (mean age at diagnosis 58 ± 11 years; 62% male) and 292 healthy controls (mean age 63 ± 14 years; 35% male). Table 1 summarizes the main values of these patients and controls. All the patients were recruited in the period January 2002–December 2010 by neurologists from the movement disorder units of five reference hospitals: Central de Asturias, Gijón, Mieres and Avilés (Asturias cohort), and Clínica Universitaria de Navarra (Navarre cohort). PD was diagnosed according to the UK Parkinson’s Disease Society Brain Bank clinical criteria (Hughes et al. 1992). Patients with age at onset <50 or ≥50 years were classified as early-onset (EO) and late-onset (LO) PD, respectively.
The controls were healthy spouses of the patients and healthy donors from the general population. None of the controls presented PD at the age of enrollment. This study was approved by the ethical committee of Hospital Universitario Central de Asturias and the University of Navarra Ethical Committee. All the patients and controls signed an informed consent to participate in the project.
SNCA 3′UTR Analysis
The SNCA 3′UTR was amplified in the 727 patients and 480 controls from the Asturias cohort in four overlapping fragments, which were subjected to Single Strand Conformation Analysis (SSCA) to identify nucleotide changes (Supplementary Table 1). Fragments with atypical electrophoresis patterns were sequenced to characterize the nucleotide changes (Supplementary Figure 1).
SNP rs356165 Genotyping
SNP rs356165 was also genotyped through polymerase chain reaction followed by Restriction Fragment Length Polymorphism (PCR-RFLP) with TaiI and electrophoresis in 4% agarose gels to visualize the two alleles (Supplementary Figure 1). To confirm the accuracy of the genotyping method, we sequenced several individuals representative of each genotype as standard samples.
LRRK2 Mutations
Genomic DNA was obtained from blood and the three common LRRK2 mutations (R1441G and R1441C in exon 31 and G2019S in exon 41) were determined in all the patients through direct sequencing of PCR fragments, as reported (Mata et al. 2006).
Statistical Analysis
Differences between allelic and genotype frequencies were compared through a Chi-squared test. The Chi-squared test was also used to determine whether the observed genotype frequencies differed from those expected under the Hardy–Weinberg equilibrium. The Student’s T test was used to compare the mean age between two groups (risk alleles and risk genotypes between patients and controls or EO vs. LO patients). The effect of each variable (genotype, sex, and age) on PD risk was calculated through multiple logistic regressions. A p < 0.01 was considered as statistically significant.
Results
SNCA 3′UTR Genetic Variation
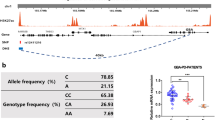
To determine whether 3′ UTR variants could be associated with PD, we screened the SNCA region in patients and controls from Asturias (Table 1 summarizes the main characteristics of patients and controls). A total of four overlapping fragments were amplified and subjected to SSCA. After sequencing all the heterogeneous electrophoresis patterns, we identified four previously reported SNPs: c.404 C>T (missense for tyrosine136, rs76642636), c.*139 T>G (rs10024743), c.*501 C>T (rs17016074), and c.*893 A>G (rs356165) and two new variants: c.*575_579del (ATTTT deletion) and c.*502 G>A (Supplementary Figure 1). With the exception of rs356165, the minor allele frequencies were <0.01 (Table 2). Allele rs356165 G was significantly more frequent in the patients group (p < 0.0001; OR = 1.42; 95%CI = 1.19–1.69).
Replication of the Association Between rs356165 and PD
SNP rs356165 was initially genotyped through SSCA in the Asturias PD patients and controls. We confirmed the patients and controls genotypes through PCR-RFLP and genotyped an additional cohort from a different population (Navarre). We confirmed the association between this SNP and PD, with a significantly higher frequency of the rs356165 G allele among the patients. Rs356165 AG and GG frequencies were higher among the patients (AA vs. AG + GG p < 0.0001, OR = 1.37, 95%CI = 1.19–1.58), suggesting a dominant effect for this allele on PD risk (Table 3). After performing a multiple logistic regression including age at onset, sex, and rs356165 as covariates, this SNP remained significantly associated with PD (p < 0.0001, OR = 1.34, 95%CI = 1.09–1.65).
SNP rs356165 is a Modifier of Age at Onset
The disease age at onset was significantly different between rs356165 genotypes, carriers of the rs356165 G allele showed a significantly lower disease age at onset compared to AA homozygote (57.6 ± 12.5 vs. 60.10 ± 12.8; p = 0.003). This effect can be also observed when comparing the genotype frequencies between the EO and LO groups (Table 4).
We also analyzed the effect of rs356165 on the age at onset in patients with one of the three common LRRK2 mutations. We found a lower mean age at onset among rs356165 G carriers (53.4 ± 13.2 vs. 58.8 ± 11.7) and a higher frequency of AG + GG among the LRRK2+ EO patients (Table 4). However, these were no significant differences maybe because they were based only on 34 patients.
Discussion
SNCA mutations have been linked to familial autosomal dominant PD (A53T, A30P, E46K) (Polymeropoulos et al. 1997; Kruger et al. 1998; Zarranz et al. 2004). In an analysis of 140 patients (37 with a positive family history of PD and 103 sporadic cases), we did not find mutations in the five coding SNCA exons (Alvarez et al., unpublished results). In addition to rare mutations, common SNCA polymorphisms have been associated with the most common sporadic forms of PD (Edwards et al. 2010; Mizuta et al. 2006; Mueller et al. 2005; Pals et al. 2004; Pastor et al. 2001; Satake et al. 2009; Simon-Sanchez et al. 2009). The mechanism by which these SNCA variants lead to PD has not yet been established, although an effect on α-synuclein expression has been proposed for some of them (McCarthy et al. 2011). SNP rs356165 is in the 3′UTR of SNCA and has been linked to PD risk among European and Japanese (Mizuta et al. 2006; Mueller et al. 2005; Myhre et al. 2008). This SNP was also associated with the disease in the two Spanish populations included in our study, with the strongest risk among individuals homozygous for the rs356165 G allele and an OR value similar to the one reported by others. Other SNPs in LD with rs356165 have been associated with PD risk. Mata et al. found an association between rs356219 and PD in the populations here reported (Mata et al. 2011). However, this SNP mapped 3' downstream of SNCA, and the association to PD could thus be attributed to LD with rs356165.
Our study also supported a significant effect of this SNP on the age at onset. Homozygous for the G allele had a mean age at onset 4 years lower and were significantly more frequent among patients with early-onset PD (<50 years) compared to homozygous for the A allele. This effect on the age at onset has not been reported by other studies that found a significant association between rs356165 and PD. The G allele was also more frequent among patients with one of the three LRRK2 mutations and an early age at onset, although the comparison based on a reduced number of patients was not significant results.
A recent study showed a correlation between the rs356165 G allele and the expression of a SNCA isoform that lacks exon 5 (SNCA112) and results in a protein prone to aggregation. Frontal cortex of homozygous for the risk G allele showed higher SNCA112 levels compared to AA homozygous. LD between rs356165 and some variants that affected pre-mRNA splicing could explain the association between this 3′ UTR SNP and the level of the SNCA112 isoform and its effect on PD risk (McCarthy et al. 2011). Several mechanisms could also explain a direct effect of this SNP on PD risk. First, it could affect mRNA length. SNCA contains several polyadenilation sites, and transcripts with different 3′UTR lengths have been found (Sotiriou et al. 2009). An effect on the length of the SNCA transcript was unlikely because this SNP was out of the polyadenylation signals.
The rs356165 could also affect SNCA expression through differential binding of miRNAs. MiRNAs are small (approximately 20 nucleotides long) non-coding RNAs that bind to the 3′ UTR of mRNAs and regulate expression at post-transcriptional level. Through this process, miRNAs regulate many cellular processes, and the deregulation of miRNA pathways could result in neurodegeneration and PD (Hebert and De Strooper 2007). Some miRNAs have been implicated in the post-transcriptional regulation of SNCA (Doxakis 2010). DNA variants at the 3′UTRs could create or destroy miRNA target sites, affecting gene expression. This could explain the association of some 3′ UTR polymorphisms to common diseases, including PD (Wang et al. 2008). An in silico analysis predicted that rs356165 would not affect the binding of known miRNAs, but in vitro studies with reporter plasmids containing the two 3′ UTR sequences should be necessary to determine whether this SNP has some effect on mRNA stability. This SNP could have also an effect on RNA editing (the conversion of adenosine to inosine in double-stranded RNA). An online search in the RNA editing database (www.cgen.com/research/Publications) did not find an effect of this SNP on SNCA edition. Adenosine deamination on 3′UTR regions could modify the binding of miRNAs (Borchert et al. 2009). Because inosine is chemically and functionally similar to guanosine, the rs356165 would not modify the miRNA-binding profile of the natural A/G variation.
Finally, it is also possible that rs356165 was in LD with some functional variant in the 3′ UTR. In addition to this common SNP, we found several 3′UTR rare variants but none of them was predicted to affect the binding of known miRNAs. Furthermore, the minor allele frequencies for these polymorphisms were <0.01, suggesting a limited contribution to PD risk in our population. A limitation of our study was the use of SSCA to the search for DNA variation, an indirect technique with a low rate for false negatives. Thus, we could not exclude that some SNCA minor variants were not detected. LD with SNPs at nearby genes could also explain the association between rs356165 and PD. In this regard, SNCA is flanked 3′ by the multimerin 1 (MMRN1) gene and 5′ by the G protein-regulated inducer of neurite outgrowth-3 (GRIN3) genes. MMRN1 encodes a factor V/Va binding protein that might function as a carrier protein for platelet factor V, and an effect on PD is unlikely. However, GRIN3 belongs to a family of proteins that participates in the regulation of synapsis transmission and energy metabolism, two processes that could be impaired in PD.
In conclusion, we confirmed the association between the common SNCA rs356165 SNP and PD in two independent populations. We also provided evidence for a significant effect of this SNP on the age at onset. The absence of other 3′UTR variants with PD relevance in our population suggests that this variant could have a direct role on PD. More functional studies to determine its effect on SNCA expression should be of special interest.
References
Borchert GM, Gilmore BL, Spengler RM, Xing Y, Lanier W, Bhattacharya D, Davidson BL (2009) Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum Mol Genet 18:4801–4807
de Lau LM, Breteler MM (2006) Epidemiology of Parkinson’s disease. Lancet Neurol 5:525–535
Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I (2009) Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 8:1150–1157
Doxakis E (2010) Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem 285:12726–12734
Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, Wang L, Zuchner S, Konidari I, Wang G, Singer C, Nahab F, Scott B, Stajich JM, Pericak-Vance M, Haines J, Vance JM, Martin ER (2010) Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 74:97–109
Hebert SS, De Strooper B (2007) Molecular biology. miRNAs in neurodegeneration. Science 317:1179–1180
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55:181–184
Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM (2009) Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci USA 106:13052–13057
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18:106–108
Mata IF, Ross OA, Kachergus J, Huerta C, Ribacoba R, Moris G et al (2006) LRRK2 mutations are a common cause of Parkinson’s disease in Spain. Eur J Neurol 13:391–394
Mata IF, Yearout D, Alvarez V, Coto E, de Mena L, Ribacoba R, Lorenzo-Betancor O, Samaranch L, Pastor P, Cervantes S, Infante J, Garcia-Gorostiaga I, Sierra M, Combarros O, Snapinn KW, Edwards KL, Zabetian CP (2011) Replication of MAPT and SNCA, but not PARK16–18, as susceptibility genes for parkinson’s disease. Mov Disord 5:819–823
McCarthy JJ, Linnertz C, Saucier L, Burke JR, Hulette CM, Welsh-Bohmer KA, Chiba-Falek O (2011) The effect of SNCA 3′ region on the levels of SNCA-112 splicing variant. Neurogenetics 12:59–64
Mizuta I, Satake W, Nakabayashi Y, Ito C, Suzuki S, Momose Y, Nagai Y, Oka A, Inoko H, Fukae J, Saito Y, Sawabe M, Murayama S, Yamamoto M, Hattori N, Murata M, Toda T (2006) Multiple candidate gene analysis identifies alpha-synuclein as a susceptibility gene for sporadic Parkinson’s disease. Hum Mol Genet 15:1151–1158
Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, Illig T, Berg D, Wullner U, Meitinger T, Gasser T (2005) Multiple regions of alpha-synuclein are associated with Parkinson’s disease. Ann Neurol 57:535–541
Myhre R, Toft M, Kachergus J, Hulihan MM, Aasly JO, Klungland H, Farrer MJ (2008) Multiple alpha-synuclein gene polymorphisms are associated with Parkinson’s disease in a Norwegian population. Acta Neurol Scand 118:320–327
Pals P, Lincoln S, Manning J, Heckman M, Skipper L, Hulihan M, Van den Broeck M, De Pooter T, Cras P, Crook J, Van Broeckhoven C, Farrer MJ (2004) Alpha-synuclein promoter confers susceptibility to Parkinson’s disease. Ann Neurol 56:591–595
Pastor P, Munoz E, Ezquerra M, Obach V, Marti MJ, Valldeoriola F, Tolosa E, Oliva R (2001) Analysis of the coding and the 5′ flanking regions of the alpha-synuclein gene in patients with Parkinson’s disease. Mov Disord 16:1115–1119
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41:1303–1307
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41:1308–1312
Sotiriou S, Gibney G, Baxevanis AD, Nussbaum RL (2009) A single nucleotide polymorphism in the 3′UTR of the SNCA gene encoding alpha-synuclein is a new potential susceptibility locus for Parkinson disease. Neurosci Lett 461:196–201
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840
Wang G, van der Walt JM, Mayhew G, Li YJ, Zuchner S, Scott WK, Martin ER, Vance JM (2008) Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. Am J Hum Genet 82:283–289
Warner TT, Schapira AH (2003) Genetic and environmental factors in the cause of Parkinson’s disease. Ann Neurol 53(Suppl 3):S16–S23, discussion S23–15
Yu L, Xu P, He X, Hu F, Lin Z, Zhu M, Liu Z, He L, Xu Y (2010) SNP rs7684318 of the α-synuclein gene is associated with Parkinson’s disease in the Han Chinese population. Brain Res 1346:262–265
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173
Acknowledgments
Authors thank the “Fundacion Parkinson Asturias” and “Obra Social Cajastur” for their support. This work was supported by grants from the Spanish “Fondo de Investigaciones Sanitarias-Fondos FEDER” European Union (FIS-05/008 and 08/0915). LFC and LDM are predoctoral fellowships of FICYT-Principado de Asturias. LS held a “Torres Quevedo” fellowship from the Spanish Ministry of Science and Technology, co-financed by the European Social Fund. This study was supported by a grant from the Spanish Ministry of Education and Science (SAF2006-10126, 2006–2009) by the project 061131 from the “Fundació La Marató de TV3” and by the UTE project FIMA, Spain to PP.
Disclosure
The authors report no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Primers designed to amplify the four 3´UTR SNCA fragments. SNP rs356165 was genotyped through amplification with fragment 4 primers, followed by digestion with TaiI and electrophoresis on 4% agarose gel to visualize the two alleles: A 273 bp and G 199 + 74 bp (DOC 32 kb)
Supplementary Fig. 1
a PCR-RFLP genotypes for the rs356165 SNP. b SSCA patterns of heterozygotes (arrows) for the c.404 C>T and the c.*139 T>G (SNP rs10024743). c DNA sequences showing the c.404 C>T and the c.*575_579 deletion (DOC 79 kb)
Rights and permissions
About this article
Cite this article
Cardo, L.F., Coto, E., de Mena, L. et al. A Search for SNCA 3′ UTR Variants Identified SNP rs356165 as a Determinant of Disease Risk and Onset Age in Parkinson’s Disease. J Mol Neurosci 47, 425–430 (2012). https://doi.org/10.1007/s12031-011-9669-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-011-9669-1