
Abstract
LXA4 methyl ester (LXA4ME), a lipoxin A4 analog, reduces ischemic insult in the rat models of transient or permanent cerebral ischemic injury. We investigated whether LXA4ME could ameliorate blood–brain barrier (BBB) dysfunction after stroke by reducing matrix metalloproteinase (MMP)-9 expression. Adult male rats were subjected to 2-h middle cerebral artery occlusion (MCAO) followed by 24-h reperfusion. Brain infarctions were detected by triphenyltetrazolium chloride (TTC) staining. BBB dysfunction was determined by examining brain edema and Evans Blue extravasation. Temporal expression of MMP-9 was determined by zymography and Western blot. The presence of tissue inhibitors of metalloproteinase-1 (TIMP-1) was also determined by Western blot in tissue protein sample. Brain edema and Evans Blue leakage were significantly reduced after stroke in the LXA4ME group and were associated with reduced brain infarct volumes. MMP-9 activity and expression were inhibited by LXA4ME after stroke. In addition, LXA4ME significantly increased TIMP-1 protein levels. Our results indicate that LXA4ME reduces brain injury by improving BBB function in a rat model of MCAO, and that a relationship exists between BBB permeability and MMP-9 expression following ischemic insult. Furthermore, these results suggest that LXA4ME-mediated reduction of MMP-9 following stroke are attributed to increased TIMP-1 expression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stroke is the one of the leading causes of death and neurological dysfunctions in the world. Complex biochemical cascades are involved in the process of neuronal damage during cerebral ischemia. Intracellular and extracellular processes have been suggested to mediate the pathophysiology following a cerebral ischemic insult. In addition to intracellular pathologic events, the dysregulation of the pericellular environment such as cell-to-matrix or cell-to-cell interactions has been suggested as a possible cause of neural tissue damage during cerebral ischemia (Gu et al. 2002; Lee et al. 2008). In particular, the structural alterations of neurovascular units, such as blood–brain barrier (BBB) degradation, may contribute to the significant pathology observed following focal cerebral ischemia (Lo 2008; Lo et al. 2002; Lu et al. 2008). Destruction of the BBB is a critical event during cerebral ischemia and is followed by passive fluid diffusion into the brain parenchyma leading to vasogenic edema and secondary brain injury.
Matrix metalloproteinases (MMPs) are a family of zinc-dependent endopeptidases that are capable of degrading most components of the extracellular matrix. MMPs are upregulated and play an important role in the pathologic processes during focal cerebral ischemia (Asahi et al. 2000; Romanic et al. 1998). In particular, MMP-9, degrades matrix components of the basement membrane, which maintains the cerebral vasculature integrity, resulting in neuroinflammation, brain edema, and hemorrhagic transformation after focal cerebral ischemic episode (Romanic et al. 1998; Heo et al. 1999; Planas et al. 2001). Reductions in MMP-9 induced by genetic manipulation or pharmacological intervention have been shown to reduce neuronal damage after global and focal brain ischemic injury (Asahi et al. 2000; Lee et al. 2004).
Lipoxins (LX) are endogenous anti-inflammatory lipid-based autacoids that are generated from arachidonic acid via lipoxygenase-mediated transcellular biosynthesis. LXA4 is a principal LX generated by mammalian cells. LXA4 binds to its specific G protein-coupled receptor termed ALXR (Chiang et al. 2005), which can be cloned in neutrophils, monocytes, and macrophage. This receptor is also expressed on astrocyte, microglia and neural stem cells in the central nervous system, suggesting that these cells may be the targets of LX activity in the brain (Maddox et al. 1997; Sodin-Semrl et al. 2004; Svensson et al. 2007; Wada et al. 2006). Because LXs are rapidly biosynthesized and enzymatically inactivated, stable and more potent analogs had been constructed (Serhan et al. 1995; Chiang et al. 2000). Native LXs and their stable analogs including LXA4 methyl ester (LXA4ME) exert their biological effects in a number of major inflammatory disorders, including vascular injury, kidney disease, periodontal disease, asthma, arthritis and cystic fibrosis (Petasis et al. 2008; Serhan et al. 2008). LXs are considered endogenous inflammatory ‘stop signals’, which can downregulate or counteract the activities of proinflammatory mediators to promote resolution (Serhan and Oliw 2001).
We recently reported that LXA4ME protects the brain against cerebral ischemia–reperfusion injury and permanent cerebral ischemic infarction in a rat model (Ye et al. 2010; Wu et al. 2010). To our knowledge, however, there are no previous studies examining the effects of LXA4ME on BBB permeability and MMP activity following transient focal cerebral ischemia–reperfusion. Herein, we investigate the effects of LXA4ME on BBB permeability, MMP-9 activity and neuronal damage in a rat model of middle cerebral artery occlusion (MCAO).
Materials and Methods
Animal Model and Drug Administration
All animal experiments were carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals of Tongji Medical College, and all usage and procedures were approved by the committee of experimental animals of Tongji Medical College. The experiments were carried out on 200–250 g adult male Sprague–Dawley rats. The rats were housed with a 12-h light/dark cycle at 22 ± 0.5°C. Pellet food and water were given ad libitum. The surgeries were performed after the rats were anesthetized with chloral hydrate (350 mg/kg, intraperitoneally). During the course of the experiment, the rats inhaled oxygen through a face mask at a flow rate of 1 L/min. Rectal temperature was maintained at 37 ± 0.5°C using a heating lamp.
MCAO was induced using the intraarterial suture occlusion method as described previously with slight modification (Longa et al. 1989). Briefly, the right common carotid, internal carotid (ICA), and external carotid (ECA) arteries were exposed and the latter was dissected distally. The ICA was isolated and the tip of 4-0 monofilament nylon was rounded by heating over a flame and coated with silicone before being introduced into the ECA lumen. The nylon was carefully advanced into the ICA lumen to occlude the MCA. A second dose of chloral hydrate (350 mg/kg, i.p.) was injected after 60 min of MCAO to maintain general anesthesia. After 2 h of MCAO, the suture was withdrawn to restore blood flow. The incision was then sutured and the rats recovered under a heating lamp to maintain rectal temperatures at 37 ± 0.5°C. Furthermore, animals were excluded from the study if there was evidence of subarachnoid hemorrhage when the brain tissue was extracted. All animals recovered from anesthesia in individual acrylic cages with sterile sawdust and received food and water ad libitum.
The LXA4ME (Cayman Chemical, Ann Arbor, MI, USA) used in this study was dissolved in the normal saline. The rats were randomly assigned to sham, MCAO, or LXA4ME groups. After MCAO, rats in the LXA4ME group received an intracerebroventricular injection of 5 μL LXA4ME (0.3 nM), as described in our previous reports (Ye et al. 2010; Wu et al. 2010). Briefly, injections were performed with a Hamilton syringe using a 27-gauge needle. The location of each injection was 1 mm posterior to bregma, 1.8 mm lateral to the midline, and 4.2 mm beneath the skull surface. Rats in the sham and MCAO groups were injected with a vehicle control (normal saline).
Cerebral Infarct Measurement
After 24 h of reperfusion, the rats were anesthetized and decapitated. Cerebral infarct volumes were measured as described previously (Ye et al. 2010). Briefly, the brains (n = 8 per group) were cut into 2-mm coronal sections on a cutting block, immediately immersed in 2% TTC (2,3,5-triphenyltetrazolium chloride, Sigma, St. Louis, MO, USA) for 20 min at 37°C, and incubated overnight in 4% paraformaldehyde. The infarct area was demarcated and analyzed by Image J software (NIH Image, Version 1.61, Bethesda, USA). The demarcation between infarcted and noninfarcted tissues was outlined, and the total volume of the infarction was calculated by integrating of the lesion areas from all six sections measured. To compensate for the effect of brain edema, a corrected infarct volume was calculated: corrected infarct area = measured infarct area × {1−[(Ipsilateral hemisphere area−contralateral hemisphere area)/contralateral hemisphere]}. The infarct size was quantified by the ratio of corrected infarct volume to the whole brain volume.
Determination of Brain Water Content in the Ischemic Hemispheres
The brain water content in the ischemic hemispheres was determined using wet/dry method (n = 8 for each group). Briefly, 24 h after reperfusion, the rats were overdosed with chloral hydrate, decapitated, and the brains were quickly removed and divided into ischemic and contralateral hemispheres. Ischemic hemisphere was weighed, incubated in an oven at 110°C for 24 h and then reweighed. The brain water content was calculated by (wet weight−dry weight)/wet weight×100%.
Quantitative Measurement of BBB Integrity
BBB integrity was assessed (n = 8 each group) by Evans blue content. Evans blue dye, which is a tracer for BBB integrity. Evans blue (2% solution 4 mL/kg) was injected into the left jugular vein 1 h before sacrifice and 24 h after reperfusion. The rats were transcardially perfused with heparinized phosphate-buffered saline to remove the intravascular dye. The brains were removed, separated into ischemic and contralateral hemispheres and weighed. For quantitative measurement, the ischemic hemisphere was homogenized in a 50% trichloroacetic acid solution to precipitate proteins and centrifuged for 20 min at 13,600×g. The supernatant was diluted with ethanol (1:3), and the fluorescence was measured (excitation 620 nm, emission 680 nm). The amount of extravasated Evans blue was quantified in micrograms per gram of brain tissue. The extravasation of Evans blue dye was also observed under a fluorescence microscope.
Preparation of Brain Tissue Extracts
Twenty-four hours after reperfusion, the rats were killed with an overdose of pentobarbital, and the ischemic cerebral cortex was quickly removed, snap-frozen in liquid nitrogen and stored at −80°C until analysis. For further analysis, brain samples were homogenized in lysis buffer (50 mM Tris base, pH 7.4; 150 mM NaCl; 1% deoxycholate; 1% sodium dodecyl sulfate; and 1% Triton X-100) including protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 2 μg/mL leupeptin) on ice. After centrifugation, the supernatant was collected, total protein concentrations were determined using the Bradford assay according to the manufacturer’s instructions (Pierce Biochemicals) and the samples were stored at −20°C until use.
MMP Zymography
The prepared protein samples (30 μg) were loaded and separated on a 10% Tris–glycine gel with 0.1% gelatin as a substrate. After separation by electrophoresis, the gels were placed in 2.5% Triton X-100 for 1 h to remove SDS and incubated at 37°C for 24 h with developing buffer (50 mmol/L Tris base, 40 mmol/L HCl, 200 mmol/L NaCl, 5 mmol/L CaCl2 and 0.2% Briji 35) on a rotary shaker. After development, the gels were stained with 0.5% Coomassie Blue R-250 for 30 min and destained. MMP-9 standards (Applygen Technologies, Inc.) were loaded into each gel for band identification and the proteolytic band intensities were quantified by scanning densitometry. For statistical analyses, all data were expressed as the ratio to optical density values of the corresponding sham control.
Western Blots
Equal amounts (30 μg) of the prepared protein samples (from the zymography samples) were mixed with 4× sample buffer, and each sample was separated by Tris–glycine SDS-polyacrylamide gel electrophoresis in reducing conditions for MMP-9 and tissue inhibitors of metalloproteinase-1 (TIMP-1). After separation, the proteins were transferred onto a nitrocellulose membrane and blocked with 5% nonfat dry milk in TBS (pH 7.4, containing 0.1% Tween 20 [TBS-T]) at 4°C overnight. Next, the membranes were incubated with the appropriate primary antibodies: MMP-9 (Abgent, 1:500) and TIMP-1 (Abgent, 1:100) diluted in blocking buffer. After washing with TBS-T, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies at room temperature for 60 min. As an internal control, the same nitrocellulose membranes were stripped and incubated with an antibody for glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Santa Cruz, 1:1,000). Immunoreactive proteins were visualized using the enhanced chemiluminescence detection system (Amersham Biosciences, Piscataway, NJ) and exposed on radiograph film (Fuji Hyperfilm, Tokyo, Japan). The protein expression for each sample was analyzed with Image J software.
Statistical Analysis
Data are presented as means ± standard error of the mean (SEM). Statistical differences between the various groups were assessed by one-way analysis of variance followed by the Tukey test. A p value less than 0.05 was considered statistically significant.
Results
Evaluation of Infarction Volume of Brain
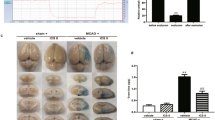
Representative coronal brain sections from all groups are shown in Fig. 1a. The white-colored areas represent the infarcted regions in these sections. No damage was observed in the sham group. However, an obvious infarcted region was observed in the cerebral hemisphere afflicted by ischemia in the MCAO group. Importantly, the administration of LXA4ME in rats that received MCAO significantly reduced the infarct volume. The infarct size was quantified by the ratio of the corrected infarct volume to the whole brain volume (Fig. 1b). As shown in Fig. 1b, LXA4ME reduced the infarct volume compared with the MCAO group (p < 0.05).

Effect of LXA4ME on cerebral infarct volume in rats. a Representative TTC-stained brain sections are shown. Rats were subjected to 2-h MCAO followed by 24-h reperfusion and intracerebroventricular injection with LXA4ME (0.3 nM). White designates the infarct area and red shows the normal area. As shown by a significant decrease in the white area, LXA4ME reduces the brain injury induced by the ischemia–reperfusion insult compared with the MCAO group. b Quantification of infarct volume 24 h after reperfusion. The ratio of the corrected infarct volume to the whole brain voume was calculated for the cerebral infarct size. The infarct volume was decreased at 24 h in the group with LXA4ME treatment. Values shown are mean ± SEM (n = 8); *p < 0.01 versus sham and #p < 0.05 versus MCAO

Brain water content in ischemic hemispheres. Brain water content increased in the ischemic hemispheres after 2-h MCAO followed by 24-h reperfusion. Brain water content were reduced in the LXA4ME group compared with the MCAO group. Values shown are mean ± SEM (n = 8); *p < 0.05, **p < 0.01 versus sham and #p < 0.05 versus MCAO
Brain Water Content in the Ischemic Hemispheres
A significant increase in brain water content was observed in the ischemic hemispheres 24 h after MCAO compared with the sham group (p < 0.01). In addition, the brain water content was significantly reduced by LXA4ME treatment (p < 0.05) (Fig 2).
Evans Blue Dye Extravasation
After 24 h of reperfusion, Evans blue was clearly observed in the ipsilateral hemisphere in whole brain coronal sections (Fig. 3). Evans blue fluoresces are in red (emission wavelength 680 nm) upon illumination in a fluorescence microscope (excitation wavelengths 470 and 540 nm). Considerable Evans blue leakage was visualized in the cortex of rats in the MCAO group; however, less Evans blue spillover was observed in the LXA4ME group (Fig. 3b). Quantitative analysis of Evans blue in the brain tissue from the sham-operated, MCAO and LXA4ME groups is shown in Fig. 3c. We observed a significant decrease in BBB permeability in the LXA4ME group compared to the MCAO group (p < 0.01).

Evans blue extravasation. a Representative samples of Evans blue-stained brain sections from rats subject to a 2-h MCAO followed by 24 h of reperfusion. Negative dye leakage was observed in the sham group, while intensive staining was seen in the ischemic area in the MCAO group. However, LXA4ME-treated rats showed weak Evans blue staining in the infarct areas. b The Evans blue staining seen in the fluorescence microscope. Considerable Evans blue leakage was visualized in the cortex of the MCAO group, but less spillover was observed in the LXA4ME group. c Statistic analysis of Evans blue extravasation severity. The amount of vascular leakage was determined by measuring the amount of brain-extracted Evan’s blue by spectrophotometry at 620 nm and expressing this in ng/g of brain tissue. The LXA4ME treatment showed reduced brain Evans blue content compared with the MCAO group (n = 8). The values shown are the mean ± SEM. *p < 0.01 versus sham and #p < 0.01 versus MCAO
MMP-9 Activity
To determine the effect of LXA4ME treatment on MMP-9 activity, zymography was performed at 24 h after MCAO. Representative MMP zymography of the ipsilateral hemisphere following MCAO is shown in Fig. 4a. Densitometric analysis of MMP-9 bands showed an increased expression of activated MMP-9 in the MCAO groups. However, LXA4ME treatment after MCAO significantly reduced the intensity of the MMP-9 band (Fig. 4b).

Gel zymograms of tissue homogenates 24 h after a 2 h transient focal cerebral ischemia. a Representative MMP zymography of the ipsilateral hemisphere following MCAO. Sham-operated animals showed very low levels of activated MMP-9, while the levels were increased in the MCAO group. LXA4ME administration reduced the MCAO-induced increase in activated in the ipsilateral hemisphere. b Bar graph of measurement of relative optical density of MMP-9 bands. The MCAO-induced increase in the optical density of the MMP-9 band was attenuated by LXA4ME administration (n = 6). Values shown are the mean ± SEM. *p < 0.01 versus sham and #p < 0.01 versus MCAO
MMP-9 and TIMP Expressions
MMP-9 was detected by Western blot in brain tissue from infarction area 24 h after reperfusion (Fig. 5). Representative band of MMP-9 was shown in Fig. 5a. Densitometric analysis of the MMP-9 bands indicated a significant increase in MMP-9 protein in the MCAO group compared to the sham group. In addition, the high MMP-9 protein levels were significantly attenuated by LXA4ME treatment (Fig. 5b).

Western blot analysis of MMP-9 in the cerebral cortex following 24 h of reperfusion. a A representative MMP-9 band is shown. GAPDH was used as a loading control. b The blots were analyzed by density and expressed as a ratio to GAPDH. The level of MMP-9 increased after MCAO, However, MMP-9 levels were attenuated following LXA4ME administration (n = 6). Values shown are mean ± SEM. *p < 0.01 versus sham and #p < 0.05 versus MCAO
TIMP-1 protein was detected by Western blot in brain tissue from infarction areas 24 h after reperfusion (Fig. 6). Representative TIMP-1 bands are shown in Fig. 6a. Densitometric analysis of the TIMP-1 bands indicated decreases in TIMP-1 in MCAO infarction tissues compared to the sham. In contrast, the low TIMP-1 levels induced by MCAO were increased by LXA4ME treatment (Fig. 6b).

Western blot analysis of TIMP-1 in the cerebral cortex after 24 h of reperfusion. a A representative TIMP-1 band is shown. GAPDH was used as a loading control. b Blots were analyzed by density and expressed as a ratio to GAPDH. The level of TIMP-1 decreased following MCAO; however, TIMP-1 levels increased after LXA4ME administration compared to the MCAO group (n = 6). Values shown are mean ± SEM. *p < 0.05, **p < 0.01 versus sham and #p < 0.05 versus MCAO
Discussion
The present study demonstrates that LXA4ME treatment effectively improves BBB integrity and reduces brain infarct volume after focal ischemia. Furthermore, MMP-9 activity and expression were reduced by LXA4ME administration in rats that were subjected to a 2 h MCAO followed by 24-h reperfusion. Moreover, the increase of TIMP-1 following LXA4ME treatment may contribute to the observed reduction in MMP-9 expression.
Disruption of the BBB is a key feature of ischemia injury to the central nervous system. Studies also suggest that endothelial cell apoptosis, gene expression changes and microenvironment alterations are essential for the initiation and propagation of the ischemic injury. In the current study, we found that BBB permeability and brain water content in the ipsilateral hemisphere following MCAO were significantly reduced by LXA4ME. Parallel to these changes, the infarct volume was also attenuated by LXA4ME administration.
Previous investigations have indicated that MMP-9 is produced in endothelial cells, microglia and astrocytes and is upregulated after the onset of permanent (Romanic et al. 1998; Gasche et al. 1999) or transient (Rosenberg et al. 1998; Heo et al. 1999; Planas et al. 2000, 2001; Wagner et al. 2003; Pfefferkorn and Rosenberg 2003) focal ischemia in experimental animals and in human patients (Clark et al. 1997; Horstmann et al. 2003). In the central nervous system, the early appearance of activated MMP-9 has been associated with changes in BBB permeability, the degradation of critical BBB components and the formation of vasogenic edema after transient focal ischemia (Gasche et al. 1999; Fujimura et al. 1999; Rosenberg et al. 1998; Heo et al. 1999; Horstmann et al. 2003). Pharmacologic inhibition of MMPs ameliorated edema after focal cerebral ischemia (Rosenberg et al. 1998; Romanic et al. 1998) and MMP-9 deficient mice showed reduced BBB disruption and edema after transient focal cerebral ischemia (Asahi et al. 2000, 2001) and traumatic brain injury (Wang et al. 2000). In the present study, LXA4ME administration ameliorated the disruption of the BBB after stroke and this correlated with reduced MMP-9 protein levels and enzyme activity. Consistent with our findings, some researches also suggested that lipoxins could potently inhibit MMP-9 activity. Indeed, LXA4-inhibited hepatocyte growth factor induced increased expression of MMP-9 in HepG2 cells (Zhou et al. 2009). Furthermore, 15-Epi-LXA4 inhibited MMP-9 activity and mRNA expression in a murine model of endometriosis (Chen et al. 2010). In addition, ATL-1, an analog of aspirin-triggered LXA4, attenuated MMP-9 increases induced by vascular endothelial growth factor in endothelial cells (Cezar-de-Mello et al. 2008). Together, these results suggest that the LXA4ME-mediated reduction in MMP-9 expression following stroke may lead to improved BBB function.
Another possible role for MMPs in the pathophysiology of cerebral ischemia is the neuronal anoikis, which is induced by the degradation of cell-to-matrix interactions in the brain parenchyma after focal ischemia (Gu et al. 2002). In particular, during transient global cerebral ischemia, the MMP-induced degradation of perineuronal matrix proteins plays a role in anoikis-type neuronal cell death (Lee et al. 2008). Importantly, our previous reports have shown that LXA4ME can inhibit neuronal apoptosis induced by transient or permanent cerebral ischemia insult (Ye et al. 2010; Wu et al. 2010). Considering these reports, we propose that the LXA4ME-mediated attenuation of neuronal apoptosis may be partly due to the inhibition of MMP-9 expression and activity.
It has also been reported that MMPs are inhibited by endogenous inhibitors called TIMPs, of which four members (TIMP 1–4) have been identified (Brew et al. 2000). TIMP-1 primarily forms complexes with MMP-9 by binding in a 1:1 stoichiometric ratio (Crocker et al. 2004). TIMP-1 inhibits MMP-9 activity through high affinity, noncovalent binding to the MMP catalytic domain (Cunningham et al. 2005; Hornebeck et al. 2005) under physiological and pathological conditions, such as stroke (Wang et al. 1998). In the present study, we observed that LXA4ME could reduce MMP-9 activity conceivably by increasing the biosynthesis of TIMP-1. Although our study did not show a direct cause-and-effect relationship for LXA4ME-induced BBB improvement in stroke, a temporal and inverse relationship between TIMP-1 and MMP-9 was clearly demonstrated and was associated with reduced brain infarction volume and improved BBB function. Moreover, previous evidence has indicated that LXA4 enhanced the synthesis of TIMP-1, which resulted in MMP-9 inhibition in human synovial fibroblasts (Sodin-Semrl et al. 2000). In addition, LXA4ME has a potent anti-inflammatory effect. Indeed, some anti-inflammatory drugs also alter MMP-9 and TIMP-1 expression and function. For example, aspirin was shown to inhibit MMP-9 activity by inducing the expression of TIMP-1 in cultured mouse celiac macrophages (Yiqin et al. 2009). Thus, the inhibition of MMP-9 by LXA4ME may be caused by enhancing the expression of TIMP-1.
Another possible mechanism underlying LXA4ME-mediated MMP-9 inhibition is the ability of LXA4ME to inhibit the activation of the NF-kappaB, p42/44 and p38 mitogen-activated protein kinase pathways. The expression of MMP-9 is tightly regulated by NF-kappaB, p42/44 and p38 (Kauppinen and Swanson 2005; Kim and Moon 2005; Wu et al. 2004). Furthermore, the inhibition of MMP-9 by NF-kappaB suppression decreases neuronal damage and infarct volumes after ischemia (Romanic et al. 1998; Koh et al. 2005). Moreover, our previous research showed that LXA4ME inhibited NF-kappaB activation following cerebral ischemia (Ye et al. 2010). p42/44 and p38 signal transduction can also be inhibited by LXA4 in endothelial cells (Wu et al. 2008). Thus, it is likely that NF-kappaB, p42/44 and p38 are involved in the LXA4ME-induced inhibition of MMP-9. Further research is necessary to clarify these possible mechanisms.
In conclusion, our results indicate that LXA4ME reduces brain injury by improving BBB function in a rat model of MCAO. This study also elucidates a relationship between BBB permeability and MMP-9 expression in response to ischemic insult. Furthermore, our results suggest that LXA4ME-mediated reduction in MMP-9 following stroke is facilitated by increased TIMP-1 expression.
References
Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH (2000) Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab 20(12):1681–1689
Asahi M, Wang X, Mori T et al (2001) Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood–brain barrier and white matter components after cerebral ischemia. J Neurosci 21(19):7724–7732
Brew K, Dinakarpandian D, Nagase H (2000) Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta 1477(1–2):267–283
Cezar-de-Mello PF, Vieira AM, Nascimento-Silva V, Villela CG, Barja-Fidalgo C, Fierro IM (2008) ATL-1, an analogue of aspirin-triggered lipoxin A4, is a potent inhibitor of several steps in angiogenesis induced by vascular endothelial growth factor. Br J Pharmacol 153(5):956–965
Chen QH, Zhou WD, Pu DM, Huang QS, Li T, Chen QX (2010) 15-Epi-lipoxin A(4) inhibits the progression of endometriosis in a murine model. Fertil Steril 93(5):1440–1447
Chiang N, Fierro IM, Gronert K, Serhan CN (2000) Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J Exp Med 191(7):1197–1208
Chiang N, Arita M, Serhan CN (2005) Anti-inflammatory circultry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot Essent Fat Acids 73(3–4):163–177
Clark AW, Krekoski CA, Bou SS, Chapman KR, Edwards DR (1997) Increased gelatinase A (MMP-2) and gelatinase B (MMP-9) activities in human brain after focal ischemia. Neurosci Lett 238(1–2):53–56
Crocker SJ, Pagenstecher A, Campbell IL (2004) The TIMPs tango with MMPs and more in the central nervous system. J Neurosci Res 75(1):1–11
Cunningham LA, Wetzel M, Rosenberg GA (2005) Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 50(4):329–339
Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH (1999) Early appearance of activated matrix metalloproteinase-9 and blood–brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res 842(1):92–100
Gasche Y, Fujimura M, Morita-Fujimura Y et al (1999) Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood–brain barrier dysfunction. J Cereb Blood Flow Metab 19(9):1020–1028
Gu Z, Kaul M, Yan B et al (2002) S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297(5584):1186–1190
Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ (1999) Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab 19(6):624–633
Hornebeck W, Lambert E, Petitfrère E, Bernard P (2005) Beneficial and detrimental influences of tissue inhibitor of metalloproteinase-1 (TIMP-1) in tumor progression. Biochimie 87(3–4):377–383
Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S (2003) Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke 34(9):2165–2170
Kauppinen TM, Swanson RA (2005) Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J Immunol 174(4):2288–2296
Kim CH, Moon SK (2005) Epigallocatechin-3-gallate causes the p21/WAF1-mediated G(1)-phase arrest of cell cycle and inhibits matrix metalloproteinase-9 expression in TNF-alphainduced vascular smooth muscle cells. Arch Biochem Biophys 435(2):264–272
Koh SH, Chang DI, Kim HT et al (2005) Effect of 3-aminobenzamide, PARP inhibitor, on matrix metalloproteinase-9 level in plasma and brain of ischemic stroke model. Toxicology 214(1–2):131–139
Lee SR, Tsuji K, Lee SR, Lo EH (2004) Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci 24(3):671–678
Lee KJ, Jang YH, Lee H, Yoo HS, Lee SR (2008) PPARgamma agonist pioglitazone reduces [corrected] neuronal cell damage after transient global cerebral ischemia through matrix metalloproteinase inhibition. Eur J Neurosci 27(2):334–342
Lo EH (2008) Experimental models, neurovascular mechanisms and translational issues in stroke research. Br J Pharmacol 153(Suppl 1):S396–S405
Lo EH, Wang X, Cuzner ML (2002) Extracellular proteolysis in brain injury and inflammation: role for plasminogen activators and matrix metalloproteinases. J Neurosci Res 69(1):1–9
Longa EZ, Weinstein PR, Carlson S, Cummins R. (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20(1):84–91
Lu A, Clark JF, Broderick JP et al (2008) Reperfusion activates metalloproteinases that contribute to neurovascular injury. Exp Neurol 210(2):549–559
Maddox JF, Hachicha M, Takano T, Petasis NA, Fokin VV, Serhan CN (1997) Lipoxin A4 stable analoges are potent mimetics that stimulate human monocytes and THP-1 cell via a G-protein linked lipoxin A4 receptor. J Biol Chem 272(11):6972–6978
Petasis NA, Keledjian R, Sun YP et al (2008) Design and synthesis of benzo-lipoxin A4 analogs with enhanced stability and potent anti-inflammatory properties. Bioorg Med Chem Lett 18(4):1382–1387
Pfefferkorn T, Rosenberg GA (2003) Closure of the blood–brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke 34(8):2025–2030
Planas AM, Solé S, Justicia C, Farré ER (2000) Estimation of gelatinase content in rat brain: effect of focal ischemia. Biochem Biophys Res Commun 278(3):803–807
Planas AM, Solé S, Justicia C (2001) Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis 8(5):834–846
Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC (1998) Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 29(5):1020–1030
Rosenberg GA, Estrada EY, Dencoff JE (1998) Matrix metalloproteinases and TIMPs are associated with blood–brain barrier opening after reperfusion in rat brain. Stroke 29(10):2189–2195
Serhan CN, Oliw E (2001) Unorthodox routes to prostanoid formation: new twists in cyclooxygenase-initiated pathways. J Clin Invest 107(12):1481–1489
Serhan CN, Maddox JF, Petasis NA et al (1995) Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry 34(44):14609–14615
Serhan CN, Chiang N, Van Dyke TE (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8(5):349–361
Sodin-Semrl S, Taddeo B, Tseng D, Varga J, Fiore S (2000) Lipoxin A4 inhibits IL-1 beta-induced IL-6, IL-8, and matrix metalloproteinase-3 production in human synovial fibroblasts and enhances synthesis of tissue inhibitors of metalloproteinases. J Immunol 164(5):2660–2666
Sodin-Semrl S, Spagnolo A, Mikus R, Barbaro B, Varga J, Fiore S (2004) Opposing regulation of interleukin-8 and NF-kappaB responses by lipoxin A4 and serum amyloid A via the common lipoxin A receptor. Int J Immunopathol Pharmacol 17(2):145–156
Svensson CI, Zattoni M, Serhan CN (2007) Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. J Exp Med 204(2):245–252
Wada K, Arita M, Nakajima A et al (2006) Leukotriene B4 and lipoxin A4 are regulatory signals for neural stem cell proliferation and differentiation. FASEB J 20(11):1785–1792
Wagner S, Nagel S, Kluge B et al (2003) Topographically graded postischemic presence of metalloproteinases is inhibited by hypothermia. Brain Res 984(1–2):63–75
Wang X, Barone FC, White RF, Feuerstein GZ (1998) Subtractive cloning identifies tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) increased gene expression following focal stroke. Stroke 29(2):516–520
Wang X, Jung J, Asahi M et al (2000) Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J Neurosci 20(18):7037–7042
Wu CY, Hsieh HL, Jou MJ, Yang CM (2004) Involvement of p42/p44 MAPK, p38 MAPK, JNK and nuclear factor-kappa B in interleukin-1beta-induced matrix metalloproteinase-9 expression in rat brain astrocytes. J Neurochem 90(6):1477–1488
Wu SH, Liao PY, Dong L, Chen ZQ (2008) Signal pathway involved in inhibition by lipoxin A(4) of production of interleukins induced in endothelial cells by lipopolysaccharide. Inflamm Res 57(9):430–437
Wu Y, Ye XH, Guo PP et al (2010) Neuroprotective Effect of lipoxin A(4) methyl ester in a rat model of permanent focal cerebral ischemia. J Mol Neurosci 42(2):226–234
Ye XH, Wu Y, Guo PP et al (2010) Lipoxin A(4) analogue protects brain and reduces inflammation in a rat model of focal cerebral ischemia reperfusion. Brain Res 1323:174–183
Yiqin Y, Meilin X, Jie X, Keping Z (2009) Aspirin inhibits MMP-2 and MMP-9 expression and activity through PPARalpha/gamma and TIMP-1-mediated mechanisms in cultured mouse celiac macrophages. Inflammation 32(4):233–241
Zhou XY, Li YS, Wu P et al (2009) Lipoxin A(4) inhibited hepatocyte growth factor-induced invasion of human hepatoma cells. Hepatol Res 39(9):921–930
Acknowledgments
This work was supported by a grant from the National Natural Science Foundation of China (No. 30700784).
Author information
Authors and Affiliations
Corresponding author
Additional information
Yan Wu and Yan-Ping Wang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Wu, Y., Wang, YP., Guo, P. et al. A Lipoxin A4 Analog Ameliorates Blood–Brain Barrier Dysfunction and Reduces MMP-9 Expression in a Rat Model of Focal Cerebral Ischemia–Reperfusion Injury. J Mol Neurosci 46, 483–491 (2012). https://doi.org/10.1007/s12031-011-9620-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-011-9620-5