
Abstract
We report different clinical expression in seven members of a large family with amyotrophic lateral sclerosis (ALS) and the G93D mutation in exon 4 of the Cu/Zn superoxide dismutase (SOD1) gene. The ALS clinical course in the proband showed an unusually fast progression of the disease compared to the paucisymptomatic presentation associated to this mutation in the two previously Italian families described. The remaining mutation carriers did not show the aggressive clinical course displayed by the proband. We selected few genes known to be ALS modifiers searching for genetic variants that could explain the wide phenotypic diversity within the family. Exclusion of causative genes such as TDP43, FUS, PGRN and VAPB was performed too. We believe that this kind of family with contrasting phenotypes of ALS may be considered an excellent human model to study the relationship between a wider genetic profile, including modifier genes, and the clinical expression of the disease. Therefore, the novelty of our approach is also represented by the study of a single family to reproduce a composite structure in which search for possible modifier genes/genetic variants linked to SOD1 mutated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS), the most common form among motoneuron diseases, is a progressive neurodegenerative disorder involving motor neurons in motor cortex, brain stem, and spinal cord. Ninety percent of patients are sporadic (SALS), and almost 10% are familial (FALS), with multiple autosomal dominant and recessive forms. Currently, three major ALS genes and five additional loci have been identified (Valdmanis and Rouleau 2008). The first ALS gene (ALS1) associated with adult-onset autosomal dominant form of the disease was mapped to the chromosome 21q22.1–22.2 and encodes for the cytoplasmic Cu/Zn superoxide dismutase (SOD1) (Rosen et al. 1993). Recently, mutations in TAR-DNA binding protein 43 (TARDBP) located on chromosome 1p36.22 have been reported to be associated to familial cases as well as sporadic cases (Kabashi et al. 2008; Sreedharan et al. 2008). In addition, mutations in the FUS/TSL gene on chromosome 16 are present in familial ALS (Kwiatkowski et al. 2009; Vance et al. 2009) as well as in sporadic cases (Corrado et al. 2010). Nevertheless, SOD1 still remains the major gene involved in the disease since approximately 20% of FALS cases and 2% of overall ALS cases have identifiable mutations. At the present time, more than 120 different mutations located in all five exons of the gene have been identified worldwide in ALS patients (http://www.alsod.org/). All SOD1 mutations are autosomal dominantly inherited except D90A and D96N. The latter was found in compound heterozygosity with D90A in a recessive FALS (Andersen et al. 1995; Robberecht et al. 1996; Hand et al. 2001).
Although some genotypes are associated with a consistent phenotype, considerable phenotypic heterogeneity, especially in terms of age at onset, duration of the disease, and severity, exists among family members carrying the same mutation (Kim et al. 2007), and it could be related to the influence of undetermined modifier genes. Recently, two Italian families have been described carrying the G93D SOD1 mutation. Both the described families presented a slowly progressive disease with a long lasting paucisymptomatic phase (Luigetti et al. 2008; Restagno et al. 2008).
We describe a family with G93D SOD1 gene mutation characterized by wide variability of disease expression among family members and with the proband showing a fast progressing motor neuron disease compared to the G93D carriers within her family and the two previously described families. In order to search for possible modifier genes explaining the clinical phenotype observed in the proband, we performed an analysis of angiogenin gene (ANG) and genetic variants (paraoxonase 1, PON1; paraoxonase 2, PON2; vascular endothelial growth factor, VEGF; methylenetetrahydrofolate reductase, MTHFR; apolipoprotein E, APOE; hemochromatosis, HFE) reported to be associated to ALS (Figlewicz and Orrell 2003). Other genes known to be causative of ALS such as TDP43, FUS, progranulin (PGRN), and vesicle-associated membrane protein-associated protein B and C (VAPB) were also analyzed to exclude their possible role (Millecamps et al. 2010).
Patients and Methods
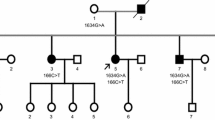
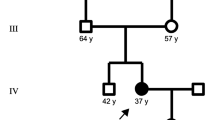
Pedigree of the family is shown in Fig. 1. We studied ten family members from a two-generation pedigree with three members affected by FALS: a 60-year-old female (III:10), her uncle (II:3 died and referred to be affected) and her niece (IV:7), respectively. ALS diagnosis related to (II:3), for whom clinical records were not available, was obtained from his descendent.

Pedigree of the SOD1 G93D Italian ALS family. All individuals carrying the mutation, with or without phenotype, are filled in. Arrow indicates index patient; diagonal line, individual deceased; horizontal double line, consanguineous marriage
Genetic Analysis
DNA was extracted from peripheral blood leukocyte (Miller et al. 1988). Written informed consent was obtained from all the participants. Genetic analysis was performed with two different approaches: direct sequencing and single nucleotide polymorphisms (SNPs) analysis.
DNA Sequencing
SOD1, ANG, TDP43, FUS, PGRN and VAPB genes were amplified by PCR with a specific subset of primers described elsewhere (Leverenz et al. 2007; Millecamps et al. 2010). Direct DNA sequencing included coding regions as well as splice site regions; the 600-bp promoter region analysis was included for the ANG gene (Corrado et al. 2007). The analysis was performed with an ABI 3730 automated sequencer (Applied Biosystems, UK).
Genetic Variants Analysis
The following genetic variants were analyzed through specific PCR amplifications followed by enzymatic restriction analysis: PON1 L55M (rs854560) and Q192R (rs662); PON2 S311C (rs6954345); and VEGF promoter variants −2578C/A (rs699947), −1154A/G (rs1570360), and −634C/G (rs2010963) (Ricci et al. 2010). We applied the LightCycler technology (Roche Molecular Biochemicals, Milan) for the typing of the following genetic variants according to PCR protocol already set out: C677T (rs1801133) mutation in the MTHFR; combinations of the codons 112 Cys/Arg (rs429358) and 158 Cys/Arg (rs7412) in the APOE gene determining the haplotype E2/E3/E4; and genetic variants C282Y, H63D, and S65C in the HFE gene.
Results
Molecular Analysis
The obtained results are summarized in Tables 1 and 2. We identified seven G93D (included the index patient) and three G93G (equal to wild type) subjects within the family. The whole SOD1 gene, screened in all the tested subjects, identified the IVS3 + 34 A/C in both the mutated and not mutated cases (IV:11 and IV:9, respectively): this variant has been already reported not associated to ALS (Battistini et al. 2005). The ANG gene analysis revealed the presence of three variants, the G110G (rs11701) in the coding region, and the IVS1+27C/T plus −127T/C in the regulatory regions. The IVS+27C/T variant was found in the index patient and in her father (IV:7 and III:8, respectively; see Table 1). Allele frequency of ANG variations has been previously determined in a healthy local population (Corrado et al. 2007). TDP43, FUS, PGRN and VAPB genes were negative for mutation screening, as expected as causative genes.
The other genetic variants analyzed (PONs, VEGF promoter, HFE, MTHFR, and APOE) showed a wide distribution among the family member tested (see Table 2).
Combination of single genotypes revealed the presence of a unique haplotype in the index patient (Table 1).
Clinical Characteristics of Symptomatic G93D SOD1 Mutation Carriers
Case IV:7
A previously healthy 33-year-old pregnant woman, unipara, presented with subacute, severe, left lower limb pain the day after the normal delivery, followed by relentless progressive weakness and wasting of the left leg. At birth, the newborn was healthy. Later, she developed a progressive worsening of the weakness with involvement of her four limbs and bulbar district, and complained a severe respiratory failure after 2 years. After 5 years, the patient is still alive, wheelchair-bound, in ventilatory assistance by tracheostomy and in enteral nutrition by gastrostomy. Her family, which included her 55-year-old mother (III:9), 60-year-old father (III:8), and a 26-year-old brother (IV:8), had positive history for motoneuron disease (III:10 and II:3); no clinical information were available for the proband’s grandfather (II:7). On June 2008, the patient was admitted at NEMO and her neurologic examination revealed severe weakness (ranging from grade 1/5 to 2/5 at the Medical Research Council (MRC) scale) of both upper and lower limb muscles; all deep tendon reflexes were reduced, and plantar reflexes were flexor bilaterally. Hoffmann sign was negative bilaterally, and jaw jerk reflexes were normal. MRI of the spine was negative. Needle electromyography (EMG) showed increased insertional activity, spontaneous activity (fibrillation potentials and positive sharp waves), increased amplitude, and long duration motor unit action potentials, associated with reduced recruitment in all limbs. Fasciculation potentials were diffusely present. The study was interpreted as consistent with lower motor neuronopathy. Comprehensive laboratory studies were normal, except mild creatine kinase (CK) level increment (175 U/L; normal value, 30–150 U/L). ALS Functional Rating Scale-revised (ALSFRS-r) score was 11.
Case III:10
A 60-year-old female was admitted to our center for a 4-year history of fasciculations in all four limbs and bilateral hand weakness. Neurological examination of the patient revealed diffuse fasciculations in both upper and lower limbs with bilateral muscular weakness (grade 3/5 on the MRC scale) and atrophy of both hands; tendon reflexes in upper and lower limbs were reduced. Hoffmann, Babinski, or Chaddock signs were not present. Laboratory tests revealed mild CK level increment (160 U/L). EMG of tibialis anterior, biceps brachii, and first dorsal interosseous bilaterally showed a neurogenic pattern: high frequency large motor unit potentials, sporadic denervation (fibrillation potentials, positive waves), and fasciculations. Transcranial magnetic stimulation of the motor cortex revealed normal central conduction time. Forced vital capacity as percent predicted was 96%. ALSFRS-r score was 45.
Clinical characteristics of all G93D SOD1 mutation carriers are summarized in Table 3.
Discussion
We report a detailed profile of clinical and genetics findings of members of an Italian family carrying the G93D SOD1 mutation with different phenotypic expression. In particular, to explore the phenotypic heterogeneity within the family, we investigated the possible relationship between the SOD1 mutation and some modifier genes reported to be involved in ALS pathogenesis. Various genes/genetic variants could have been tested; however, we decided to focus on a few of them. Since the involvement of angiogenic factors in ALS, albeit controversial, is still attractive (Lambrechts et al. 2006), we selected ANG and VEGF SNPs for this study. Considering the important role of oxidative stress in motor neuron degeneration (Barber and Shaw 2010), we looked for HFE gene variants, in particular for H63D allele found associated with ALS (Restagno et al. 2007; Sutedja et al. 2007). PON genes have been studied as susceptibility factor to sporadic ALS (Slowik et al. 2006). However, association studies between PON genetic variants and the risk for SALS in different populations reported conflicting results (Wills et al. 2009). We have recently studied PON polymorphisms in an Italian population negative for SOD1 mutations and no significant association was observed between the examined SNPs and the risk of ALS (Ricci et al. 2010). Despite that, point mutations in PON genes have been identified in FALS and SALS (Ticozzi et al. 2010). MTHFR C677T variant was selected because linked to an increased level of homocysteine (Hcy) and, in preliminary studies, ALS subjects showed higher median Hcy levels compared to age- and sex-matched controls (Valentino et al. 2010); higher Hcy levels were also correlated with a possible marker of disease progression (Zoccolella et al. 2010). Finally, alleles E2, E3, and E4 of APOE gene were included in our analysis because of their association with some neurodegenerative disorders, such as ALS (Zetterberg et al. 2008).
Results of our study did not disclose haplotypes common to G93D mutation carriers or to wild-type subjects. As unique characteristic of the proband (IV:7), we identified the ANG IVS1+27 variant in heterozygous state C/T. This genetic variation has been previously described, although no association with ALS has been reported in a SOD1 negative population (Corrado et al. 2007). To date, little information about the role of ANG variants in a SOD1 mutated genetic context are available. In the present family, the proband (IV:7) manifested the disease after the delivery that physiologically could determine a hypoxic condition: ANG, like VEGF, is expressed in response to hypoxia (Campo et al. 2005; Nakamura et al. 2006) and plays a neuroprotective role in motoneurons (Subramanian et al. 2008). Thus, we might speculate that the patient’s genetic framework, including the G93D mutation and the IVS1+27 variation, together with the hypoxic status linked to the delivery facilitated the anticipation of onset age and contributed to the aggressive clinical course observed.
The G93D SOD1 mutation has been previously described in two Italian families (Luigetti et al. 2008; Restagno et al. 2008) and associated with a slowly developing motor neuron disease; this mutation was reported to confer a high propensity of protein aggregation too (Stathopulos et al. 2006). On the other hand, the G93A SOD1 mutation has been shown to cause the protein exclusion from the nucleus with consequent reduction of DNA protection from oxidative damage, thus underlying the important role of the amino acidic position at 93 in the protein (Sau et al. 2007).
The innovation of our approach consists in studying a family as a model to search for possible modifier genes/genetic variants linked to SOD1 mutations. A similar approach was introduced by Broom WJ et al. (Broom et al. 2006), which looked for variants in candidate ALS modifier genes linked to Cu/Zn superoxide dismutase explaining divergent survival phenotypes. DNA of four genes within the haplotypic region shared in A4V and D90A ALS patients was sequenced, and 15 novel variants were identified, but none resulted specifically associated with SOD1 D90A/D90A or SOD1 A4V ALS.
In conclusion, the current study describes ten family members, seven of which with G93D SOD1 mutation, and represents an attempt at using the whole family group as human model to identify putative modifier genes responsible for different intrafamilial phenotypic expression.
References
Andersen PM, Nilsson P, Ala-Hurula V et al (1995) Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn superoxide dismutase. Nat Genet 10:61–66
Barber SC, Shaw PJ (2010) Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med 48:629–641
Battistini S, Giannini F, Greco G et al (2005) SOD1 mutations in amyotrophic lateral sclerosis. Results from a multicenter Italian study. J Neurol 252:782–788
Broom WJ, Russ C, Sapp PC et al (2006) Variants in candidate ALS modifier genes linked to Cu/Zn superoxide dismutase do not explain divergent survival phenotypes. Neurosci Lett 392:52–57
Campo L, Turley H, Han C et al (2005) Angiogenin is up-regulated in the nucleus and cytoplasm in human primary breast carcinoma and is associated with markers of hypoxia but not survival. J Pathol 205:585–591
Corrado L, Battistini S, Penco S et al (2007) Variations in the coding and regulatory sequences of the angiogenin (ANG) gene are not associated to ALS (amyotrophic lateral sclerosis) in the Italian population. J Neurol Sci 258:123–127
Corrado L, Del Bo R, Castellotti B et al (2010) Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J Med Genet 47:190–194
Figlewicz DA, Orrell RW (2003) The genetics of motor neuron diseases. Amyotroph Lateral Scler Other Mot Neuron Disord 4:225–231
Hand CK, Mayeux-Portas V, Khoris J et al (2001) Compound heterozygous D90A and D96N SOD1 mutations in a recessive amyotrophic lateral sclerosis family. Ann Neurol 49:267–271
Kabashi E, Valdmanis PN, Dion P et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Kim W, Kim JS, Lee KS, Gwoun YJ, Kim JM, Lee KH (2007) Anticipation and phenotypic heterogeneity in korean familial amyotrophic lateral sclerosis with superoxide dismutase 1 gene mutation. J Clin Neurol 3:38–44
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Lambrechts D, Lafuste P, Carmeliet P, Conway EM (2006) Another angiogenic gene linked to amyotrophic lateral sclerosis. Trends Mol Med 12:345–347
Leverenz JB, Yu CE, Montine TJ et al (2007) A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain 130:1360–1374
Luigetti M, Madia F, Conte A et al (2008) SOD1 G93D mutation presenting as paucisymptomatic amyotrophic lateral sclerosis. Amyotroph Lateral Scler 31:1–4
Millecamps S, Salachas F, Cazeneuve C et al (2010) SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J Med Genet 47:554–560
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res 16:1215
Nakamura M, Yamabe H, Osawa H et al (2006) Hypoxic conditions stimulate the production of angiogenin and vascular endothelial growth factor by human renal proximal tubular epithelial cells in culture. Nephrol Dial Transplant 21:1489–1495
Restagno G, Lombardo F, Ghiglione P et al (2007) HFE H63D polymorphism is increased in patients with amyotrophic lateral sclerosis of Italian origin. J Neurol Neurosurg Psychiatry 78:327
Restagno G, Lombardo F, Sbaiz L et al (2008) The rare G93D mutation causes a slowly progressing lower motor neuron disease. Amyotroph Lateral Scler 9:35–39
Ricci C, Battistini S, Cozzi L et al (2010) Lack of association of PON polymorphisms with sporadic ALS in an Italian population. Neurobiol Aging (in press)
Robberecht W, Aguirre T, Van Den Bosch L, Tilkin P, Cassiman JJ, Matthijs MG (1996) D90A heterozygosity in the SOD1 gene is associated with familial and apparently sporadic amyotrophic lateral sclerosis. Neurology 47:1336–1339
Rosen DR, Siddique T, Patterson D et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Sau D, De Biasi S, Vitellaro-Zuccarello L et al (2007) Mutation of SOD1 in ALS: a gain of a loss of function. Hum Mol Genet 16:1604–1618
Slowik A, Tomik B, Wolkow PP et al (2006) Paraoxonase gene polymorphisms and sporadic ALS. Neurology 67:766–770
Sreedharan J, Blair IP, Tripathi VB et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Stathopulos PB, Rumfeldt JA, Karbassi F et al (2006) Calorimetric analysis of thermodynamic stability and aggregation for apo and holo amyotrophic lateral sclerosis-associated Gly-93 mutants of superoxide dismutase. J Biol Chem 281:6184–6193
Subramanian V, Crabtree B, Acharya KR (2008) Human angiogenin is a neuroprotective factor and amyotrophic lateral sclerosis associated angiogenin variants affect neurite extension/pathfinding and survival of motor neurons. Hum Mol Genet 17:130–149
Sutedja NA, Sinke RJ, Van Vught PW et al (2007) The association between H63D mutations in HFE and amyotrophic lateral sclerosis in a Dutch population. Arch Neurol 64:63–67
Ticozzi N, Leclerc AL, Keagle PJ et al (2010) Paraoxonase gene mutations in amyotrophic lateral sclerosis. Ann Neurol 12(68):102–107
Valdmanis PN, Rouleau GA (2008) Genetics of familial amyotrophic lateral sclerosis. Neurology 70:144–152
Valentino F, Bivona G, Butera D et al (2010) Elevated cerebrospinal fluid and plasma homocysteine levels in ALS. Eur J Neurol 17:84–89
Vance C, Rogelj B, Hortobágyi T et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1210
Wills AM, Cronin S, Slowik A et al (2009) A large-scale international meta-analysis of paraoxonase gene polymorphisms in sporadic ALS. Neurology 73:16–24
Zetterberg H, Jacobsson J, Rosergren L, Blennow K, Andersen PL (2008) Association of APOE with age at onset of sporadic amyotrophic lateral sclerosis. J Neurol Sci 273:67–69
Zoccolella S, Bendotti C, Beghi E, Logroscino G (2010) Homocysteine levels and amyotrophic lateral sclerosis: a possible link. Amyotroph Lateral Scler 11:140–147
Acknowledgments
The authors would like to thanks the index patient and her family for participation in this study and the “Fondazione Alberto Monti” for supporting LM and part of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Penco, S., Lunetta, C., Mosca, L. et al. Phenotypic Heterogeneity in a SOD1 G93D Italian ALS Family: An Example of Human Model to Study a Complex Disease. J Mol Neurosci 44, 25–30 (2011). https://doi.org/10.1007/s12031-010-9480-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-010-9480-4