
Abstract
Background and Aim
Sodium valproate (SV), a novel class of histone deacetylases (HDACs) inhibitors commonly used as an antiepileptic drug. HDAC inhibitors are known to possess anticancer potentials. In this study, we investigated the cytotoxic potential of SV in human hepatocellular carcinoma (HepG2 cells) cell line.
Methods
MTT assay was used to analyze cytotoxicity. Intracellular ROS and cytochrome c expression were analyzed by fluorescence microscopy. Morphology-related apoptosis was analyzed by dual staining with acridine orange/ethidium bromide. Caspase 3 protein expression was investigated by Western blotting analysis.
Results
Sodium valproate treatments in HepG2 cells caused significant and dose-dependent cytotoxicity. Intracellular ROS was remarkably increased in the cells which are treated with SV and caused early and late apoptosis as evidenced by dual staining. SV-treated cells expressed cytochrome c and caspase 3 protein expression.
Conclusion
These results suggest the cytotoxic potentials of SV in HepG2 cells. This study may give an important clue for the inclusion of SV as an adjuvant along with standard anticancer agents after necessary in vivo and clinical studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) prevalence is swiftly increasing globally with a high mortality rate [1]. Approximately 700,000 people die of HCC each year, making it the third leading cause of cancer-related deaths worldwide [2, 3]. Hepatitis B and C virus infections, aflatoxin-contaminated food, non-alcoholic fatty liver diseases, non-alcoholic steatohepatitis, and metabolic disorders like diabetes, obesity, and hemochromatosis are the major risk factors associated with the development of HCC [4]. Currently, the Food and Drug Administration (FDA) has approved tyrosine kinase inhibitors like sorafenib, regorafenib, nivolumab, lenvatinib, cabozatinib, and pembrolizumab for HCC patients to improve quality of life and survival [1]. The common side effects of these drugs are fatigue, diarrhea, hand-foot skin reaction, nausea, vomiting, decreased appetite, hypertension, and weight loss, etc. [5, 6]. On the other hand, chemo/radiotherapy is responsible for off-target effects like nausea, vomiting, and metallic taste [7,8,9]. Despite significant clinical management involving targeted therapies, chemo/radiotherapies, and surgical procedures, HCC remains one of the frequently responsible for cancer-related death worldwide and it is cause for concern. Therefore, the need of the hour is to identify a therapeutic compound for use in HCC patients with fewer or no side effects.
Sodium valproate (SV) is an FDA-approved anticonvulsant drug commonly used in the long-term therapy of epilepsy [10]. Sodium valproate is reported as a novel class of histone deacetylase (HDAC) inhibitors [11]. HDACs play crucial roles in transcriptional regulation and pathogenesis of cancer. In posttranslational histone modification, histone acetylation is controlled by the opposing activities of histone acetyltransferases and HDACs. By removing acetyl groups, HDACs reverse chromatin acetylation and alter transcription of oncogenes and tumor suppressor genes. Besides, HDACs deacetylate numerous non-histone cellular substrates that govern a wide array of biological processes including cancer initiation and progression [12]. HDACs are often overexpressed in a variety of cancer including HCC, gastric, colorectal, bladder, breast, and esophageal squamous cell carcinomas [13,14,15,16,17,18], and therefore, HDACs inhibitors seem to be promising anti-cancer drugs particularly as an adjuvant in combination with other anti-cancer drugs and/or radiotherapy [19,20,21]. In previous studies, HDACs inhibitors have been shown to have anticancer effects via inhibition of cancer cell proliferation and induction of cell cycle arrest and apoptosis [22,23,24]. Therefore, it is reasonable to assume that as an HDACs inhibitor, SV may have the cytotoxic potential in cancer cells. Further, SV has not been studied previously for its cytotoxic potential against hepatocellular cancer cells. Therefore, in the present study, we investigated the cytotoxic potential of SV in human hepatocellular carcinoma (HepG2) cells.
Methods
Chemicals
Dulbecco’s minimum essential low glucose medium (DMEM), penicillin, streptomycin, dimethylsulfoxide (DMSO), tryspin-ethylenediaminetetraacetic acid (EDTA), and fetal bovine serum (FBS) were obtained from GIBCO BRL (Gaithersburg, MD). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from Sigma Chemicals (Chennai, India). Sodium valproate EP (Batch No. BSW 026) was obtained as gratis from Anjan Drug Private Limited (Chennai, India). All other chemicals were of analytical grade.
Cell Culture and Maintenance
The HepG2 cell line was procured from National Centre for Cell Science (Pune, Maharashtra, India). The cells were cultured in 25 cm2 flask using DMEM with low glucose containing 10% FBS with penicillin and streptomycin with 5% CO2 at 37 °C. Cells were cultured for a couple of passages for acclimatization and then used for experiments. Cells were collected after reaching enough confluence using trypsin-EDTA solution (0.25%) and were seeded for experiments. SV was dissolved in 0.1% DMSO.
MTT Assay
Cytotoxicity was evaluated in HepG2 cells by MTT assay [25]. Cells were seeded at 1 × 104 cells/well and were left for cell attachment for 24 h. The existing medium was then changed with medium containing SV at 0.5, 1, 1.5, 2, and 2.5 mM/mL and incubated for 24 h. After 24 h, the media was aspirated and cells were washed once with PBS and then cells were incubated with 50 μL of MTT (0.5 mg/mL) for 4 h inside the CO2 incubator. MTT was discarded and DMSO was added to dissolve the colored formazan crystals produced by the viable cells. The purple-blue formazan formed was measured using Perkin Elmer Multimode Reader (USA) at 570 nm. The optical density of each sample was compared with control optical density and graphs were plotted.
Dichloro-dihydro-fluorescein Diacetate (DCFH-DA) Staining
The ROS expression analysis was investigated by DCFH-DA staining in SV-treated HepG2 cells [7]. At the end of treatments, 8 × 106 cells/mL from control and SV treatments were taken and were made up to 2 mL using PBS (pH 7.4). Then, 100 μL of DCFH-DA (10 μM) was added to 1 mL of the cell suspension and incubated for 30 min at 37 °C and images were captured using Nikon Eclipse Ti fluorescence microscope (Nikon Instruments Inc., NY, USA).
Acridine Orange/Ethidium Bromide (AO/EB) Staining and Fluorescent Microscopy
Apoptosis-related morphological damage was investigated by AO/EB staining [26]. Cells were plated at a density of 1 × 104 in 48-well plates. At the end of the treatment period, the culture medium was aspirated and cells were washed once with PBS at room temperature. Then, 100 μL of dye mixture (1:1) was mixed with equal volume of cells from control and experimental groups and viewed immediately under Nikon inverted fluorescence microscope (Nikon Instruments Inc., NY, USA). A minimum of 200 cells was counted in each sample at 5 different fields. The percentage of apoptotic cells was determined by [% of apoptotic cells = (total number of apoptotic cells/total number of cells counted) × 100].
Cytochrome c Immunofluorescence
HepG2 cells (5 × 104) were seeded in 12-well plates, and treatments were done using different concentrations of SV for 24 h. After 24 h, cells were washed with PBS and fixed in 4% formaldehyde for 10–15 min. Cells fixed on slides were rehydrated, blocked with 5% normal goat serum, and permeabilized with 0.5% Tween 20 and then incubated with monoclonal cytochrome C primary antibody (ab13575), followed by probing with goat anti-mouse IgG (ab150115) secondary antibody for 2 h, and then images were captured using Nikon Eclipse Ti inverted fluorescence microscope (Nikon Instruments Inc., NY, USA).
Western Blot Analysis of Caspase 3
Control and SV-treated cells were lysed with RIPA buffer and phosphatase inhibitor cocktails. Protein was estimated using bovine serum albumin (BSA) as standard. Total protein extracts were subjected to electrophoresis and electroblotted onto polyvinylidene difluoride membrane. Then, the membrane was blocked with 5% BSA and incubated overnight with anti-cleaved (activated) caspase-3 antibody (monoclonal, IgG1, 1:100, Cell signaling technology, #9669) at 4 °C and 2 h with corresponding secondary antibodies at room temperature. The enhanced chemiluminescence with protein A-horseradish peroxidase was used to detect the immunoreactive bands.
Statistical Analysis
Data were expressed as mean ± S.E.M and analyzed by one-way ANOVA followed by Dunnett’s multiple comparison test to determine the significant differences between groups. p < 0.05 was considered significant (Graph Pad prism 7.0. CA, USA).
Results
Sodium Valproate Treatments Induced Cytotoxicity in HepG2 Cells
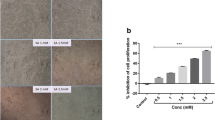
In this study, we investigated the cytotoxic potential of SV in HepG2 cells. The morphology of SV-treated HepG2 cells is presented in Fig. 1a. The HepG2 cells were treated with SV for 24 h, and cytotoxicity was investigated by MTT assay. Different concentrations of SV (0.5, 1, 1.5, 2, and 2.5 mM/mL) treatments caused a significant (p < 0.001) and a dose-dependent cytotoxicity in HepG2 cells (Fig. 1b). The IC50 of SV in HepG2 cells is reported to be found at 2 mm/mL. Therefore, further studies were carried out with 2 and 4 mM/mL of SV.

Sodium valproate (SV) induced changes in the proliferation of HepG2 cells. a Morphology of control and SV-treated HepG2 cells. b Cytotoxicity analysis by MTT assay. n = 3 ***p < 0.001 vs control
Sodium Valproate Treatments Caused ROS Accumulation in HepG2 Cells
In order to delineate the reason behind the cytotoxicity, we investigated ROS inducing potentials of SV in HepG2 cells. SV treatments at two different concentrations, i.e., 2 and 4 mM/mL for 24 h in HepG2 cells, caused remarkable intracellular accumulation of ROS as compared with control cells. The ROS inducing potential of SV has seemed to be as a dose-dependent (Fig. 2).

Reactive oxygen species inducing potentials of sodium valproate (SV) by 2′,7′-dichlorodihydrofluorescein diacetate immunofluorescence staining
Sodium Valproate Treatments Caused Apoptosis in HepG2 Cells
Increased intracellular accumulation of ROS is often caused apoptosis-related morphological damage in cancer cells. Therefore, to investigate whether the accumulation of ROS is responsible for apoptosis, we investigated morphological changes induced by SV in HepG2 cells by AO/EB staining. Control cells did not show any fluorescent signal. SV in low-concentration caused early and late apoptosis as evidenced by the presence of bright green–colored and red-colored nuclei respectively. At the same time, high concentration of SV treated cells caused predominantly late apoptosis as evidenced by the presence of red-colored nuclei (Fig. 3a). The number of apoptotic cell presence was counted after SV treatments show significant (p < 0.001) and a dose-dependent increase as compared with control (Fig. 3b).

a Morphological analysis of apoptosis by acridine orange/ethidium bromide dual staining. b Quantification of early and late apoptotic cells. n = 3. ***p < 0.001 vs control
Sodium Valproate Treatments Induced Cytochrome c Expression
Cytosolic release of cytochrome c from mitochondria into the cytosol is an important and earlier molecular event associated with an intrinsic mitochondrial pathway of apoptosis. Therefore, to find out the role of mitochondria in SV induced apoptosis, we investigated the cytochrome c expression. The SV-treated HepG2 cells were expressed cytochrome c in their cytoplasm as compared with control cells (Fig. 4).

Cytochrome c dislocation analysis by immunofluorescence staining in control and sodium valproate (SV)–treated HepG2 cells
Sodium Valproate Treatments Induced Caspase 3 Expression
Caspase 3 is an important downstream molecular target of cytochrome c, and its activation plays a pivotal role in the execution of intrinsic apoptosis. Therefore, we investigated the caspase protein expression by Western blotting. SV especially at the high dose used in this study induced caspase 3 expressions as compared with control. The maximum expression was observed with the dose of 4 mM/mL of SV (Fig. 5a). The densitometry analysis also confirmed the significant (p < 0.001) increase in caspase 3 expressions upon with the high dose of SV in HepG2 cells (Fig. 5b). β actin was used as an internal control for normalization.

a Western blot expression of caspase 3 and its cleaved fraction. b Quantification of caspase protein expression by densitometry analysis. n = 3. ***p < 0.001 vs control
Discussion
Histone deacetylases plays an imperative role in regulating the transcription of many genes involved in the progression of HCC, and therefore, HDACs inhibitors are emerging as a promising therapeutic drug candidate for HCC [27, 28]. The present study showed the cytotoxic potential of SV, a HDACs inhibitor in HepG2 cells. The inhibition of proliferation could be attributed to the HDAC inhibiting ability of SV. In previous studies, SV has been shown to have cytotoxic potential against cervical [29], breast [30, 31], colon [32], pancreas [33], cancer cell lines, and our current results are in agreement with these reports.
Ample evidence suggests that accumulation of intracellular ROS affects cancer cell homeostasis, and it plays a critical role in cancer cell death via induction of cytotoxicity [7, 34, 35]. Studies are showing that HDAC inhibitors can induce cancer cell death via intracellular ROS accumulation [22, 36]. Further, in a previous study, HDACs knockdown induced the accumulation of intracellular ROS in gastric cancer cells [37]. SV also has the potential to accumulate intracellular ROS-mediated cytotoxicity in cancer cells [29, 38, 39]. In light of the above studies, it is reasonable to assume that the cytotoxicity induced by SV could be due to the intracellular ROS accumulation and inhibition of HDACs.
An excessive accumulation of intracellular ROS can cause damage to biological macromolecules, cell membrane, and cell organelle like mitochondria [9, 40]. Cancer cells are highly vulnerable to ROS-induced cytotoxicity, and an increased ROS level induces apoptosis-related morphological damage in various cancer cells [7, 8, 41, 42]. In this study, we observed remarkable early and late apoptosis in lower and higher doses of SV treatments respectively. Excessive intracellular ROS could be related to cell membrane damage and apoptosis-related morphological changes in HepG2 cells.
Histone deacetylase inhibitors are shown to promote apoptosis in human liver cancer cells [43]. Apoptosis is programmed cell death, which is regulated by the complex process that can be triggered by external or internal stimuli, which activate the extrinsic or the intrinsic apoptotic pathway [44]. Mitochondrion is a highly sensitive intracellular organ for pro-apoptotic agents that induce ROS [45, 46]. ROS accumulation can cause mitochondrial membrane toxicity, which can lead to the loss of mitochondrial membrane potential (MMP) and the release of cytochrome c into the cytosol [47]. Therefore, cytochrome c is considered a key molecule for apoptosis-mediated cell death, and its release from mitochondria upon MMP loss is used to interpret the mitochondrial toxicity due to ROS-mediated oxidative stress and intrinsic mitochondrial pathway of apoptosis [47]. In cytosol, cytochrome c involves in the apoptosome formation and apoptosis induction via activation of executioner caspase 3 [48]. In this study, our immunofluorescence analysis confirmed the expression of cytochrome c in the cytosol and caspase 3 protein expression upon SV treatments in HepG2 cells indicating the possibility of MMP loss and apoptosis induction mainly through the intrinsic apoptotic pathway.
Interestingly, in experimetnal studies SV enhances the anticancer potentials of standard chemotherapeutic drugs [21, 49, 50]. To conclude, the present study also suggests that SV can be included as an adjuvant in the anticancer chemotherapeutic regimen along with standard cancer chemotherapeutic drugs for HCC patients. However, further detailed experimental studies in vivo and clinical studies are warranted on the safety and mechanism of action of SV.
References
Jindal A, Thadi A, Shailubhai K. Hepatocellular carcinoma: etiology and current and future drugs. J Clin Exp Hepatol. 2019;9:221–32.
Ezhilarasan D. Lead compounds with the potentials for the treatment of chronic liver diseases. In: Egbuna C, Kumar S, Ifemeje J, Ezzat S, Kaliyaperumal S, editors. Phytochemicals as lead compounds for new drug discovery. 1st ed. Amsterdam: Elsevier; 2019. p. 195–210.
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917.
Madduru D, Ijaq J, Dhar S, Sarkar S, Poondla N, Das PS, et al. Systems challenges of hepatic carcinomas: a review. J Clin Exp Hepatol. 2019;9:233–44.
Rimassa L, Danesi R, Pressiani T, Merle P. Management of adverse events associated with tyrosine kinase inhibitors: improving outcomes for patients with hepatocellular carcinoma. Cancer Treat Rev. 2019;77:20–8.
Granito A, Marinelli S, Negrini G, Menetti S, Benevento F, Bolondi L. Prognostic significance of adverse events in patients with hepatocellular carcinoma treated with sorafenib. Ther Adv Gastroenterol. 2016;9:240–9.
Gheena S, Ezhilarasan D. Syringic acid triggers reactive oxygen species-mediated cytotoxicity in HepG2 cells. Hum Exp Toxicol. 2019;38:694–702.
Shebi S, Ezhilarasan D, Thomas J, Chandrasekaran N, Mukherjee A. Gracilaria foliifera (Forssk.) Børgesen ethanolic extract triggers apoptosis via activation of p53 expression in HepG2 cells. Phcog Mag. 2019;15:259–63.
Ezhilarasan D. Herbal therapy for cancer. In: Prakash Srinivasan, Timiri Shanmugam., editors. Understanding cancer therapies, CRC Press; 2018. pp. 129–166.
Brookes RL, Crichton S, Wolfe CDA, Yi Q, Li L, Hankey GJ, et al. Sodium valproate, a histone deacetylase inhibitor, is associated with reduced stroke risk after previous ischemic stroke or transient ischemic attack. Stroke. 2018;49:54–61.
Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–78.
Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6:a026831.
Schizas D, Mastoraki A, Naar L, Tsilimigras DI, Katsaros I, Fragkiadaki V, et al. Histone Deacetylases (HDACs) in gastric cancer: an update of their emerging prognostic and therapeutic role. Curr Med Chem. 2019. https://doi.org/10.2174/0929867326666190712160842.
Ishikawa D, Takasu C, Kashihara H, Nishi M, Tokunaga T, Higashijima J, et al. The significance of MicroRNA-449a and its potential target HDAC1 in patients with colorectal cancer. Anticancer Res. 2019;39:2855–60.
Buckwalter JM, Chan W, Shuman L, Wildermuth T, Ellis-Mohl J, Walter V, et al. Characterization of histone deacetylase expression within in vitro and in vivo bladder cancer model systems. Int J Mol Sci. 2019;20:E2599.
Linares A, Assou S, Lapierre M, Thouennon E, Duraffourd C, Fromaget C, et al. Increased expression of the HDAC9 gene is associated with antiestrogen resistance of breast cancers. Mol Oncol. 2019;13:1534–47.
Ma S, Liu T, Xu L, Wang Y, Zhou J, Huang T, et al. Histone deacetylases inhibitor MS-275 suppresses human esophageal squamous cell carcinoma cell growth and progression via the PI3K/Akt/mTOR pathway. J Cell Physiol. 2019;234:22400–10.
Wang Z, Wang H, Shen P, Xie R. Expression of HDAC4 in stage B hepatocellular carcinoma and its influence on survival. Ann Clin Lab Sci. 2019;49:189–92.
Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18:E1414.
Suraweera A, O'Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front Oncol. 2018;8:92.
Gavrilov V, Lavrenkov K, Ariad S, Shany S. Sodium valproate, a histone deacetylase inhibitor, enhances the efficacy of vinorelbine-cisplatin-based chemoradiation in non-small cell lung cancer cells. Anticancer Res. 2014;34:6565–72.
Sohaib M, Ezhilarasan D. Carbamazepine, a histone deacetylase inhibitor induces apoptosis in human colon adenocarcinoma cell line HT-29. J Gastrointest Cancer. 2019:1–7. https://doi.org/10.1007/s12029-019-00286-x.
Gong P, Wang Y, Jing Y. Apoptosis induction by histone deacetylase inhibitors in cancer cells: role of Ku70. Int J Mol Sci. 2019;20:E1601.
Bao L, Diao H, Dong N, Su X, Wang B, Mo Q, et al. Histone deacetylase inhibitor induces cell apoptosis and cycle arrest in lung cancer cells via mitochondrial injury and p53 up-acetylation. Cell Biol Toxicol. 2016;32:469–82.
PonselviInduja M, Ezhilarasan D, Ashok VN. Evolvulusalsinoidesmethanolic extract triggers apoptosis in HepG2 cells. Avicenna J Phytomed. 2018;8:504–12.
Lakshmi T, Ezhilarasan D, Vijayaragavan R, Bhullar SK, Rajendran R. Acacia catechu ethanolic bark extract induces apoptosis in human oral squamous carcinoma cells. J Adv Pharm Technol Res. 2017;8:143–9.
Tsilimigras DI, Ntanasis-Stathopoulos I, Moris D, Spartalis E, Pawlik TM. Histone deacetylase inhibitors in hepatocellular carcinoma: a therapeutic perspective. Surg Oncol. 2018;27:611–8.
Liu KY, Wang LT, Hsu SH. Modification of epigenetic histone acetylation in hepatocellular carcinoma. Cancers (Basel). 2018;10:E8.
Han BR, You BR, Park WH. Valproic acid inhibits the growth of HeLa cervical cancer cells via caspase-dependent apoptosis. Oncol Rep. 2013;30:2999–3005.
Ma XJ, Wang YS, Gu WP, Wang Y, Zhou J, Huang T, et al. The role and possible molecular mechanism of valproic acid in the growth of MCF-7 breast cancer cells. Croat Med J. 2017;58:349–57.
Sargazi S, Kooshkaki O, Zavar Reza J, Saravani R, Zarei Jaliani H, Mirinejad S, et al. Mild antagonistic effect of valproic acid in combination with AZD2461 in MCF-7 breast cancer cells. Med J Islam Repub Iran. 2019;33:29.
Ghecham A, Senator A, Pawlowska E, Bouafia W, Błasiak J. Epigenetic modifiers 5-aza-2'-deoxycytidine and valproic acid differentially change viability, DNA damage and gene expression in metastatic and non-metastatic colon cancer cell lines. Acta Biochim Pol. 2019;66(3):355–60.
Li H, Zhang Z, Gao C, Wu S, Duan Q, Wu H, et al. Combination chemotherapy of valproic acid (VPA) and gemcitabine regulates STAT3/Bmi1 pathway to differentially potentiate the motility of pancreatic cancer cells. Cell Biosci. 2019;9:50.
Ezhilarasan D, Apoorva VS, Ashok Vardhan N. Syzygium cumini extract induced reactive oxygen species-mediated apoptosis in human oral squamous carcinoma cells. J Oral Pathol Med. 2019;48:115–21.
Rohit Singh T, Ezhilarasan D. Ethanolic extract of Lagerstroemia Speciosa (L.) Pers., induces apoptosis and cell cycle arrest in HepG2 cells. Nutr Cancer. 2020;72:146–56.
Rivera-Del Valle N, Cheng T, Irwin ME, Donnella H, Singh MM, Chandra J. Combinatorial effects of histone deacetylase inhibitors (HDACi), vorinostat and entinostat, and adaphostin are characterized by distinct redox alterations. Cancer Chemother Pharmacol. 2018;81:483–95.
Lee JH, Jeong EG, Choi MC, Kim SH, Park JH, Song SH, et al. Inhibition of histone deacetylase 10 induces thioredoxin-interacting protein and causes accumulation of reactive oxygen species in SNU-620 human gastric cancer cells. Mol Cells. 2010;30:107–12.
Cornago M, Garcia-Alberich C, Blasco-Angulo N, Vall-Llaura N, Nager M, Herreros J, et al. Histone deacetylase inhibitors promote glioma cell death by G2 checkpoint abrogation leading to mitotic catastrophe. Cell Death Dis. 2014;5:e1435.
Tseng JH, Chen CY, Chen PC, Hsiao SH, Fan CC, Liang YC, et al. Valproic acid inhibits glioblastoma multiforme cell growth via paraoxonase 2 expression. Oncotarget. 2017;8:14666–79.
Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 1863;2016:2977–92.
Wang L, Hu T, Shen J, Zhang L, Li LF, Chan RL, et al. Miltirone induced mitochondrial dysfunction and ROS-dependent apoptosis in colon cancer cells. Life Sci. 2016;151:224–34.
Wang Y, Luo Q, He X, Wei H, Wang T, Shao J, et al. Emodin induces apoptosis of colon cancer cells via induction of autophagy in a ROS-dependent manner. Oncol Res. 2018;26:889–99.
Gong D, Zeng Z, Yi F, Wu J. Inhibition of histone deacetylase 11 promotes human liver cancer cell apoptosis. Am J Transl Res. 2019;11:983–90.
Cavalcante GC, Schaan AP, Cabral GF, Santana-da-Silva MN, Pinto P, Vidal AF, et al. A cell’s fate: an overview of the molecular biology and genetics of apoptosis. Int J Mol Sci. 2019;20:E4133.
Jin H, Ko YS, Park SW, Chang KC, Kim HJ. 13-Ethylberberine induces apoptosis through the mitochondria-related apoptotic pathway in radiotherapy-resistant breast cancer cells. Molecules. 2019;24:E2448.
Heimer S, Knoll G, Schulze-Osthoff K, Ehrenschwender M. Raptinal bypasses BAX, BAK, and BOK for mitochondrial outer membrane permeabilization and intrinsic apoptosis. Cell Death Dis. 2019;10:556.
Kanipandian N, Li D, Kannan S. Induction of intrinsic apoptotic signaling pathway in A549 lung cancer cells using silver nanoparticles from Gossypium hirsutum and evaluation of in vivo toxicity. Biotechnol Rep (Amst). 2019;23:e00339.
Vairavel M, Devaraj E, Shanmugam R. An eco-friendly synthesis of Enterococcus sp.-mediated gold nanoparticle induces cytotoxicity in human colorectal cancer cells. Environ Sci Pollut Res Int. 2020. https://doi.org/10.1007/s11356-019-07511-x.
Tong XH, Zheng C, Jiang GJ, Dong SY. Sodium valproate enhances doxorubicin cytotoxicity in breast cancer cells in vitro. Nan Fang Yi Ke Da Xue Xue Bao. 2015;35:62–5.
Killick-Cole CL, Singleton WGB, Bienemann AS, Asby DJ, Wyatt MJ, Boulter LJ, et al. Repurposing the anti-epileptic drug sodium valproate as an adjuvant treatment for diffuse intrinsic pontine glioma. PLoS One. 2017;12:e0176855.
Acknowledgments
Authors thank M/s. Anjan Drug Private Limited, Chennai, Tamil Nadu, India, for providing sodium valproate EP as gratis. Special thanks to Dr. S. Gheena, Department of Oral Pathology, Saveetha Dental College and Hospitals, Chennai, India, for the drug procurement procedures.
Author information
Authors and Affiliations
Contributions
PR and ED performed the research. PR reviewed the literature and drafted the manuscript, and ED corrected the manuscript, designed the figures, and submitted the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rithanya, P., Ezhilarasan, D. Sodium Valproate, a Histone Deacetylase Inhibitor, Provokes Reactive Oxygen Species–Mediated Cytotoxicity in Human Hepatocellular Carcinoma Cells. J Gastrointest Canc 52, 138–144 (2021). https://doi.org/10.1007/s12029-020-00370-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12029-020-00370-7