
Abstract
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most malignant tumors with a dismal prognosis. Although our understanding of the carcinogenesis of the disease increases continuously, no effective conservative therapeutic strategies exist. Therefore, novel targets have to be defined at the experimental level. Histone deacetylases (HDACs), especially the class I isoenzymes HDAC1, 2, and 3, are highly expressed in PDAC.
Conclusion
This article summarizes the expression and functions of HDAC isoenzymes in PDAC, with a special focus on their promoter-specific mode of action. Although we have gained some molecular insight into the HDAC function in PDAC, less is known about the relevance of histone acetyltransferases (HATs) in PDAC. As an example, we will summarize function of the HAT p300, for which promoter-specific functions were described recently. Increasing the molecular insights into the functions of the acetylating and deacetylating machineries in PDAC are important, since this will lead to novel rationally based therapeutic strategies in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Over the last decade, increased knowledge concerning the molecular basis of pancreatic ductal adenocarcinoma (PDAC) has accumulated. Therefore, a far better understanding of the disease has evolved [1–4]. We now know that the proto-oncogene Kras, which is mutated in the majority of human PDACs [5], can initiate pancreatic carcinogenesis in genetically engineered mouse models. Furthermore, these elegant in vivo models are now used to develop new therapeutic strategies, which will hopefully increase the survival of patients suffering from this dismal disease [6, 7]. In addition to the in vivo models, deep sequencing data demonstrate genetic alterations in 12 core signaling pathways altered in 67% to 100% of PDACs [8]. However, PDAC-relevant signaling pathways engaged by oncogenic Kras and other mutated genes are still ill-defined.
Oncogenes, like Kras, activate signaling pathways, which are ultimately integrated by transcription factors to change the transcriptome and to finally drive carcinogenesis in the pancreas. However, transcription factors are no soloists and they work in stereo-specific nucleoprotein-complexes in concert with co-regulators. These modify and epigenetically remodel chromatin structures bridging transcription factors to the basal transcriptional machinery. Therefore, co-regulators, which promote (co-activator) or attenuate (co-repressors) target gene transcription, confer a second level of specificity of an oncogenic transcriptional response. Furthermore, co-regulator complexes contain enzymatic activities, which can be classified as posttranslational histone modifying enzymes, like histone deacetylases (HDACs)/histone acetylases (HATs) and DNA methylases/DNA demethylases, and the family of ATP-dependent remodeling complexes, like the SWI/SNF complex, which are responsible for the sliding of the nucleosome needed for active transcription [9].
The enzymatic activities of co-regulator complexes target histones. This group of evolutionarily conserved proteins is critically involved in chromatin condensation and building of a second code, the histone code [9, 10]. The histone code is encrypted by several posttranslational modifications of the histones, like acetylation or methylation [10]. Transcriptionally inactive, repressed chromatin (heterochromatin) is compacted with predominant binding of hypoacetylated histones, whereas transcriptional active chromatin (euchromatin) is characterized by the binding of hyperacetylated histones and acetylation of histone H3 lysine 9 and lysine 14 is a mark for active genes. The central packaging unit of the DNA is the nucleosome consisting out of two copies of histones H2A, H2B, H3, and H4, and ~146 base pairs of DNA wrapped around each histone core particle. The basal transcriptional machinery inefficiently transcribes nucleosomal DNA implicating that the nature of the DNA-nucleosome interaction is highly dynamic and flexible to assure highly accurate transcriptional regulation. Although the sum of the posttranslational modifications of histones decides whether DNA is condensed and transcriptional inactive or open and transcriptional active, we will describe here especially the deacetylation machinery, the HDACs, since they are the most intensively studied enzymes within the group of chromatin modifiers in PDAC. Many molecular functions of HDACs in PDAC were elucidated by the use of HDAC inhibitors (HDACI). Since this was summarized recently [11], we will focus on the function of HDAC isoenzymes in the context of transcriptional regulation in PDAC.
HDACs—Dual Regulators of Transcription
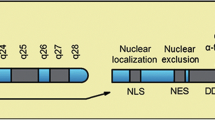
Phylogenetic analyses and sequence homology leads to a classification of HDACs into class I to IV enzymes (Fig. 1). HDACs 1, 2, 3, and 8 with homology to the yeast Rpd3 homologues represent class I and the yeast Hda1 homologues enzymes HDAC 4, 5, 6, 7, 9, and 10 represent class II HDACs. The class IV enzyme HDAC11 shares homologies with class I as well with class II HDACs. Whereas class I, II, and IV enzymes use zinc-dependent catalysis, the class III deacetylases (SIRT1-7), homologues of the yeast SIR2 enzyme, use NAD+ as co-factor [12, 13] (Fig. 1). SIRT specific functions in PDAC are unknown so far.

HDACs. HDACs are classified with respect to their homology in the catalytic domain into class I (HDAC1, 2, 3, and 8), class II (HDAC4, 5, 6, 7, 9, and 10), and class IV (HDAC11) enzymes. Class II is subdivided depending onto the presence of one (class IIa) or two (class IIb) catalytic domains. The NAD+-dependent sirtuin protein deacetylases, SIRT1-7, represent class III and are not illustrated. aa amino acids
HDACs deacetylate the ε-amino group of lysines located at the N-terminal tail of histones, leading to a repressive chromatin formation (heterochromatin) and the suppression of gene expression (Fig. 2a). In contrast, HATs counteract histone deacetylation, which generates an open chromatin structure (euchromatin), enabling transcription factors to activate their target genes (Fig. 2b). In addition to histones, HDACs as well as HATs have been found to acetylate a growing number of non-histone substrates. These proteins are often transcription factors, like p53, NFκB, or signal transducers and activators of transcription (STATs) [14, 15]. Therefore, changes in the transcriptome mediated by the deacetylation machinery can be due to alteration in transcription factor activity or the histone code [11]. Since HDACs are involved in the control of cancer cell proliferation, apoptosis, differentiation, migration, and angiogenesis in cancer [16], they represent attractive therapeutic targets [17], and rational HDACI-based combination therapies are currently tested in pre-clinical and clinical settings for the treatment of PDAC [11].

Acetylation and the chromatin. Simplified illustration of the molecular action of HDACs and HATs. a Repressive transcription factors (TF) recruit co-repressor complexes containing HDACs to a gene-specific promoter, leading to the deacetylation of the ε-amino group of lysines located at the N-terminal tail of histones. This process contributes to a repressive chromatin formation (heterochromatin) and the suppression of gene expression. b Activating transcription factors (TF) recruit a co-activator complex containing HATs to a gene-specific promoter. Acetylation (Ac) of the ε-amino group of lysines located at the N-terminal tail of histones permits an open chromatin structure (euchromatin) and gene transcription
HDAC Expression in PDAC
As in other gastrointestinal cancers, PDAC is characterized by overexpression of class I HDACs, especially HDAC1, 2, and 3. Fifty-six percent of PDACs show positive immunohistochemical staining for HDAC1, and the coexpression of HDAC1 and HIF1α was found to remarkably correlate with poor prognosis [18]. In a recent study with 54 PDAC patients, high HDAC1 expression was significantly correlated with the proliferative activity, degree of tumor differentiation, higher TNM staging, and survival [19]. In tissue microarrays, we detected overexpression of HDAC2 especially in moderately differentiated (G2) and undifferentiated (G3) PDACs [20]. These findings were recently confirmed in a cohort of 81 patients with PDAC [21]. In addition, this study demonstrated a strong nuclear immunoreactivity of HDAC3 in 79% of PDACs [21]. This is mirrored in the murine KrasG12D-dependent model of PDAC, were we observed high expression of HDAC1, 2, and 3 (unpublished data). Furthermore, increased expression of HDAC7 was recently demonstrated in 11 well to moderately differentiated PDACs [22]. These expression data together with the observation that HDACI reduce proliferation and induce apoptosis in pre-clinical models of PDAC [11] points to an important functions of HDACs in the carcinogenesis of PDAC. However, HDAC specific functions at the level of transcriptional control are ill-defined and we will focus now on examples where such data exist.
HDAC Isoenzyme Function in PDAC
HDAC2 Controls the Gene Locus of the BH3-Only Protein NOXA
A hallmark of cancer is the evasion of apoptosis, and PDAC is no exception [23]. Apoptosis or programmed cell death is a central regulator of normal tissue homeostasis, with the apoptotic program being essential for the elimination of redundant, damaged, or infected cells. Typically, two well-characterized pathways initiate apoptosis. Both lead to the activation of executioner caspases-3, -6, and -7. The extrinsic pathway is triggered by death-receptors, like the CD95 receptor or tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor. Activation by their corresponding ligands results in the activation of the initiator caspase 8 via the death-inducing signaling complex and subsequent activation of effector caspases. The intrinsic or mitochondrial pathway of apoptosis is regulated by pro- (e.g., BH3-only proteins like NOXA, PUMA, BID, BIM) and anti-apoptotic members (e.g., MCL1 and BCLXL) of the BCL-2 protein family. BH3-only proteins are sensors of various cellular stresses. These molecules induce apoptosis by counteracting survival functions of the BCL-2 members. These anti-apoptotic proteins prevent cytochrome C efflux from mitochondria and subsequent activation of the cell death initiator caspase-9 [24].
Apoptotic resistance translates into therapeutic resistance and one mechanism is the imbalanced expression of pro- and anti-apoptotic BCL2 family members favoring survival [23]. Using RNA interference we recently observed that depletion of HDAC2, but not HDAC1, markedly increased the sensitivity of PDAC cell towards the topoisomerase II inhibitor etoposide [20] and UV-light (unpublished observation), both inducers of the DNA- Damage response pathway. Transcriptome profiling after HDAC2 depletion in PDAC cells revealed upregulation of the death-inducing BH3-only protein NOXA and we were able to demonstrate that NOXA is essentially mediating HDAC2-dependent sensitization of PDAC cells towards DNA-damage induced apoptosis [20]. NOXA was first identified as a phorbol ester-inducible and p53-regulated gene that is involved in apoptosis upon genotoxic stress and is known to lead to activation of the mitochondrial apoptosis pathway via neutralizing anti- apoptotic functions of the BCL2 family member MCL1 [25]. At the molecular level, we observed in chromatin immunoprecipitation (ChIP) assays binding of HDAC2 together with deacetylated histone H3 to the NOXA promoter. This suggests that one function of HDAC2 in PDAC cells is to silence the death-inducing NOXA gene, thereby allowing survival under genotoxic stress, a condition that commonly occurs in cancer cells. Upon depletion of HDAC2 we observed acetylation of lysine 9 and lysine 14 in the N-terminal tail of histone H3 and recruitment of RNA polymerase II to the NOXA promoter as a correlate of an open chromatin and active transcription. Subsequently, after the depletion of HDAC2 the NOXA promoter can be easily targeted by a transcription factors activated by the DNA-damage response pathway, leading to an efficient transcriptional induction of the NOXA gene to threshold levels needed to induce apoptosis [20]. Additionally, HDAC2 counteracts apoptosis induced by TRAIL in PDAC cells [26]. Therefore HDAC2 is an attractive therapeutic target, especially when HDAC2-dependent sensitization effects to other therapeutics and in other tumors are considered (summarized in [27]). Of note, certain HDACI not only target the catalytic activity of HDACs but equally the stability of particularly HDAC2 [15, 20, 27–29]
HDAC3 and Proliferation—the SKP2 Connection
Aberrant activity of the transcription factor family NFκB is linked to cancer development and resistance against chemotherapy in PDAC [30]. Investigating the molecular function of this pathway, we observed that one of the NFκB-activating kinases, IκB kinase α (IKKα), promoted G1- to S-phase progression and proliferation of PDAC cells by activating the transcription of the S-phase kinase associated protein 2 (SKP2) gene [31]. The cell cycle is controlled by the highly coordinated interplay between cyclin-dependent kinases (CDKs) and cyclin-dependent kinase inhibitors (CDKIs), like p16INK4a, p21Cip1, or p27Kip1. Unrestrained proliferation of cancer cells is amongst others due to aberrantly low expression of CDKIs. PDAC cells express little p27Kip1, suggesting contribution of this pan-CDKI to pancreatic carcinogenesis [32]. One main mechanism controlling p27Kip1 expression affects its protein turnover. The cyclinE/CDK2 complex phosphorylates p27Kip1 and tags the protein for recognition by SKP2, an F-box protein that functions as a receptor component of the SCF ubiquitin ligase complex, resulting in p27Kip1 ubiquitination and proteasomal degradation [33]. High levels of SKP2 expression were observed in PDAC specimens and this was correlated with a worse patient outcome [34].
After depletion of IKKα with RNAi, we observed a transcriptional downregulation of SKP2 in PDAC cells with a loss of acetylated histone H3 and histone H4 bound to the SKP2 gene promoter [31]. Concomitant with the binding of deacetylated histones, a complex containing HDAC1 and the co-repressor silencing mediator for retinoid and thyroid receptors (SMRT) was recruited to the SKP2 promoter after the interference with IKKα activity, arguing for the contribution of this complex in silencing the SKP2 locus after the depletion of IKKα in PDAC cells (Fig. 3). SMRT builds a docking interface that is needed to tether components of the repressive machinery, like HDACs, via transcription factors to repressed genes [35]. More interestingly, we observed binding of HDAC3 to the SKP2 promoter (Fig. 3) when the gene was activated. Hence, this data suggest that HDAC3 is needed to maintain SKP2 promoter activity. To test this, we synchronized the pancreatic cancer cell line MiaPaCa2 in the G0- (SKP2 transcription off) or early S-phase (SKP2 transcription on). We observed recruitment of HDAC3 together with lysine 9 and lysine 14 acetylated histone H3 and RNA polymerase II to the SKP2 promoter in early S-phase, pointing again to an activating function of HDAC3 for SKP2 transcription [36]. This demonstrates that binding of a HDAC to a promoter and acetylation of histones is not exclusive at specific promoters. Consistent with an activator function of HDAC3 for the SKP2 gene in early S-phase, downregulation of HDAC3 in PDAC cells using RNA interference results in impaired proliferation and transcriptional inactivation of SKP2 [36]. Together, these data demonstrate that transcription of SKP2 depends on HDAC3 activity. However, the transcription factors regulated by HDAC3-dependent deacetylation needed to activate SKP2 in early S-phase in PDAC cells are not known presently. Furthermore, acetylation-independent HDAC3-mediated mechanisms can account for the molecular effects observed.

IKKα depletion leads to recruitment of a HDAC1/SMRT repressor complex to the SKP2 gene promoter in PDAC cells. Transfection of IKKα siRNAs (IKKα +89 and +129) and ChIP assay were done as described recently [31]. The figure demonstrates the recruitment of a HDAC1/SMRT containing repressor complex to the SKP2 gene promoter after interference with the mitogenic IKKα pathway in the PDAC cell lines MiaPaCa2 and DanG. HDAC3 is recruited to the SKP2 gene promoter in random cycling, proliferating PDAC cells and disappears after depletion of IKKα
A HDAC1/HDAC2 Complex Contributes to Epithelial to Mesenchymal Transition and Controls the CDH1 Promoter
Although most patients suffering from PDAC present with metastatic disease causing the vast majority of pancreatic cancer deaths, molecular mechanisms driving the metastatic cascade are ill-defined [32]. Metastasis is a highly organ-specific process, which requires multiple steps and interactions between tumor cells and the host. These include detachment of tumor cells from the primary tumor, intravasation into lymph and blood vessels, survival in the circulation, extravasation into target organs, and subsequent proliferation and induction of angiogenesis [37]. One early mechanism of metastasis is the change from highly differentiated epithelial cell morphology to a mesenchymal phenotype [38]. This process is called epithelial to mesenchymal transition (EMT). During EMT, an epithelial cell loses polarity and intercellular adhesions and acquires a fibroblastoid phenotype. Furthermore, the transcriptome and proteome profiles are changed, leading to expression of mesenchymal markers such as Vimentin, N-cadherin, Snail, and Twist, whereas epithelial markers, such as E-Cadherin, are lost [39]. Loss of the adherens junction protein E-cadherin, a single-span transmembrane glycoprotein that maintains intercellular contacts and cellular polarity in epithelial tissues, is a main indicator of the epithelial/mesenchymal phenotype switch. Reduced expression of E-cadherin is associated with tumor invasiveness, metastatic dissemination, and poor prognosis for cancer patients [40].
By serial in vivo passaging of parental pancreatic cancer cells with low metastatic potential, we selected for cells with high metastatic potential. Interestingly, we observed in contrast to the epithelial morphology of the parental cell line a clear mesenchymal morphology in the in vivo passaged cell line together with a mesenchymal transcriptome, arguing that EMT contributes to metastasis [41]. Since EMT was correlated with downregulation of E-cadherin and dominant-negative E-cadherin induces EMT and metastasis in vivo in the used pancreatic cancer model, we investigated regulation of the CDH1 (E-cadherin) gene in molecular detail. During EMT, the CDH1 promoter can be repressed by the action of the zinc-finger transcription factors Snail or Slug, the basic helix-loop-helix factors E47 or Twist, or the two-handed zinc factors ZEBl and ZEB2. In ChIP assays, we observed recruitment of Snail together with HDAC1 and HDAC2 to the proximal CDH1 promoter in the highly metastatic cell line. Recruitment of the Snail/HDAC1/HDAC2 complex was correlated with binding of deacetylated histone H4, arguing that the repressor complex is active [41]. These experimental findings seem to be relevant for the human disease, since Snail and E-cadherin expression scores inversely correlate in pancreatic cancer specimens [42]. Although a HDAC2/SIN3a co-repressor complex functionally represses the TGF beta receptor II (TGFB2R) gene in pancreatic cancer cells [43], we were not able to demonstrate the recruitment of the SIN3a scaffolding protein and co-repressor [44] to the CDH1 promoter in the mesenchymal pancreatic cancer cells investigated. To elucidate the mode of repression of the CDH1 gene, it will be important to determine repressor complex composition in future experiments.
The TGF Beta Receptor II—a Tumor Suppressor Gene Repressed by HDACs
Transforming growth factor beta (TGFβ)-induced signaling controls several cellular processes, including proliferation, differentiation, and apoptosis. Although this cytokine acts tumor suppressive in early stages of epithelial carcinogenesis, TGFβ promotes tumor progression in advanced stages, by inducing tumor growth, EMT, invasion, evasion of immune surveillance, and metastasis [45, 46]. TGFβ signals through the serine–threonine receptor kinases type I (TGFBR1) and type II (TGFBR2) and binding of TGFβ induces canonical SMAD signaling. The activated TGFBR1 phosphorylates the SSXS motif of the receptor-regulated SMADs (R-SMADs), like SMAD2 and SMAD3. Phosphorylated R-SMADs bind to their common partner SMAD (co-SMAD) SMAD4, translocate into the nucleus, and control transcription of target genes in concert with co-operating transcription factors [45, 46].
The TGFβ signaling pathway is inactivated in PDAC cells by several mechanisms. Deletion of the tumor suppressor SMAD4 is the most prominent example [5]. In addition to SMAD4, approximately 50% of PDACs express no or low amounts of the TGFB2R [47]. The tumor suppressor function of this gene is best demonstrated in the murine KrasG12D-dependent mouse model. Herein, the conditional deletion of the Tgfb2r gene dramatically accelerates KrasG12D-dependent carcinogenesis in the murine pancreas [48]. The demonstration that the TGFB2R gene is affected at the genetic level in only 9% of PDACs, points to the contribution of the epigenetic machinery to the downregulation of the TGFB2R gene in PDAC [49]. Indeed, upregulation of the TGFB2R gene was observed in PDAC cells upon treatment with the HDAC inhibitor trichostatin A. At the molecular level, it was shown that the zinc-finger transcription factor Sp1 recruits HDAC1 to the TGFB2R promoter, to repress the gene [50, 51]. In addition, KLF14, belonging to the Sp/KLF family of transcription factors, was shown to repress the TGFB2R gene by recruiting a SIN3a/HDAC2 complex to the promoter [43].
HATs—Unclear Role in PDAC
Whereas clear expression and functional data demonstrate an important role of HDACs in PDAC, the function of HATs is unclear in the moment. HATs are crudely classified into two different groups, type A HATs are nuclear and type B HATs are cytoplasmic functioning to modify newly synthesized histones before the assembly [14, 52]. Nuclear HATs, which acetylate nuclear histones and chromatin-associated proteins, fall into four main families, according to homology in the HAT domain: the GNAT family with the first identified HAT, the general control of amino acid synthesis protein 5 (GCN5), and p300/CBP-associated factor (PCAF); the MYST family named according to the founding members MOZ (monocytic leukemia zinc-finger protein), YBF2 (yeast binding factor 2)/SAS3 (something about silencing 3), SAS2, TIP60 (Tat interactive protein-60); the p300/CREB binding protein (CBP) family; and the Rtt109 (Regulator of Ty1 Transposition gene product 109) family [53]. In addition, several other nuclear proteins, like the steroid receptor co-activators family (like AIB1), the general transcription factor TAF250, or the transcription factor ATF-2, possess HAT activities. HATs counteract HDAC activities leading to an open transcriptionally active chromatin (Fig. 2b). Furthermore, similar to HDACs, HATs substrates are not exclusively histones, and several transcription factors, e.g., p53, p73, E2F1, c-Jun, are directly regulated and controlled by HATs [52]. Although a clear link between HATs and cancer exists [54], HATs are even less studied than HDACs in PDAC. Furthermore, the existing data are conflicting with respect to the overall assessment, which is best illustrated by considering data available for p300.
A recent study characterizing the metastatic behavior of several orthotopic implanted PDAC cell lines revealed upregulation of specific miRNAs, like miR-194, -200b, -200c, and -429 in human PDAC cell lines with the highest capacity to metastasize [55]. miRNAs are small non-coding single-stranded RNAs, 18–23 nucleotides long, which bind to the three untranslated region of target mRNAs with perfect or near-perfect complementarity, resulting in degradation or inhibition of the target mRNA [56]. In silico, p300 was determined as a potential target of the upregulated miRNAs. In line, the cell lines with the highest metastatic capacity revealed the lowest p300 protein expression levels, arguing for a direct involvement of p300 in regulating metastasis [55]. Although only three PDAC specimens were investigated, a very low p300 expression was observed [55]. This, together with the observation of somatic p300 mutations in two out of 11 investigated PDAC cell lines [57] argue for a metastasis suppressive role of p300 in the carcinogenesis of PDAC. In contrast to this metastasis suppressive functions, growth-promoting function of p300 were recently demonstrated. In a very detailed molecular study, p300 was recently demonstrated to be involved in the activation of the c-myc promoter in PDAC cells [58]. The basic helix-loop-helix leucine-zipper transcription factor c-myc dimerizes with Max to bind to specific E-box sequences in genes directly responsible for controlling cell growth and proliferation [59]. In PDAC, high frequencies of c-myc overexpression ranging from 43.5% to 70.2% of primary PDACs were demonstrated [60, 61]. This high expression level in PDAC is due to amplification of the c-myc gene [62], stabilization of the c-myc protein by the phosphatidylinositol 3-kinase/AKT/glycogen synthase kinase-3 signaling pathway [63], and c-myc promoter activation by the nuclear factor of activated T cells (NFAT) transcription factors [64]. Using starvation release experiments, Dr. Ellenrieder’s group detected activation of the calcineurin/NFAT signaling pathway in PDAC cells [58]. This signaling pathway was necessary to activate the c-myc promoter and to drive G1- to S-phase progression. In ChIP experiments, a sequential course of c-myc promoter activation during early G1-phase of the cell cycle was figured out. NFAT was recruited to the proximal c-myc promoter leading to binding of p300 and consecutive acetylation of histones. Afterwards, the open c-myc locus can be bound by the transcription factor ELK1, who synergizes with NFAT to activate the promoter. Furthermore, the function of p300 was proven at the genetic level. RNA interference, targeting p300, led to a failure to acetylate histones and to recruit ELK1 to the c-myc promoter [58]. Such data propose that the HAT p300 mediates growth-promoting functions in PDAC. Together, these data demonstrate that we need further studies, especially at the genetic level, to investigate HAT functions in the carcinogenesis of PDAC.
Conclusion
Reversible acetylation is an important epigenetic transcriptional control mechanism surely involved in the carcinogenesis of PDAC. Loss of lysine 16 acetylation of histone H4 [65] is a common marker of cancer and the acetylation status of histones predicts outcome of certain cancers [66]. Although recent evidence also demonstrate in PDAC that low levels of lysine 18 acetylated histone H3 was a predictor of poor survival [67], the enzymes conducting acetylation and deacetylation are underinvestigated in PDAC. Therefore, we need further detailed molecular analysis to determine the function of individual members of the acetylating/deacetylating machinery at the expression level and at the transcriptome level. Furthermore, various signaling pathways and transcription factors relevant for PDAC are controlled by acetylation. We need further detailed analysis to decipher the signaling pathways controlled by individual members of the acetylating/deacetylating machinery. These increasing insights into the actions of HDACs and HATs will ultimately generate new ideas for rationally and molecularly based therapeutic approaches for this dismal disease.
References
Rustgi AK. The molecular pathogenesis of pancreatic cancer: clarifying a complex circuitry. Genes Dev. 2006;20:3049–53.
Schneider G, Siveke JT, Eckel F, Schmid RM. Pancreatic cancer: basic and clinical aspects. Gastroenterology. 2005;128:1606–25.
Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–49.
Koorstra JB, Hustinx SR, Offerhaus GJ, Maitra A. Pancreatic carcinogenesis. Pancreatology. 2008;8:110–25.
Schneider G, Schmid RM. Genetic alterations in pancreatic carcinoma. Mol Cancer. 2003;2:15.
Olive KP, Tuveson DA. The use of targeted mouse models for preclinical testing of novel cancer therapeutics. Clin Cancer Res. 2006;12:5277–87.
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61.
Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6.
Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–28.
Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80.
Schneider G, Krämer OH, Fritsche P, Schüler S, Schmid RM, Saur D. Targeting histone deacetylases in pancreatic ductal adenocarcinoma. J Cell Mol Med. 2010;14:1255–63.
Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–18.
Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42.
Spange S, Wagner T, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41:185–98.
Buchwald M, Krämer OH, Heinzel T. HDACi—targets beyond chromatin. Cancer Lett. 2009;280:160–7.
Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–32.
Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem. 2009;107:600–8.
Miyake K, Yoshizumi T, Imura S, Sugimoto K, Batmunkh E, Kanemura H, et al. Expression of hypoxia-inducible factor-1alpha, histone deacetylase 1, and metastasis-associated protein 1 in pancreatic carcinoma: correlation with poor prognosis with possible regulation. Pancreas. 2008;36:e1–9.
Wang W, Gao J, Man XH, Li ZS, Gong YF. Significance of DNA methyltransferase-1 and histone deacetylase-1 in pancreatic cancer. Oncol Rep. 2009;21:1439–47.
Fritsche P, Seidler B, Schüler S, Schnieke A, Göttlicher M, Schmid RM, et al. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut. 2009;58:1399–409.
Lehmann A, Denkert C, Budczies J, Buckendahl AC, Darb-Esfahani S, Noske A, et al. High class I HDAC activity and expression are associated with RelA/p65 activation in pancreatic cancer in vitro and in vivo. BMC Cancer. 2009;9:395.
Ouaissi M, Sielezneff I, Silvestre R, Sastre B, Bernard JP, Lafontaine JS, et al. High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann Surg Oncol. 2008;15:2318–28.
Hamacher R, Schmid RM, Saur D, Schneider G. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol Cancer. 2008;7:64.
Adams JM, Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol. 2007;19:488–96.
Häcker G, Weber A. BH3-only proteins trigger cytochrome c release, but how? Arch Biochem Biophys. 2007;462:150–5.
Schüler S, Fritsche P, Diersch S, Arlt A, Schmid RM, Saur D, et al. HDAC2 attenuates TRAIL-induced apoptosis of pancreatic cancer cells. Mol Cancer. 2010;9:80.
Krämer OH. HDAC2: a critical factor in health and disease. Trends Pharmacol Sci. 2009;30:647–55.
Krämer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003;22:3411–20.
Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, et al. Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia. Cancer. 2005;104:2717–25.
Sebens S, Arlt A, Schäfer H. NF-kappaB as a molecular target in the therapy of pancreatic carcinoma. Recent Results Cancer Res. 2008;177:151–64.
Schneider G, Saur D, Siveke JT, Fritsch R, Greten FR, Schmid RM. IKKalpha controls p52/RelB at the skp2 gene promoter to regulate G1- to S-phase progression. EMBO J. 2006;25:3801–12.
Schneider G, Hamacher R, Eser S, Friess HM, Schmid RM, Saur D. Molecular biology of pancreatic cancer—new aspects and targets. Anticancer Res. 2008;28:1541–50.
Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–81.
Einama T, Kagata Y, Tsuda H, Morita D, Ogata S, Ueda S, et al. High-level Skp2 expression in pancreatic ductal adenocarcinoma: correlation with the extent of lymph node metastasis, higher histological grade, and poorer patient outcome. Pancreas. 2006;32:376–81.
Stanya KJ, Kao HY. New insights into the functions and regulation of the transcriptional corepressors SMRT and N-CoR. Cell Div. 2009;4:7.
Schneider G, Reichert M, Saur D, Hamacher R, Fritsch R, Schmid RM. HDAC3 is linked to cell cycle machinery in MiaPaCa2 cells by regulating transcription of skp2. Cell Prolif. 2007;40:522–31.
Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–95.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial–mesenchymal transitions in development and disease. Cell. 2009;139:871–90.
Kalluri R, Weinberg RA. The basics of epithelial–mesenchymal transition. J Clin Invest. 2009;119:1420–8.
Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–32.
von Burstin J, Eser S, Paul MC, Seidler B, Brandl M, Messer M, et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology. 2009;137:361–71. 371 e361-365.
Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG. Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res. 2007;13:4769–76.
Truty MJ, Lomberk G, Fernandez-Zapico ME, Urrutia R. Silencing of the transforming growth factor-beta (TGFbeta) receptor II by Kruppel-like factor 14 underscores the importance of a negative feedback mechanism in TGFbeta signaling. J Biol Chem. 2009;284:6291–300.
Grzenda A, Lomberk G, Zhang JS, Urrutia R. Sin3: master scaffold and transcriptional corepressor. Biochim Biophys Acta. 2009;1789:443–50.
Massague J. TGFbeta in cancer. Cell. 2008;134:215–30.
Ellenrieder V. TGFbeta regulated gene expression by Smads and Sp1/KLF-like transcription factors in cancer. Anticancer Res. 2008;28:1531–9.
Venkatasubbarao K, Ahmed MM, Mohiuddin M, Swiderski C, Lee E, Gower Jr WR, et al. Differential expression of transforming growth factor beta receptors in human pancreatic adenocarcinoma. Anticancer Res. 2000;20:43–51.
Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20:3147–60.
Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, Kern SE. Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res. 1998;58:5329–32.
Zhao S, Venkatasubbarao K, Li S, Freeman JW. Requirement of a specific Sp1 site for histone deacetylase-mediated repression of transforming growth factor beta Type II receptor expression in human pancreatic cancer cells. Cancer Res. 2003;63:2624–30.
Huang W, Zhao S, Ammanamanchi S, Brattain M, Venkatasubbarao K, Freeman JW. Trichostatin A induces transforming growth factor beta type II receptor promoter activity and acetylation of Sp1 by recruitment of PCAF/p300 to a Sp1.NF-Y complex. J Biol Chem. 2005;280:10047–54.
Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120.
Marmorstein R, Trievel RC. Histone modifying enzymes: structures, mechanisms, and specificities. Biochim Biophys Acta. 2009;1789:58–68.
Keppler BR, Archer TK. Chromatin-modifying enzymes as therapeutic targets—part 2. Expert Opin Ther Targets. 2008;12:1457–67.
Mees ST, Mardin WA, Wendel C, Baeumer N, Willscher E, Senninger N, et al. EP300—a miRNA-regulated metastasis suppressor gene in ductal adenocarcinomas of the pancreas. Int J Cancer. 2010;126:114–24.
Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69.
Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, et al. Mutations truncating the EP300 acetylase in human cancers. Nat Genet. 2000;24:300–3.
König A, Linhart T, Schlengemann K, Reutlinger K, Wegele J, Adler G, et al. NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells. Gastroenterology. 2009;138(1189–99):e1–2.
Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22:2755–66.
Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U. c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol. 2002;15:462–9.
Li YJ, Wei ZM, Meng YX, Ji XR. Beta-catenin up-regulates the expression of cyclinD1, c-myc and MMP-7 in human pancreatic cancer: relationships with carcinogenesis and metastasis. World J Gastroenterol. 2005;11:2117–23.
Mahlamaki EH, Barlund M, Tanner M, Gorunova L, Hoglund M, Karhu R, et al. Frequent amplification of 8q24, 11q, 17q, and 20q-specific genes in pancreatic cancer. Genes Chromosom Cancer. 2002;35:353–8.
Schild C, Wirth M, Reichert M, Schmid RM, Saur D, Schneider G. PI3K signaling maintains c-myc expression to regulate transcription of E2F1 in pancreatic cancer cells. Mol Carcinog. 2009;48:1149–58.
Buchholz M, Schatz A, Wagner M, Michl P, Linhart T, Adler G, et al. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006;25:3714–24.
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400.
Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–6.
Manuyakorn A, Paulus R, Farrell J, Dawson NA, Tze S, Cheung-Lau G, et al. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J Clin Oncol. 2010;28:1358–65.
Acknowledgments
The authors are supported by Deutsche Forschungsgemeinschaft (Grant SCHN 959/1-2), SFB456, SFB824, Else Kröner-Fresenius-Stiftung, Novartis-Stiftung für therapeutische Forschung, Fritz-Thyssen Stiftung, Bayerische Forschungsstiftung, and Deutsche Krebshilfe. We would like to apologize for not citing any relevant reports due to lack of space, the need to selectively choose examples, or an oversight on our part.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schneider, G., Krämer, O.H., Schmid, R.M. et al. Acetylation as a Transcriptional Control Mechanism—HDACs and HATs in Pancreatic Ductal Adenocarcinoma. J Gastrointest Canc 42, 85–92 (2011). https://doi.org/10.1007/s12029-011-9257-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12029-011-9257-1