
Abstract
A growing body of clinical literature emphasizes the impact of cerebral edema in early brain injury following aneurysmal subarachnoid hemorrhage (aSAH). Aneurysm rupture itself initiates global cerebral edema in up to two thirds of cases. Although cerebral edema is not a universal feature of aSAH, it portends a poor clinical course, with quantitative analysis revealing a direct correlation between cerebral edema and poor outcome, including mortality and cognitive deficits. Mechanistically, global cerebral edema has been linked to global ischemia at the time of aneurysm rupture, dysfunction of autoregulation, blood breakdown products, neuroinflammation, and hyponatremia/endocrine abnormalities. At a molecular level, several culprits have been identified, including aquaporin-4, matrix metalloproteinase-9, SUR1-TRPM4 cation channels, vascular endothelial growth factor, bradykinin, and others. Here, we review these cellular and molecular mechanisms of global cerebral edema formation in aSAH. Given the importance of edema to the outcome of patients with aSAH and its status as a highly modifiable pathological process, a better understanding of cerebral edema in aSAH promises to hasten the development of medical therapies to improve outcomes in this frequently devastating disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
An emerging focus of research in aneurysmal subarachnoid hemorrhage (aSAH) is the concept of early brain injury, the notion that injury to the brain immediately following aSAH forms the pathophysiologic foundation responsible for both late neurologic decline and poor outcome in this patient population. Concurrently, a growing body of clinical literature emphasizes the role of cerebral edema, particularly early edema, as both a negative prognostic feature and direct pathologic actor in the days following aSAH. Despite the ubiquity of cerebral edema in both experimental studies of early brain injury and in clinical evaluations of aSAH patients, no unified theory of its pathophysiology, particularly at a molecular level, exists. This review provides an overview of the current understanding of cerebral edema by summarizing cerebral edema as a general phenomenon, discussing clinical evidence for its importance in aSAH, situating edema as a direct result of several pathophysiological processes inherent to aSAH, and, finally, discussing some of the better studied molecular mediators of cerebral edema in aSAH.
Pathophysiology of Cerebral Edema and Brain Swelling
Cerebral edema and brain swelling refer to related pathophysiologic phenomena, whereby the failure of mechanisms of fluid and ion regulation within various physiologic compartments leads to edema––an abnormal accumulation of fluid within a perfused tissue––that causes the tissues to undergo volumetric expansion, i.e., to swell. The basic principles of edema formation were first enunciated by Starling [1] in the late nineteenth century. Starling posited that flow from the capillary to the interstitial space is the result of two independent pressure differentials, the net hydrostatic pressure, as determined by the difference between intravascular and interstitial pressures, and the net osmotic pressure, as determined by the difference in osmotically active particles in the blood and in the extracellular space. A modern understanding of this process in the brain recognizes differences in permeability of capillaries to hydrostatic and osmotic pressure differentials [2, 3]. Therefore, two separate permeability constants, which reflect the ability of fluid to flow across the blood–brain barrier through extracellular and intracellular routes, modulate these pressure differentials (Fig. 1). Under normal physiologic conditions, tight junctions between capillary endothelial cells limit the extracellular flow of fluid into the brain. As such, in the healthy brain, osmotic forces predominantly control fluid homeostasis.

Starling’s equation. Net fluid movement (J v) results from differences in both hydrostatic (ΔP) and osmotic (Δπ) pressures. Although the differences in pressures drive fluid movement, the rate of flow is limited by the permeability of the capillary to each pressure type, K H and K O, for hydrostatic and osmotic pressure, respectively. Starling’s original equation considered the K H and the K O terms to be identical, but modern studies of the blood–brain barrier suggest that these terms differ from each other and thus should be considered separately [2, 3]. In the healthy brain, the K H term is very small, essentially allowing flow to be governed by the osmotic term. In the injured brain, values for both K H and K O are increased above baseline
Cerebral edema and brain swelling occur through two primary mechanisms (Fig. 2) [2, 3]. The first “ionic edema” follows after a failure of cellular homeostasis in which osmotically active particles and water accumulate within cells, a phenomenon called cellular (or cytotoxic) edema. Intracellular ions and water accumulate at the expense of the extracellular compartment, setting up an osmotic gradient between the intravascular and extracellular compartments. This gradient drives the net flow of ions and water into the tissues, which is called ionic edema, and is a direct cause of tissue swelling. This mechanism of tissue swelling occurs with an intact blood–brain barrier (NB: a previous classification combined cellular and ionic edema together under the term cytotoxic edema). The second mechanism, “vasogenic edema,” represents a failure of the blood–brain barrier, either due to endothelial cell malfunction or disruption of the tight junctions between these cells. The increase in permeability across the capillary allows for increased flow of protein-rich plasma driven by the hydrostatic gradient between the intravascular and extracellular spaces. Both ionic and vasogenic edema can occur as part of the same pathologic process.

Schema of edema formation following aSAH. Transient ischemia due an abrupt rise in intracranial pressure caused by aSAH may lead to homeostatic failure of CNS cells, including neurons and astrocytes, resulting in cellular (cytotoxic) edema. The resulting depletion of ions and water from the interstitial space leads to secondary transcapillary flow of ions and water from the vascular compartment, resulting in interstitial ionic edema despite an intact blood–brain barrier. A variety of post-hemorrhagic environmental factors can lead to failure of the blood–brain barrier with late formation of vasogenic edema, including inflammation-mediated endothelial cell apoptosis, as well as destruction of membrane components of the blood–brain barrier secondary to proteolytic activity of MMP-9
Global Cerebral Edema in aSAH
Although cerebral edema may derive from a number of secondary pathological processes following aneurysm rupture, including perihematomal edema from an intracerebral hemorrhage or swelling of infarcted brain secondary to vasospasm, aneurysm rupture itself initiates diffuse cerebral edema in a substantial proportion of cases. The majority of studies evaluating global edema in clinical SAH employed CT scanning. Brain swelling on CT may be due either to increased brain water content (edema) or increased blood content (hyperemia or venous congestion). However, hypoattenuation of gray matter with loss of gray–white differentiation directly correlates with increased brain water content in experimental edema [4], whereas hyperemia, confirmed by CT perfusion, may increase but does not decrease attenuation [5]. In addition, CT perfusion studies of patients with global edema diagnosed by non-contrast CT fail to demonstrate changes in cerebral blood volume when compared to patients without edema following aSAH, offering further evidence that the determination of brain edema on plain CT is possible following aSAH [6].
Early studies of global edema employing CT noted a relatively low rate of 6% of patients with diffuse edema [7]. Later studies with modern CT imaging and formal criteria for diagnosis of cerebral edema demonstrated that global cerebral edema is relatively common in aSAH, with reported rates of edema formation in 20–67% of patients following aSAH [8–10]. In certain subpopulations, CT findings of cerebral edema may be considerably higher. For example, patients presenting with cardiac arrest following aSAH virtually all develop some CT findings of diffuse edema [11]. CT scans which assess edema via relatively gross signs such as loss of gray–white differentiation or brain swelling may fail to capture subtle findings of brain edema. Indeed, evaluation of otherwise radiographically normal brains with MRI-based diffusion measurements demonstrates significant changes following aSAH, suggesting that CT and conventional MRI evaluation may under-diagnose global cerebral edema following aSAH [12].
Although cerebral edema is not a universal feature of aSAH, it portends a poor clinical course. In one large series evaluating mortality in aSAH patients, cerebral edema, and not traditional causes of mortality such as aneurysm re-rupture or delayed cerebral ischemia, was felt by physician adjudicators to cause the majority of deaths in the first thirty days following aSAH [13]. Another large series identified cerebral edema as a frequent physician-identified primary cause of in-hospital death following aSAH. Compellingly, cerebral edema, but not clinical deterioration due to delayed cerebral ischemia, predicted in-hospital mortality on multivariate analysis [8]. Another study evaluating admission factors associated with prognosis identified global brain hypodensity on CT, along with age and WFNS grade on admission, as predictors of mortality [14], with global cerebral hypodensity being the only prognostic variable potentially amenable to treatment. A quantitative evaluation of global cerebral edema (selective sulcal volume) revealed a direct correlation between significant cerebral edema and poor outcome [15].
The impact of cerebral edema in aSAH is not limited to survival. Strikingly, in a multivariate analysis of cognitive outcomes in aSAH, global edema emerged as one of the few secondary pathologies predicting worse cognitive outcome [16]. These consistent correlations between cerebral edema and both death and poor cognition following aSAH suggest that timely and effective treatment of global cerebral edema might significantly improve outcomes following aSAH.
The development of global cerebral edema following aSAH demonstrates a complex time course. A significant proportion of patients exhibit global edema on their initial CT scan, with 8–67% of patients demonstrating so-called early global cerebral edema [9, 10]. Evaluation of patients later in their course demonstrates a second group of patients who develop edema within two weeks of hemorrhage, with 12% of patients developing edema in this delayed fashion [9]. This latter group is heterogeneous and encompasses both true de novo edema formation and progression of previously unrecognized early edema not recognized on initial CT. A number of animal studies deepen these insights regarding the timing of edema formation following aSAH. In cases of severe aSAH, edema formation appears to be a very early event. MRI studies show evidence of decreased ADC signal on MRI, a sign of cellular swelling, as early as 2 min following experimental aSAH induced by endovascular perforation in rats [17]. MRI studies using a canine double hemorrhage model of aSAH demonstrate that edema is a progressive phenomenon following aSAH, with radiographic evidence of worsening edema on days 2 and 7 following hemorrhage [18]. Although direct measurement of brain water content in rats following aSAH induced by endovascular perforation demonstrates detectable edema formation at 24 h following aSAH [19], experimental studies using higher-order mammals demonstrate detectable increases in water content at earlier time points, 6 h in cats [20] and 2 h in baboons [21].
Causes of Edema Formation Following aSAH
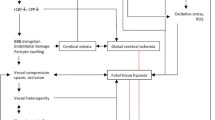
A number of processes are thought to cause edema following aSAH. Given that edema in aSAH appears to be a biphasic phenomenon, and given the complexity of pathological processes following aSAH, the emergence of a single cause for edema formation following aSAH seems unlikely. Nevertheless, good evidence supports the formation of early cerebral edema as a direct result of early ischemic injury at time of the initial hemorrhage. Subsequent delayed edema appears to result from failure of the blood–brain barrier, subsequent to a variety of pathologic processes such as neuroinflammation, autoregulatory dysfunction, blood products decomposition, and endocrine abnormalities. Although all of these pathologic processes are potentially edemagenic, both clinical and experimental data suggest that ischemic injury and autoregulatory dysfunction may be particularly relevant to edema formation in aSAH, with further research needed to establish to what degree, if any, other factors play a role.
Ischemic Injury
Experimental and clinical evidence suggests that aneurysm rupture can generate a transient but severe elevation of intracranial pressure with subsequent cerebral hypoperfusion and, in some cases, complete arrest of intracranial circulation [19, 20, 22, 23]. Ischemic brain injury is a well-established cause of cerebral edema in other conditions, including cardiac arrest and stroke. Ischemic injury mediates brain swelling through a variety of mechanisms, including, but not limited to, ion channel dysfunction with cellular edema [24]. Multiple lines of evidence, both clinical and experimental, suggest that ischemic injury from hypoperfusion is a key step in edema formation following aSAH. In a multivariate analysis of risk factors for cerebral edema formation, loss of consciousness on ictus (a surrogate for cerebral hypoperfusion) and Hunt–Hess grade emerged as the only factors predictive of early global edema [9]. In aSAH patients with a known period of cerebral hypoperfusion due to cardiac arrest, radiographic signs of cerebral edema develop in the vast majority of patients [11]. Finally, radiographic evaluation of patients with aSAH and perfusion imaging demonstrates an association between CT perfusion deficits and edema [25]. Animal models lend further credence to the hypothesis that early ischemic injury causes cerebral edema. In a cat model of aSAH, findings of cerebral edema following experimental aSAH correlated with the degree of cerebral hypoperfusion. Animals without significant changes in cerebral perfusion failed to develop edema in these experiments [20]. Evaluation of cerebral perfusion at multiple time points following aSAH in rats demonstrates a close correlation between prolonged hypoperfusion and edema formation [26]. Conversely, improvement in microvascular perfusion via inhaled nitric oxide in a murine aSAH model leads to a reduction in edema formation [27]. Based on these clinical and experimental findings, cerebral hypoperfusion with subsequent ischemic injury following aSAH seems highly relevant to the formation of cerebral edema, particularly early global cerebral edema, following aSAH.
Dysfunction of Autoregulation
Cerebral blood flow is maintained over a wide range of physiologic pressures, a phenomenon termed cerebral autoregulation. Autoregulatory dysfunction, as is hypothesized to occur with severe hypertension in posterior reversible encephalopathy syndrome (PRES), can potentially lead to increased cerebral blood flow, endothelial injury, blood–brain barrier breakdown, and subsequent vasogenic edema formation [28]. Given that both clinical and experimental evidence supports the disruption of cerebral autoregulation following aSAH [21, 29–31], and given that induced hypertension is used as part of “triple H” therapy for vasospasm, cerebral autoregulatory dysfunction is a plausible cause of vasogenic edema following aSAH. Clinical evidence supports this hypothesis. In a review of risk factors for the delayed development of cerebral edema, use of vasopressors significantly predicted subsequent development of vasogenic edema [9]. While the use of vasopressors could potentially serve as a surrogate for severity of vasospasm, the same study failed to identify an association between cerebral edema and vasospasm as defined by clinical, angiographic, or Doppler criteria, lending credence to the hypothesis that delayed edema results from autoregulatory dysfunction in conjunction with pressor use, rather than vasospasm-mediated ischemia. Bolstering this hypothesis are a number of cases in the literature reporting the development of a PRES-like syndrome with cerebral edema formation in the context of induced hypertension for aSAH [32]. Abnormal increases in cerebral blood flow due to autoregulatory dysfunction may therefore underlie a substantial proportion of edema resulting from aSAH, particularly vasogenic edema.
Hemorrhagic Blood Products
In the days following aneurysm rupture, blood in the subarachnoid space begins to degrade, increasing CSF levels of potentially toxic blood degradation products such as hemoglobin derivatives [33] and thrombin [34]. Besides their toxic effects, certain blood product derivatives, such as methemoglobin, initiate potent immune activation via direct binding of cellular receptors [35]. In other hemorrhagic diseases of the brain, these substances are key mediators of secondary edema [36], leading to blood–brain barrier disruption through a variety of mechanisms. Given their global distribution via CSF flow and their edemagenic potential, these blood products are potential mediators of global edema following aSAH. Rat studies present evidence in line with this hypothesis. In one study evaluating the role of thrombin in an endovascular perforation model of aSAH, treatment with argatroban, a thrombin inhibitor, was found to attenuate blood–brain barrier disruption and edema formation [37]. Binding of thrombin with thrombomodulin had similar effects [38]. In a prechiasmatic cistern injection model of aSAH, deferoxamine, an iron scavenger, demonstrated efficacy against the development of edema following aSAH [39]. In human aSAH, although radiographic quantification of subarachnoid and intraventricular blood on initial CT scan predicted edema formation on univariate analysis, multivariate analysis failed to confirm these findings [9], suggesting that other factors may modulate the edemagenic potential of blood products. Although hemorrhagic blood products may play a role in edema formation following aSAH, further research is needed to clarify its extent.
Neuroinflammation
aSAH initiates a significant inflammatory response in the brain, resulting in activation of the brain’s endogenous microglial compartment and infiltration of leukocytes, which may contribute to secondary injury following the initial insult. Neuroinflammation initiates both vasogenic edema due to blood–brain barrier disruption and cellular (cytotoxic) edema due to direct toxic effects on cells. Of note, in aSAH, inflammation appears to represent a source of edema independent of ischemic injury: soluble epoxide hydrolase knockout mice, which mount a defective vascular inflammatory response following experimental aSAH due to inhibition of NF-κB translocation, demonstrate a reduction in white matter edema despite experiencing an equivalent period of brain hypoperfusion as wild-type mice [40]. Multiple lines of evidence suggest that blunting of the inflammatory response represents a valid therapeutic strategy in aSAH. Modulation of microglial activation from a pro- to an anti-inflammatory phenotype via mTOR inhibition prevents edema formation in a rat aSAH model [41]. Similarly, pharmacologic blunting of NF-κB activation via SUR1 inhibition leads to a significant reduction in vasogenic edema and a concurrent increase blood–brain barrier integrity following experimental aSAH [42]. Blockade of IL-1β, a key microglial inflammatory cytokine, significantly reduces brain edema formation following aSAH [43]. As such, inflammation may represent an important cause of edema formation following aSAH.
Hyponatremia/Endocrine Abnormalities
Besides its effects on the brain, aSAH induces a number of systemic pathologies. One pathologic change of particular relevance is the induction of hyponatremia, due to either cerebral salt wasting or syndrome of inappropriate antidiuretic hormone [44]. Because of the osmotic gradient therefore generated, hyponatremia may lead to accumulation of water in brain tissues [45], a form of “reverse ionic edema.” Surprisingly, the development of hyponatremia does not consistently correlate with cerebral edema in clinical studies of aSAH, suggesting that the two pathologies either are not strictly related or that therapeutic measures to counteract hyponatremia are sufficient to prevent the development of edema [9].
Hyponatremia following aSAH commonly results from inappropriate secretion of vasopressin [44]. Given that vasopressin mediates cerebral edema in other conditions such as stroke [46] and cardiac arrest [47], vasopressin also may play a role in the development of edema following aSAH. Although human studies of the interaction between global edema and inappropriate vasopressin secretion are lacking in aSAH, several studies in rats suggest a complex relationship between the two pathologies. One study of brain edema following aSAH in vasopressin-deficient rats found a reduction in early brain edema following aSAH, but a paradoxical increase in brain edema at later times [48]. Alternatively, blockade of the V2 subclass of vasopressin receptors following experimental aSAH in rats effectively reduced edema formation [49]. However, given the multiple physiologic roles of vasopressin, including its actions as a pressor, interpretation of the effects of vasopressin blockade on edema formation should proceed cautiously. One study effectively demonstrated a reduction in early brain edema following endovascular perforation in rats treated with a vasopressin antagonist. However, vasopressin treatment had a number of other effects, including a significant reduction in episodes of re-bleeding via control of post-hemorrhage hypertension, preventing any simple interpretation of these experiments with regard to edema formation [50]. Of note, low-dose vasopressin is used as a pressor in patients with aSAH without apparent exacerbation of edema, arguing against an edemagenic role [51].
Molecular Mechanisms
Although molecular mediators of edema in aSAH remain less well studied than physiologic causes, several molecular candidates appear to mediate edema formation following aSAH either through altering membrane permeability or directly disrupting the blood–brain barrier.
Aquaporin 4
The aquaporins represent a ubiquitous class of water-specific channels that allow for passive diffusion of water through the cell membrane [52]. As passive channels, aquaporins cannot cause edema without some form of underlying osmotic gradient. However, by altering the permeability of cell membranes to water flow, they can exacerbate edema formation secondary to these forces. Although several aquaporins are expressed in the brain and may contribute to various disease states (such as aquaporin 1 expression by choroid plexus in hydrocephalus) [53], aquaporin 4 appears to be the most relevant to cerebral edema formation, with aquaporin 4 knockout mice demonstrating significant reductions in cerebral edema following both stroke and water intoxication [54]. Although upregulation of aquaporin 4, as demonstrated in human and rat tissues following aSAH [55, 56], clearly plays a role in post-aSAH edema formation, studies of this role demonstrate conflicting results. One study of aSAH-associated cerebral edema in aquaporin 4-deficient mice found a paradoxical increase in edema, ICP, and mortality following aSAH, which the authors attributed to a reduced ability of brain tissues to eliminate water. No difference was found in other parameters such as blood–brain barrier permeability or hydrocephalus between knockout and wild-type mice [57]. Conversely, antibody blockade of AQP-4 in rats following prechiasmatic injection of blood effectively reduces cerebral edema formation [58], again despite having no effect on blood–brain barrier permeability. Although differences in injection pressure and time points of edema evaluation could explain the conflicting results between the two reports, these studies caution against a simple understanding of aquaporin 4 as an edemagenic molecule in post-aSAH edema formation.
MMP-9
The matrix metalloproteinases are zinc endopeptidases responsible for degrading the extracellular matrix. Although essential for normal organism function, inflammatory activation of these enzymes can promote tissue destruction. Disruption of the proteins forming tight junctions between capillary endothelial cells leads to blood–brain barrier breakdown and vasogenic edema. Subarachnoid hemorrhage significantly upregulates both the expression and activity of matrix metalloproteinase 9 (MMP-9) in blood and CSF [59–61]. Several different experimental models suggest that MMP-9 activity mediates blood–brain barrier breakdown and subsequent vasogenic edema following aSAH. Genetic deletion of MMP-9 both protects the integrity of the blood–brain barrier [62] and reduces edema formation [63] in mouse models of aSAH. Pharmacologic inhibition of MMP-9 using either minocycline [58, 64] or a specific MMP-9 inhibitor [65] both restores blood–brain barrier integrity and reduces brain edema in rat models of aSAH. Thus, MMP-9-mediated degradation of the blood–brain barrier presents both a clear proximal molecular mechanism for vasogenic edema formation and a potential target to prevent it.
SUR1–TRPM4 Cation Channels
The sulfonylurea receptor, SUR1, is a membrane protein that associates with and regulates pore-forming proteins to form either SUR1–Kir6.2 (KATP) or SUR1–TRPM4 channels. The SUR1–Kir6.2 channel is responsible for metabolic sensing and insulin secretion in pancreatic β cells and other functions in neurons. A growing body of research situates the SUR1–TRPM4 channel in all CNS cells in a variety of brain pathologies [66]. TRPM4 is a non-selective cationic channel with physiologic functions rooted in calcium homeostasis. ATP depletion as well as excess intracellular calcium, both of which are commonly found in various neuropathological conditions, results in persistent opening of SUR1–TRPM4 channels.
The SUR1–TRPM4 channel contributes to edema formation in a number of ways. On the cellular level, persistent opening of the channel allows for massive sodium influx into the cells of the CNS, most notably astrocytes and neurons, with the resulting influx of sodium driving cellular (cytotoxic) and ionic edema [24]. Pathologic activation of SUR1–TRPM4 in endothelial cells disrupts the integrity of the blood–brain barrier and results in vasogenic edema via disruption of the interendothelial tight junctions, most likely due to cytoskeletal changes resulting from sodium influx or cell swelling [42]. Blockade of SUR1–TRPM4 also results in significant reductions in ischemia-associated MMP-9 [67–69]. As such, SUR1–TRPM4 may contribute to cellular, ionic, and vasogenic edema following aSAH. Studies of SUR1–TRPM4 confirm significant upregulation of channel expression following aSAH in both humans and rats [70]. Pharmacologic blockade of SUR1–TRPM4 in a rat model of aSAH prevented endothelial cell swelling and subsequent reorganization of the actin cytoskeleton, leading to improved integrity of the blood–brain barrier and reduced formation of vasogenic edema, as assessed by capillary permeability to immunoglobulin as well as immunohistochemical evaluation of endothelial tight junctions [42]. Furthermore, SUR1–TRPM4 blockade demonstrates anti-inflammatory effects in aSAH; as inflammatory cytokines also induce cytoskeletal rearrangements, blockade of their effects may also prevent blood–brain barrier disruption and vasogenic edema formation following aSAH [42, 70]. Although formal evaluation of the role SUR1–TRPM4 plays in the development of early global cerebral edema following aSAH remains to be performed, its established role in edema formation in general renders it a highly plausible mediator of early global cerebral edema as well.
VEGF
Under conditions of hypoxia, cellular degradation of the transcription factor HIF-1 (hypoxia inducible factor 1) ceases, resulting in upregulation of the angiogenic factor VEGF (vascular endothelial growth factor), as well as upregulation of the aforementioned SUR1–TRPM4 channel [71]. Although originally studied in oncology for its role in angiogenesis, recent studies in the field of neuro-oncology have highlighted the important edemagenic role of VEGF in brain tumors [72]. As part of the angiogenic process, VEGF potently induces the formation of capillary fenestrations, allowing for leakage of fluid and large molecules [73]. Given that VEGF production in the brain is significantly upregulated following aSAH, VEGF-mediated blood–brain barrier disruption forms a highly plausible mechanism for edema formation following aSAH [74, 75]. Consistent with this hypothesis, inhibition of VEGF activity, either through antibodies directed at the molecule itself or one of its receptors (VEGFR2), significantly ameliorates blood–brain barrier disruption and cerebral edema following experimental aSAH [76]. Several other studies exploring blockade of HIF-1, the transcription factor responsible for VEGF production, demonstrate concurrent reductions in both VEGF expression and brain edema [58, 77, 78]. Given that a number of anti-VEGF directed therapies, such as the monoclonal antibody bevacizumab, are in clinical use, these strategies merit further exploration for the treatment of cerebral edema in aSAH.
Bradykinin
The kinin–kallikrein system is an important signaling pathway connecting inflammation to vasodilation and vascular permeability, primarily through the actions of bradykinin [79]. Bradykinin receptor activation induces edema and increases vascular permeability both through inflammatory mechanisms and direct endothelial signaling. Studies of patients following aSAH demonstrate increased levels of bradykinin in plasma and CSF [80, 81]. Importantly, these levels appear to correlate with edema formation. Mice genetically deficient in bradykinin signaling due to bradykinin receptor 2 knockout demonstrate reduced edema formation following experimental aSAH [82]. Of note, bradykinin receptor 1-deficient mice did not enjoy a similar degree of protection. Although the authors of the former study speculate as to the downstream effects of bradykinin blockade on edema formation in SAH, a mechanistic evaluation of these effects is lacking. Bradykinin-mediated edema following aSAH appears to be a relatively early signaling event, as inhibition of bradykinin signaling even 30 min following aSAH fails to prevent edema formation [83], suggesting that bradykinin inhibition to prevent edema formation may be clinically impractical.
Conclusion
The accumulated evidence of a relationship between cerebral edema and poor outcomes following aSAH argues for the importance of a specific understanding of cerebral edema as a pathologic entity in aSAH. Although a large number of candidate therapies are under investigation, the lack of a unified molecular theory of cerebral edema formation hampers their development, leading to paradoxical or inconsistent findings. While good clinical evidence supports ischemic injury as the primary mediator of early global cerebral edema, molecular studies are needed to provide a precise framework for how this edema develops at a mechanistic level. A better understanding of molecular mechanisms of early edema formation in aSAH could accelerate the translation of experimental therapies to patients. Conversely, although experimental models support a number of mechanisms of late edema formation, including inflammation and autoregulatory dysfunction, further clinical studies are needed to determine which of these mechanisms predominate in clinical practice so as to help focus experimental work. Given the importance of edema to the outcome of patients with aSAH and its status as a highly modifiable pathological process, a better understanding of cerebral edema in aSAH would not only hasten the development of medical therapies, but also likely improve outcomes in this frequently devastating disease.
References
Starling EH. On the absorption of fluids from the connective tissue spaces. J Physiol. 1896;19:312–26.
Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V. Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 2007;6:258–68.
Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab. 2016;36:513–38.
Rieth KG, Fujiwara K, Di Chiro G, et al. Serial measurements of CT attenuation and specific gravity in experimental cerebral edema. Radiology. 1980;135:343–8.
Na DG, Kim EY, Ryoo JW, et al. CT sign of brain swelling without concomitant parenchymal hypoattenuation: comparison with diffusion-and perfusion-weighted MR imaging. Radiology. 2005;235:992–8.
Ivanidze J, Kallas ON, Gupta A, et al. Application of blood–brain barrier permeability imaging in global cerebral edema. AJNR Am J Neuroradiol. 2016;37:1599–603.
Kassell NF, Torner JC, Haley EC Jr, Jane JA, Adams HP, Kongable GL. The international cooperative study on the timing of aneurysm surgery. part 1: overall management results. J Neurosurg. 1990;73:18–36.
Lantigua H, Ortega-Gutierrez S, Schmidt JM, et al. Subarachnoid hemorrhage: who dies, and why? Crit Care. 2015;19:309.
Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke. 2002;33:1225–32.
Zetterling M, Hallberg L, Ronne-Engstrom E. Early global brain oedema in relation to clinical admission parameters and outcome in patients with aneurysmal subarachnoid haemorrhage. Acta Neurochir (Wien). 2010;152:1527–33 (discussion 33).
Inamasu J, Nakatsukasa M, Hayashi T, Kato Y, Hirose Y. Early CT signs of hypoxia in patients with subarachnoid hemorrhage presenting with cardiac arrest: early CT signs in SAH patients presenting with CA. Acta Neurochir Suppl. 2013;118:181–4.
Liu Y, Soppi V, Mustonen T, et al. Subarachnoid hemorrhage in the subacute stage: elevated apparent diffusion coefficient in normal-appearing brain tissue after treatment. Radiology. 2007;242:518–25.
Beseoglu K, Holtkamp K, Steiger HJ, Hanggi D. Fatal aneurysmal subarachnoid haemorrhage: causes of 30-day in-hospital case fatalities in a large single-centre historical patient cohort. Clin Neurol Neurosurg. 2013;115:77–81.
Lagares A, Gomez PA, Lobato RD, Alen JF, Alday R, Campollo J. Prognostic factors on hospital admission after spontaneous subarachnoid haemorrhage. Acta Neurochir (Wien). 2001;143:665–72.
Choi HA, Bajgur SS, Jones WH, et al. Quantification of cerebral edema after subarachnoid hemorrhage. Neurocrit Care. 2016;25:64–70.
Kreiter KT, Copeland D, Bernardini GL, et al. Predictors of cognitive dysfunction after subarachnoid hemorrhage. Stroke. 2002;33:200–8.
Busch E, Beaulieu C, de Crespigny A, Moseley ME. Diffusion MR imaging during acute subarachnoid hemorrhage in rats. Stroke. 1998;29:2155–61.
Jadhav V, Sugawara T, Zhang J, Jacobson P, Obenaus A. Magnetic resonance imaging detects and predicts early brain injury after subarachnoid hemorrhage in a canine experimental model. J Neurotrauma. 2008;25:1099–106.
Thal SC, Sporer S, Klopotowski M, et al. Brain edema formation and neurological impairment after subarachnoid hemorrhage in rats. Lab Investig J Neurosurg. 2009;111:988–94.
Shigeno T, Fritschka E, Brock M, Schramm J, Shigeno S, Cervos-Navarro J. Cerebral edema following experimental subarachnoid hemorrhage. Stroke. 1982;13:368–79.
Handa Y, Takeuchi H, Kabuto M, et al. Blood–brain barrier disruption caused by impairment of cerebral autoregulation during chronic cerebral vasospasm in primates. Acta Neurochir Suppl (Wien). 1990;51:338–40.
Grote E, Hassler W. The critical first minutes after subarachnoid hemorrhage. Neurosurgery. 1988;22:654–61.
Kamiya K, Kuyama H, Symon L. An experimental study of the acute stage of subarachnoid hemorrhage. J Neurosurg. 1983;59:917–24.
Simard JM, Chen M, Tarasov KV, et al. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12:433–40.
Baradaran H, Fodera V, Mir D, et al. Evaluating CT perfusion deficits in global cerebral edema after aneurysmal subarachnoid hemorrhage. AJNR Am J Neuroradiol. 2015;36:1431–5.
Westermaier T, Stetter C, Raslan F, Vince GH, Ernestus RI. Brain edema formation correlates with perfusion deficit during the first six hours after experimental subarachnoid hemorrhage in rats. Exp Transl Stroke Med. 2012;4:8.
Terpolilli NA, Feiler S, Dienel A, et al. Nitric oxide inhalation reduces brain damage, prevents mortality, and improves neurological outcome after subarachnoid hemorrhage by resolving early pial microvasospasms. J Cereb Blood Flow Metab. 2016;36:2096–107. doi:10.1177/0271678X15605848.
Servillo G, Bifulco F, De Robertis E, et al. Posterior reversible encephalopathy syndrome in intensive care medicine. Intensive Care Med. 2007;33:230–6.
Springborg JB, Ma X, Rochat P, et al. A single subcutaneous bolus of erythropoietin normalizes cerebral blood flow autoregulation after subarachnoid haemorrhage in rats. Br J Pharmacol. 2002;135:823–9.
Voldby B, Enevoldsen EM, Jensen FT. Cerebrovascular reactivity in patients with ruptured intracranial aneurysms. J Neurosurg. 1985;62:59–67.
Tran Dinh YR, Lot G, Benrabah R, Baroudy O, Cophignon J, Seylaz J. Abnormal cerebral vasodilation in aneurysmal subarachnoid hemorrhage: use of serial 133Xe cerebral blood flow measurement plus acetazolamide to assess cerebral vasospasm. J Neurosurg. 1993;79:490–3.
Muhammad S, Guresir A, Greschus S, Scorzin J, Vatter H, Guresir E. posterior reversible encephalopathy syndrome as an overlooked complication of induced hypertension for cerebral vasospasm: systematic review and illustrative case. Stroke. 2016;47:519–22.
Yin W, Tibbs R, Tang J, Badr A, Zhang J. Haemoglobin and ATP levels in CSF from a dog model of vasospasm. J Clin Neurosci. 2002;9:425–8.
Suzuki M, Kudo A, Otawara Y, Hirashima Y, Takaku A, Ogawa A. Extrinsic pathway of blood coagulation and thrombin in the cerebrospinal fluid after subarachnoid hemorrhage. Neurosurgery. 1999;44:487–93 (discussion 93-4).
Kwon MS, Woo SK, Kurland DB, et al. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int J Mol Sci. 2015;16:5028–46.
Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–93.
Sugawara T, Jadhav V, Ayer R, Chen W, Suzuki H, Zhang JH. Thrombin inhibition by argatroban ameliorates early brain injury and improves neurological outcomes after experimental subarachnoid hemorrhage in rats. Stroke. 2009;40:1530–2.
Xu T, Zhang WG, Sun J, et al. Protective effects of thrombomodulin on microvascular permeability after subarachnoid hemorrhage in mouse model. Neuroscience. 2015;299:18–27.
Yu ZQ, Jia Y, Chen G. Possible involvement of cathepsin B/D and caspase-3 in deferoxamine-related neuroprotection of early brain injury after subarachnoid haemorrhage in rats. Neuropathol Appl Neurobiol. 2014;40:270–83.
Siler DA, Berlow YA, Kukino A, et al. Soluble epoxide hydrolase in hydrocephalus, cerebral edema, and vascular inflammation after subarachnoid hemorrhage. Stroke. 2015;46:1916–22.
You W, Wang Z, Li H, et al. Inhibition of mammalian target of rapamycin attenuates early brain injury through modulating microglial polarization after experimental subarachnoid hemorrhage in rats. J Neurol Sci. 2016;367:224–31.
Simard JM, Geng Z, Woo SK, et al. Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2009;29:317–30.
Sozen T, Tsuchiyama R, Hasegawa Y, et al. Role of interleukin-1beta in early brain injury after subarachnoid hemorrhage in mice. Stroke. 2009;40:2519–25.
Sherlock M, O’Sullivan E, Agha A, et al. The incidence and pathophysiology of hyponatraemia after subarachnoid haemorrhage. Clin Endocrinol (Oxf). 2006;64:250–4.
Cuesta M, Hannon MJ, Thompson CJ. Diagnosis and treatment of hyponatraemia in neurosurgical patients. Endocrinol Nutr. 2016;63:230–8.
Zeynalov E, Jones SM, Seo JW, Snell LD, Elliott JP. Arginine-vasopressin receptor blocker conivaptan reduces brain edema and blood–brain barrier disruption after experimental stroke in mice. PLoS ONE. 2015;10:e0136121.
Nakayama S, Amiry-Moghaddam M, Ottersen OP, Bhardwaj A. Conivaptan, a selective arginine vasopressin v1a and v2 receptor antagonist attenuates global cerebral edema following experimental cardiac arrest via perivascular pool of aquaporin-4. Neurocrit Care. 2016;24:273–82.
Doczi T, Laszlo FA, Szerdahelyi P, Joo F. Involvement of vasopressin in brain edema formation: further evidence obtained from the Brattleboro diabetes insipidus rat with experimental subarachnoid hemorrhage. Neurosurgery. 1984;14:436–41.
Laszlo FA, Varga C, Doczi T. Cerebral oedema after subarachnoid haemorrhage. pathogenetic significance of vasopressin. Acta Neurochir (Wien). 1995;133:122–33.
Hockel K, Scholler K, Trabold R, Nussberger J, Plesnila N. Vasopressin V(1a) receptors mediate posthemorrhagic systemic hypertension thereby determining rebleeding rate and outcome after experimental subarachnoid hemorrhage. Stroke. 2012;43:227–32.
Muehlschlegel S, Dunser MW, Gabrielli A, Wenzel V, Layon AJ. Arginine vasopressin as a supplementary vasopressor in refractory hypertensive, hypervolemic, hemodilutional therapy in subarachnoid hemorrhage. Neurocrit Care. 2007;6:3–10.
Agre P. The aquaporin water channels. Proc Am Thorac Soc. 2006;3:5–13.
Wang D, Nykanen M, Yang N, et al. Altered cellular localization of aquaporin-1 in experimental hydrocephalus in mice and reduced ventriculomegaly in aquaporin-1 deficiency. Mol Cell Neurosci. 2011;46:318–24.
Manley GT, Fujimura M, Ma T, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–63.
Yan JH, Khatibi NH, Han HB, et al. p53-induced uncoupling expression of aquaporin-4 and inwardly rectifying K+4.1 channels in cytotoxic edema after subarachnoid hemorrhage. CNS Neurosci Ther. 2012;18:334–42.
Badaut J, Brunet JF, Grollimund L, et al. Aquaporin 1 and aquaporin 4 expression in human brain after subarachnoid hemorrhage and in peritumoral tissue. Acta Neurochir Suppl. 2003;86:495–8.
Tait MJ, Saadoun S, Bell BA, Verkman AS, Papadopoulos MC. Increased brain edema in aqp4-null mice in an experimental model of subarachnoid hemorrhage. Neuroscience. 2010;167:60–7.
Wang Z, Meng CJ, Shen XM, et al. Potential contribution of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 to blood–brain barrier disruption and brain edema after experimental subarachnoid hemorrhage. J Mol Neurosci. 2012;48:273–80.
Chou SH, Feske SK, Simmons SL, et al. Elevated peripheral neutrophils and matrix metalloproteinase 9 as biomarkers of functional outcome following subarachnoid hemorrhage. Transl Stroke Res. 2011;2:600–7.
Horstmann S, Su Y, Koziol J, Meyding-Lamade U, Nagel S, Wagner S. MMP-2 and MMP-9 levels in peripheral blood after subarachnoid hemorrhage. J Neurol Sci. 2006;251:82–6.
Chou SH, Lee PS, Konigsberg RG, et al. Plasma-type gelsolin is decreased in human blood and cerebrospinal fluid after subarachnoid hemorrhage. Stroke. 2011;42:3624–7.
Egashira Y, Zhao H, Hua Y, Keep RF, Xi G. White matter injury after subarachnoid hemorrhage: role of blood–brain barrier disruption and matrix metalloproteinase-9. Stroke. 2015;46:2909–15.
Feiler S, Plesnila N, Thal SC, Zausinger S, Scholler K. Contribution of matrix metalloproteinase-9 to cerebral edema and functional outcome following experimental subarachnoid hemorrhage. Cerebrovasc Dis. 2011;32:289–95.
Sherchan P, Lekic T, Suzuki H, et al. Minocycline improves functional outcomes, memory deficits, and histopathology after endovascular perforation-induced subarachnoid hemorrhage in rats. J Neurotrauma. 2011;28:2503–12.
Guo Z, Sun X, He Z, Jiang Y, Zhang X, Zhang JH. Matrix metalloproteinase-9 potentiates early brain injury after subarachnoid hemorrhage. Neurol Res. 2010;32:715–20.
Simard JM, Woo SK, Schwartzbauer GT, Gerzanich V. Sulfonylurea receptor 1 in central nervous system injury: a focused review. J Cereb Blood Flow Metab. 2012;32:1699–717.
Kimberly WT, Battey TW, Pham L, et al. Glyburide is associated with attenuated vasogenic edema in stroke patients. Neurocrit Care. 2014;20:193–201.
Simard JM, Geng Z, Silver FL, et al. Does inhibiting Sur1 complement rt-PA in cerebral ischemia? Ann N Y Acad Sci. 2012;1268:95–107.
Sheth KN, Elm JJ, Molyneaux BJ, et al. Safety and efficacy of intravenous glyburide on brain swelling after large hemispheric infarction (GAMES-RP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2016;15:1160–9.
Tosun C, Kurland DB, Mehta R, et al. Inhibition of the Sur1–Trpm4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage. Stroke. 2013;44:3522–8.
Woo SK, Kwon MS, Geng Z, et al. Sequential activation of hypoxia-inducible factor 1 and specificity protein 1 is required for hypoxia-induced transcriptional stimulation of Abcc8. J Cereb Blood Flow Metab. 2012;32:525–36.
Hou J, Kshettry VR, Selman WR, Bambakidis NC. Peritumoral brain edema in intracranial meningiomas: the emergence of vascular endothelial growth factor-directed therapy. Neurosurg Focus. 2013;35:E2.
Roberts WG, Palade GE. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci. 1995;108(Pt 6):2369–79.
Josko J, Gwozdz B, Hendryk S, Jedrzejowska-Szypulka H, Slowinski J, Jochem J. Expression of vascular endothelial growth factor (VEGF) in rat brain after subarachnoid haemorrhage and endothelin receptor blockage with BQ-123. Folia Neuropathol. 2001;39:243–51.
Josko J. Cerebral angiogenesis and expression of VEGF after subarachnoid hemorrhage (SAH) in rats. Brain Res. 2003;981:58–69.
Liu L, Fujimoto M, Kawakita F, et al. Anti-vascular endothelial growth factor treatment suppresses early brain injury after subarachnoid hemorrhage in mice. Mol Neurobiol. 2016;53:4529–38.
Xu W, Xu R, Li X, Zhang H, Wang X, Zhu J. Downregulating hypoxia-inducible factor-1alpha expression with perfluorooctyl-bromide nanoparticles reduces early brain injury following experimental subarachnoid hemorrhage in rats. Am J Transl Res. 2016;8:2114–26.
Wu C, Hu Q, Chen J, et al. Inhibiting HIF-1alpha by 2ME2 ameliorates early brain injury after experimental subarachnoid hemorrhage in rats. Biochem Biophys Res Commun. 2013;437:469–74.
Albert-Weissenberger C, Mencl S, Hopp S, Kleinschnitz C, Siren AL. Role of the kallikrein-kinin system in traumatic brain injury. Front Cell Neurosci. 2014;8:345.
Kasuya H, Shimizu T, Okada T, Takahashi K, Summerville T, Kitamura K. Activation of the coagulation system in the subarachnoid space after subarachnoid haemorrhage: serial measurement of fibrinopeptide A and bradykinin of cerebrospinal fluid and plasma in patients with subarachnoid haemorrhage. Acta Neurochir (Wien). 1988;91:120–5.
Kunz M, Nussberger J, Holtmannspotter M, Bitterling H, Plesnila N, Zausinger S. Bradykinin in blood and cerebrospinal fluid after acute cerebral lesions: correlations with cerebral edema and intracranial pressure. J Neurotrauma. 2013;30:1638–44.
Scholler K, Feiler S, Anetsberger S, Kim SW, Plesnila N. Contribution of bradykinin receptors to the development of secondary brain damage after experimental subarachnoid hemorrhage. Neurosurgery. 2011;68:1118–23.
Thal SC, Sporer S, Schmid-Elsaesser R, Plesnila N, Zausinger S. Inhibition of bradykinin B2 receptors before, not after onset of experimental subarachnoid hemorrhage prevents brain edema formation and improves functional outcome. Crit Care Med. 2009;37:2228–34.
Acknowledgements
This work was supported by grants to JMS from the National Institute of Neurological Disorders and Stroke (NINDS) (NS060801; NS061808) and the National Heart, Lung and Blood Institute (HL082517).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
Dr. Simard holds a US patent (7,285,574), a novel non-selective cation channel in neural cells and methods for treating brain swelling. Dr. Simard is a member of the scientific advisory board and holds shares in Remedy Pharmaceuticals. No support, direct or indirect, was provided to Dr. Simard, or for this project, by Remedy Pharmaceuticals. All other authors report no conflicts.
Rights and permissions
About this article

Cite this article
Hayman, E.G., Wessell, A., Gerzanich, V. et al. Mechanisms of Global Cerebral Edema Formation in Aneurysmal Subarachnoid Hemorrhage. Neurocrit Care 26, 301–310 (2017). https://doi.org/10.1007/s12028-016-0354-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-016-0354-7