
Abstract
Background
Apolipoprotein E has previously been demonstrated to modulate acute brain injury responses, and administration of COG1410, an apoE-mimetic peptide derived from the receptor-binding region of apoE, improves outcome in preclinical models of acute neurological injury. In the current study, we sought to establish the optimal dose and timing of peptide administration associated with improved functional outcome in a murine model of intracerebral hemorrhage (ICH).
Methods
Ten to twelve-week-old C57/BL6 male mice were injured by collagenase-induced ICH and randomly selected to receive either vehicle or one of four doses of COG1410 (0.5, 1, 2, or 4 mg/kg) via tail vein injection at 30 min after injury and then daily for 5 days. The injured mice were euthanized at various time points to assess inflammatory mediators, cerebral edema, and hematoma volume. Over the first 5 days following injury, vestibulomotor function was tested via Rotorod (RR) latency. After an optimal dose was demonstrated, a final cohort of animals was injured with ICH and randomly assigned to receive the first dose of COG1410 or vehicle at increasingly longer treatment initiation times after injury. The mice were then assessed for functional deficit via RR testing over the first 5 days following injury.
Results
The mice receiving 2 mg/kg of COG1410 after injury demonstrated reduced functional deficit, decreased brain concentrations of inflammatory proteins, and less cerebral edema, although hematoma volume did not vary. The improved RR performance was maintained when peptide administration was delayed for up to 2 h after ICH.
Conclusions
COG1410 administered at a dose of 2 mg/kg within 2 h after injury improves functional recovery in a murine model of ICH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Intracerebral hemorrhage (ICH) is a devastating and relatively common form of cerebrovascular disease associated with a 30-day mortality rate of approximately 50% and significant disability in many survivors. Despite this devastating personal and societal impact, ICH remains an understudied disease with little improvement in patient outcomes over the last 20 years [1]. Preclinical data from our and other laboratories [2, 3] illuminate a number of inflammatory mechanisms that may play a crucial role in the development of secondary neuronal injury, suggesting the development of targeted anti-inflammatory approaches may be readily translatable into human patients [4, 5]. However, at present, no intervention has demonstrated improvement in function and mortality in humans [6].
Apolipoprotein E (apoE) is the primary apolipoprotein synthesized in the brain in response to injury where it exerts neuroprotective effects via multiple mechanisms including antioxidant, anti-inflammatory, anti-excitotoxic, and neurotrophic properties [7–17]. Peptides derived from apoE-binding region mimic the anti-inflammatory properties of the intact holoprotein, cross the blood–brain barrier (BBB), and are protective in preclinical models of traumatic brain injury, [18–20] stroke, [21] subarachnoid hemorrhage, [22, 23] and intracerebral hemorrhage [2]. In addition, the molecular mechanism(s) by which apoE modifies glial activation and the central nervous system (CNS) response to injury is beginning to become understood [24].
In the current study, we use a preclinical model of ICH to determine the dose–response and optimal timing of peptide administration for COG1410, an apoE-mimetic peptide, previously shown to reduce neuroinflammation and improve neurologic recovery in this model [2]. We also sought to further address its effects on neuroinflammatory pathways.
Methods
All animal procedures were designed to minimize animal discomfort and numbers, conformed to international guidelines on the use of animals, and were approved by the Duke University Institutional Animal Care and Use Committee.
Experimental Groups
Group 1
Physiologic measurements for arterial blood gas and glucose at 15 min before and 1 h after COG1410 (2 mg/kg) administration via intravenous (IV) tail vein injection with concomitant arterial blood pressure, and rectal temperature recorded continuously (n = 6/group).
Group 2
Determination of therapeutic window through functional performance of the mice treated with COG1410 (2 mg/kg) or vehicle administered via IV injection at 30 min, 1 h, 2, or 4 h after ICH and continued daily via IV injection for 5 days; an additional cohort of mice received COG1410 (4 mg/kg) via IV injection at 1 h after injury and continued daily IV injection for 5 days to determine if increased dose could extend the therapeutic window; testing included RR every other day over first 5 days (n = 12/group).
Group 3
Measurement of brain water content and hematoma lesion volume at 24 h after ICH in the mice treated with COG1410 (2 mg/kg) or vehicle administered via IV injection at 30 min after injury (n = 8/group).
Group 4
Analysis of intracellular signaling proteins using Western blot at 2 and 6 h after ICH in the mice treated with COG1410 (2 mg/kg) or vehicle administered via IV injection at 30 min after injury (n = 3/group).
Group 5
Quantitative stereology for F4/80 (microglia) and Flouro Jade B (FJB; degenerating neurons) positive cells at 5 days after ICH in the mice treated with COG1410 (2 mg/kg) or vehicle administered via IV injection at 30 min after injury and continued daily for 5 days (n = 6/group).
Group 6
Functional performance of the mice treated with COG1410 (0.5, 1, 2, or 4 mg/kg) or vehicle administered via IV injection at 30 min after ICH and continued daily for 5 days; testing included Rotorod (RR) latencies every other day over the first 5 days (n = 12/group).
Physiologic Measurements
The mice were anesthetized in a chamber with 5% isoflurane in 30% O2/70% N2. The trachea was intubated with a 20-gauge Insyte-W intravenous catheter (Becton–Dickinson, Sandy, UT). The inspired isoflurane concentration was reduced to 1.6%, and the lungs were mechanically ventilated at a rate of 105 breaths/min with a delivered tidal volume of 0.75 ml. Rectal temperature was monitored and servoregulated with a surface heating/cooling system to a target of 37.0 ± 0.2°C. The right femoral artery was cannulated (PE10 catheter; Becton–Dickinson, Sparkes, MD) to monitor arterial blood pressure and to collect arterial blood samples. The right jugular vein was cannulated with a PE10 catheter for drug delivery. Arterial blood gases and glucose were measured at 15 min before and at 1 h after drug delivery by ABL800 FLEX (Radiometer copenhagen, Denmark). Arterial blood pressure and rectal temperature were recorded continuously before, during, and after treatment for 1 h using Chart V3.3.8 program (MacLab, ADInstruments Pty Ltd, Australia).
Intracerebral Hemorrhage Model
Our murine injury model [3] was adapted from a previously described model of ICH in rats [25]. Ten to twelve-week-old male mice, C57-BL/6, were used in these experiments. The trachea was intubated after anesthesia induction with 4.6% isoflurane, and the lungs were mechanically ventilated with 1.6% isoflurane in 30% O2/70% N2. Rectal temperature was maintained at 37 ± 0.2°C by underbody warming system. The animal’s head was secured in a stereotactic frame, local anesthetic injected, and the scalp incised. After exposure of the skull, a burr hole was created 2 mm left lateral to bregma, and a 0.5 μl syringe needle (Hamilton, Reno, NV, USA) was advanced to a depth of 3 mm from cortex. Type IV-S Clostridial collagenase (Sigma, St. Louis, MO, USA) was injected over 5 min (0.1 U in 0.4 μl NS) with the needle being held motionless for an additional 5 min. After slowly withdrawing the needle, the incision was closed, and animals were allowed to recover spontaneous ventilation with subsequent extubation. Following recovery in a warm non-stimulating environment, the mice were allowed free access to food and water.
Preparation and Administration of COG1410
COG1410 were synthesized by Polypeptide Labs (San Diego, CA) to a purity of 95%. COG1410 is acetyl-AS-Aib-LRKLAib-KRLL-amide, which is derived from apoE residues 138–149 with Aib (amino isobutyric acid) substitutions at positions 140 and 145. For all experiments, peptides were dissolved in sterile saline immediately before use, and the mice were injected via the tail vein with varying doses of COG1410 peptide in 100 μl of sterile saline or 100 μl of sterile saline (vehicle) at predefined time points after injury, and then at 24 h intervals for 5 days. The mice were randomized to treatment or vehicle groups at the time of their first dose.
Neurological Functional Testing
An automated RR (Ugo Basile, Comerio, Italy) was used to assess vestibulomotor function [26]. On the day before ICH induction, the mice underwent two consecutive conditioning trials at a set rotational speed (16 revolutions/min) for 60 s followed by three additional trials with an accelerating rotational speed. The average time to fall from the rotating cylinder in the final three trials was recorded as baseline latency. The mice underwent testing with three trials of accelerating rotational speed (inter-trial interval of 15 min) on days 1, 3, and 5 after injury. The average latency to fall from the rod was recorded. The mice that were unable to grasp the rotating rod were given a latency value of 0 s.
Neuroseverity scoring was assessed on days 1–7 after injury by means of a neurobehavioral examination (scoring scale 7–21) as previously described [27]. Motor score [4–12] was derived from spontaneous activity, symmetry of limb movements, climbing, balance, and coordination. Sensory score [3–9] was derived from body proprioception, vibrissae, visual, and tactile responses. Sensory tests examined function from both cerebral hemispheres.
Measurement of Hematoma Volume
The mice were anesthetized with 4.6% isoflurane in 30% O2/70% N2 and euthanized. The brains were removed and frozen at −20°C. Coronal sections of 20 μm thickness were taken at 320-μm intervals over the rostral–caudal extent of the lesion. The sections were stained with hematoxylin and eosin, and lesion volume measured by digitally sampling stained sections with an image analyzer. Lesion volumes (mm3) were computed as running sums of lesion area multiplied by the known interval (e.g., 320 μm) between sections over the extent of the lesion expressed as an orthogonal projection.
Measurement of Brain Water Content
The mice were anesthetized with 4.6% isoflurane in 30% O2/70% N2 and euthanized. Brains were sectioned in the mid-sagittal plane, and each hemisphere weighed immediately (“wet” weight). Hemispheres were allowed to dehydrate over 24 h at 40°C and then re-weighed (“dry” weight). Inter-hemispheric water weight differences were then recorded and used for analysis [28]. To compare among the treatment groups, the percentage of water weight due to cerebral edema of the injured hemisphere was determined and used for analysis.
Western Analysis
Frozen mouse brain samples were ground to a fine powder using a mortar and pestle cooled with liquid nitrogen. Extracts were prepared by suspending 100 μg of brain powder in NP40 buffer and performing a freeze–thaw cycle. Equal amounts of cellular protein were added to 4× laemmli buffer and heated to 60°C for 10 min. Proteins were separated by SDS PAGE and blotted onto nitrocellulose membranes (Bio-Rad). After the proteins were transferred to nitrocellulose membranes, the membranes were blocked using Odyssey Blocking Buffer (LiCor, Lincoln, NE) for 3 h and incubated overnight at 4°C in a phospho-p38 or total p38 antibodies (cell signaling). The membranes were washed thoroughly and incubated with IRDye®-labeled secondary antibodies (LiCor) and protein bands were visualized and quantified using an Odyssey Infrared scanner (LiCor). Similar blots were incubated with phospho-nuclear factor kappa B (NFκB) or total-NFκB antibodies (cell signaling).
Tissue Processing and Histological Methods
After induction of anesthesia and intracardiac perfusion with phosphate-buffered saline (PBS), mouse brains were rapidly removed and dissected in the mid-sagittal plane. Hemispheres were sectioned sagittally and flash-frozen in liquid nitrogen and stored at −80°C; the other hemisphere was immersion-fixed in 10% formaldehyde for 24 h, transferred into 1× PBS and stored at 4°C. Frozen sagittal sections (40 μm) were collected on a freezing sliding microtome. For immunostaining, tissue was incubated in 1% hydrogen peroxide, permeabilized by 0.1% saponin, and blocked with 10% goat serum. When possible, to minimize background resulting from the use of monoclonal antibodies and for antigen retrieval purposes, brain sections were first microwaved in SSC and antigen retrieval buffer (Vector Labs, Burlingame, CA, USA) according to manufacturer’s instructions. Brain sections were mounted on slides, dehydrated, cleared, and cover-slipped using DPX mounting media (Fluka, Milwaukee, WI, USA). Fluoro-Jade B (FJB) staining [29] and anti-rat F4/80 antibody specific for activated microglia and cells of the monocyte lineage (rat monoclonal, 1:10,000; Serotec, Raleigh, NC) [30] were performed using standard protocols.
Stereological Analysis
Cell counting was conducted using a Nikon 218912 light microscope interfaced with the StereoInvestigator software package (MicroBrightField, Williston, VT, USA). The number of stained cells per mm3 of hippocampus was estimated by the optical fractionator method [31]. The optical fractionator is an unbiased counting method, which is independent of the size, shape, and orientation of the cells to be counted. The parameters of the fractionator-sampling scheme were established in a pilot experiment and were uniformly applied to all animals. Before counting, all the slides were coded to avoid experimenter bias. As determined by StereoInvestigator, we chose six sagittal sections (40 μm thick) spaced eight sections apart along the dorsal hippocampal formation by systematic random sampling. This number of sections proved sufficient to provide a coefficient of error (CE) between 0.09 and 0.11. On each section, the whole hippocampal area was delineated. For microglial quantification, the sampling grid was 399.027 (X) × 367.92 (Y) μm, and cells were counted within a probe volume defined by the counting frame (80 × 80 μm) and the dissector height (11 μm). For neuron quantification, the sampling grid was 168.36 (X) × 111.84 (Y) μm, and cells were counted within a probe volume defined by the counting frame (50 × 50 μm) and the dissector height (11 μm). Only cells within the counting frame or overlapping the right or superior border of the counting frame, and for which nuclei came into focus while focusing down through the dissector height, were counted. The total number of F4/80 or FJB immunopositive cells was calculated per hippocampal volume of 1920-μm thickness.
Statistical Analysis
RR performances and neuroseverity scoring were compared with two-way repeated-measures analysis of variance with day as the repeated variable. The F values were calculated, and if the probability distribution of F with the appropriate degrees of freedom suggested a significant group effect, pairwise testing was performed between groups using Scheffe’s post-hoc method to correct for multiple comparisons. The numbers of FJB and F4/80 cell counts were compared between groups by the Mann–Whitney U statistic. Student’s t-test was used to compare hemorrhage volume and brain water content. Neuroseverity scores on day 7 after injury and brain protein concentrations as measured by Western blots were compared by Chi-square. Statistical significance was assumed with P < 0.05. All values were expressed as mean ± standard error of the mean (SEM) and measurements were performed on JMP (v8.0.1, SAS, Cary, NC).
Results
Physiologic Effects of COG1410
To assess the physiologic impact of COG1410 treatment, we performed hemodynamic monitoring with blood sampling before and after administration of COG1410 (2 mg/kg) or vehicle. No difference was seen in pH, PaCO2, PaO2, glucose, hematocrit, or temperature before or after treatment at 15 min and 1 h. PaO2 was controlled at 193 ± 23.35 mmHg, PaCO2 at 37.3 ± 6.04 mmHg, pH at 7.36 ± 0.02, and glucose 126.33 ± 33.71 gm/dl. Treatment with COG1410 (2 mg/kg) did significantly reduce mean arterial pressure (MAP) at 10–60 min after treatment compared with baseline (P < 0.05). MAP in the COG1410 (2 mg/kg)-treated group started at 76.31 ± 2.38 mmHg before injection and decreased to 63.28 ± 2.14 mmHg at 10 min and then steadily to 59.79 ± 5.41 mmHg at 60 min after injection of collagenase, which represented a 22.4 ± 10.2% change from baseline at 60 min after injury. MAP in vehicle-treated animals started at 78.99 ± 2.44 mmHg before injection and did not change during the hour of monitoring.
COG1410 Improves Neurological Function after ICH
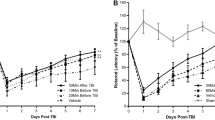
In order to inform the critical variables necessary to perform an early clinical trial using COG1410 in ICH, we investigated the optimal dose of peptide required for maximal neurological improvement after ICH in our murine model. One of the four doses of COG1410 (0.5, 1, 2, or 4 mg/kg) or vehicle was delivered by tail vein injection at 30 min after injury. Following this initial administration, additional doses were given at 24-h intervals for 5 days. The mice were then subjected to RR testing on days 1, 3, and 5 following injury. We found that administration of all doses of COG1410 resulted in improved functional performance as measured by RR latencies during all the days tested when compared with their vehicle-treated counterparts (Fig. 1; P < 0.01). On further analyses, equivalent functional improvement was demonstrated with COG1410 at doses of 0.5 or 1 mg/kg; however, the mice treated with COG1410 at doses of 2 or 4 mg/kg demonstrated functional improvement in RR latency compared with animals treated with COG1410 0.5 or 1 mg/kg (P < 0.05). Neuroseverity scoring mirrored the RR findings as the mice treated with COG1410 attained higher scores over the 7 days of testing (P < 0.05) with significant differences between COG1410-treated mice and their vehicle-treated counterparts on day 7 (COG1410 vs. vehicle: 17.4 ± 1.7 vs. 11.1 ± 3.2; P < 0.05). However, neuroseverity scores did not differ between the mice treated with different doses of COG1410. Interestingly, mortality was decreased in all COG1410 groups compared with vehicle (COG1410 vs. vehicle: three of 48 animals (6.25%) vs. three of 12 animals (25%); P < 0.05). However, mortality was not different between the COG1410 groups (one of 12 animals (8.3%) in COG1410 (0.5 mg/kg) vs. one of 12 animals (8.3%) in COG1410 (1.0 mg/kg) vs. one of 12 animals (8.3%) in COG1410 (2.0 mg/kg) vs. zero of 12 animals in COG1410 (4.0 mg/kg).

Mice treated with 0.5, 1, 2, or 4 mg/kg of COG1410 via tail vein injection at 30 min after ICH injury demonstrated increased RR latencies when compared with their vehicle-treated counterparts. (* represents P < 0.05; ** represents P < 0.01) This effect was maximal in the group treated with 2 mg/kg after ICH
COG1410 Decreases the Amount of Cerebral Edema after ICH
In order to assess the effect of the optimized dose of COG1410 (2 mg/kg) on the evolution of cerebral edema following ICH, we measured brain water content at 24 h after injury in the mice treated with COG1410 (2 mg/kg) or vehicle. Brain water content was quantified by comparison of the “wet-to-dry” weight measurements of brain hemispheres. We found that COG1410 (2 mg/kg) decreased brain water content at 24 h after ICH when compared with their vehicle-treated counterparts (COG1410 (2 mg/kg) vs. vehicle: 77.06 ± 0.01% vs. 78.96 ± 0.01%, respectively; P < 0.05). The decrease in brain water content was independent of hematoma volume measured at 24 h after injury (COG1410 (2 mg/kg) vs. vehicle: 0.577 ± 0.098 vs. 0.585 ± 0.011 mm3, respectively).
COG1410 Reduces Activation of Inflammatory Signaling Pathways and Cytokine Production after ICH
To assess whether the improvement in neurological function and associated decrease in cerebral edema after administration of COG1410 (2 mg/kg) following ICH was related to decreased inflammation, the phosphorylation status of proteins in inflammatory signal transduction pathways were analyzed by quantitative Western blotting. The mice were treated with COG1410 (2 mg/kg) or vehicle at 30 min following injury, and brains were harvested at 2 and 6 h following injury. The phosphorylation status of p38 and NFκB in brain extracts were determined using phospho- and total-antibodies. At the 2-h post-injury time point, we demonstrated significant decrease in the ratio of the phosphorylated p38 and NFκB protein divided by the total amount in the mice treated with COG1410 (2 mg/kg) relative to vehicle-treated animals (Fig. 2a, b; P < 0.05). Further, reduced phosphorylation of p38 and NFκB was associated with reduced production of IL-6 in COG1410-treated mice relative to vehicle-treated mice (Fig. 2c, P < 0.05). No differences were found between groups in these proteins at 6 h after injury.

Western blot of inflammatory proteins at 2 h after ICH in mice treated with COG1410 (2 mg/kg) or vehicle. The ratio between relative fluorescence units of the phospho-protein to the total protein for p38 (a) and NFκB (b), and IL-6 in (c) were significantly down-regulated after administration of COG1410. Phosphorylation of IΚB kinase (IΚK) (d) nearly reached statistical significance (P < 0.06). Representative blots (e) of phospho-p38 and total p38 in the ipsilateral hemisphere of a vehicle-treated mouse (Sal) and a COG1410-treated (COG) mouse and in the ipsilateral (ips) and contralateral (con) hemispheres of a vehicle-treated mouse 2 h after ICH
COG1410 Reduces Microglial Activation after ICH
To determine whether this functional effect and associated decrease in inflammatory brain protein concentration was related to microglial recruitment and activation, F4/80 staining of bilateral hippocampi was performed at 5 days after injury (Fig. 3). Though contralateral cell counts were similar, there was substantial reduction in F4/80 immunopositive cells in ipsilateral hippocampi of COG1410-treated mice at this 5-day timepoint (COG1410 (2 mg/kg) vs. vehicle: 20.72 ± 1.78 vs. 33.82 ± 1.46 cells/mm3; P < 0.001). In addition, in an attempt to quantify neuronal injury at 5 days after hemorrhage, FJB staining was performed; however, no FJB-positive cells were found in the hippocampi of either treatment group.

F4/80 immunocytochemical staining of microglia at 5 days after ICH in mice treated with vehicle (a) or COG1410 (2 mg/kg) (b). (** represents P < 0.01)
COG1410 Improves Neurological Function when Initiated at 2 h after ICH
To determine if initial administration of COG1410 could be extended beyond 30 min after ICH, we randomized the mice to receive the initial dose of COG140 (2 mg/kg) or vehicle via tail vein injection at 30 min, 1, 2, or 4 h after injury and then daily for 5 days. To determine if the therapeutic window could be further extended through administration of a higher dose of COG1410, an additional cohort of mice in the same experiment received COG1410 (4 mg/kg) via tail vein injection at 1 h after ICH and then daily for 5 days. The mice were then subjected to RR testing on days 1, 3, and 5 following injury. The mice receiving the initial dose of COG1410 (2 mg/kg) at either 30 min, 1 h, or 2 h or COG1410 (4 mg/kg) at 1 h after injury exhibited longer RR latencies over the days of testing when compared with their vehicle-treated counterparts (Fig. 4; P < 0.01) There were no differences in RR latencies between vehicle-treated animals and the mice receiving their initial dose of COG1410 (2 mg/kg) at 4 h after ICH. Finally, while initiation of COG1410 (2 mg/kg) treatment at 30 min after injury resulted in superior RR performance compared with all groups (P < 0.05), no differences in RR latencies were demonstrated between the mice receiving their initial dose of COG1410 (4 mg/kg) at 1 h or COG1410 (2 mg/kg) at 1 h or 2 h after ICH. On post-injury day 5, the mice treated with COG1410 (2 mg/kg) initiated at 30 min had equivalent RR latencies (139.85 ± 22.7 s) to the mice treated with COG1410 (2 mg/kg) initiated at 2 h after injury (117.6 ± 12.7 s) and COG1410 (4 mg/kg) initiated at 1 h after injury (106.7 ± 10.8 s) but had longer RR latencies than the mice treated with COG1410 (2 mg/kg) initiated at 1 h after injury (98.9 ± 9.3 s), COG1410 (2 mg/kg) initiated at 4 h after injury (85.0 ± 17.5 s). and vehicle-treated mice (72.8 ± 10.0 s; P < 0.05).

Mice with initiation of treatment at 30 min, 1, or 2 h after ICH with COG1410 (2 mg/kg) demonstrated improved rates of recovery when compared to vehicle-treated animals after ICH injury. (* represents P < 0.05; ** represents P < 0.01) No improvement was demonstrated in mice when COG1410 treatment was initiated at 4 h after ICH compared with vehicle-treated mice, nor was any increased efficacy demonstrated when the dose of COG1410 was increased to (4 mg/kg) compared with mice treated with COG1410 (2 mg/kg) with treatment initiation at 1 h after injury (data not shown)
Discussion
Given the paucity of therapeutic interventions for ICH, there is an important unmet clinical need for new approaches to treat this devastating condition. The present study demonstrates that COG1410, a novel apoE-derived peptide, improves neurological function in the mice injured with ICH when administered as an IV injection at a dose of 2 mg/kg given up to 2 h after injury. This improvement in neurological function was associated with decreased cerebral edema, microglial activation, and neuroinflammatory proteins concentration.
COG1410 represents a second-generation apoE-based therapeutic in which the helicity and anti-inflammatory potency of the peptide was enhanced by the introduction of two non-naturally occurring amino-isobutyric acid residues [19]. Although apoE does not cross the BBB, many of its adaptive properties appear to be mediated by signaling cascades initiated by specific interaction with cell surface receptors [32, 33]. Based on these receptor interactions, a series of peptides derived from the receptor-binding domain of apoE were created and demonstrated many of the same adaptive functional effects as the intact holoprotein [12]. These studies were extended to preclinical models of traumatic brain injury, where a peptide derived from the receptor-binding region was well tolerated, crossed the BBB, and was associated with improved functional and histological outcomes [20]. Finally, the use of apoE-mimetic peptides to improve neurological function after acute CNS injury was extended to ICH. We have previously demonstrated that COG1410 has pharmacogenomic interactions as a function of APOE genotype after ICH in the mice [2].
The current study sought to build on the basis of this body of research. Using the RR task, we performed dose-escalation and timing trials to lay the framework for translation of COG1410 into potential clinical trial. The RR task is considered to be the most sensitive and reliable test of vestibulomotor function in the acute phase (days 1–5) of post-ICH recovery [26]. Measurement of vestibulomotor function is a clinically relevant outcome as it models everyday skills relevant to human ICH patients (e.g., balance, coordination, and walking). These results validate our previous report [2] of COG1410 showing improved outcome following ICH and demonstrate that the functional improvement following COG1410 is dose and timing dependent. Furthermore, COG1410 (2 mg/kg) treatment exhibited significant reduction in cerebral edema, microglial activation, and neuroinflammatory protein concentrations in the brain after ICH in our murine model—findings that are consistent with its palliative effects in other models of acute CNS injury [20, 22, 34]. Although we utilize both a collagenase-induced and autologous blood injection models of ICH in our laboratory [5], we chose the collagenase-induced model for these experiments. Each model represents different aspects of the human condition [35], and in general, both models may be used to validate efficacy of different therapies [36]. The collagenase-induced ICH model results in a more severe neurological deficit that is of use when performing dose-finding studies, was used in prior experiments with this compound [2], and more closely represents the aspects of the human condition that are of interest with COG1410, namely neuroinflammation and cerebral edema [37].
Clinical data suggest that the inflammatory response in the CNS begins within hours of ICH and correlates with poor functional recovery of the patient [38]. The inflammatory response is characterized by activation of astrocytes and microglia, invasion of peripheral immune cells, and production of inflammatory cytokines and matrix metalloproteases [39]. In the case of ICH, inflammation is associated with disruption of the BBB and loss of neurons in the tissues at the peripheral edge of the hematoma [40]. Production of pro-inflammatory cytokines is mediated by glial cells and may be involved in secondary brain injury after ICH. As previously demonstrated, the expressions of cytokines including IL-1β and IL-6 increase in the ipsilateral striatum of collagenase-injected rats relative to sham animals that received no collagenase injection [40]. In this report, we have shown that COG1410 treatment significantly reduced the production of IL-6 in the ipsilateral hemisphere following ICH relative to vehicle-treated animals.
The production of inflammatory cytokines is at least partially regulated by activation of signal transduction cascades that involve the mitogen-activated protein kinases (MAPKs) and nuclear factor kappa B (NFκB) activation. Evidence of a role of MAPK activation in the loss of neurons was suggested by demonstration that extracellular signal-regulated kinases (ERKs) and p38 MAPKs were phosphorylated in the area of the hematoma following ICH [41]. Ohnishi et al. [42] used immunofluorescence co-localization techniques to show that both neurons and microglia within the hematoma area exhibit immunoreactivity for phospho-ERK after collagenase injection. Reduced neuronal loss upon treatment with inhibitors specific for ERK, p38, and jun N-terminal kinases (JNKs) provided further support for MAPK activation playing a critical role in neuron loss following ICH [41]. These data were also recapitulated in the clostridial collagenase model of ICH where inhibitors of MAPKs reduced the neuronal loss that normally accompanies formation of a hematoma [42].
Unfortunately, the MAPK inhibitors do not penetrate the BBB and, thus, have limited clinical utility in the management of acute CNS injury. However, we have recently reported that COG1410, related apoE-mimetic peptides, and the apoE holoprotein may bind to the SET protein, also known as inhibitor #2 of protein phosphatase 2A (I2PP2A) [43]. In a set of experiments, binding of COG1410 to SET was related to activation of PP2A activity in the brains of the mice [24]. Because the phosphorylations of p38 [44], JNK [45], ERK [46], and IκK [47] are downregulated by PP2A, activation of PP2A in the brain may result in reduction in the phosphorylations of p38 and IκK. Indeed, our results showed a significant reduction in p38 phosphorylation in the ipsilateral hemisphere of COG1410-treated mice relative to vehicle-treated animals. COG1410 treatment also reduced the phosphorylation of NFκB, which is consistent with our previous reports that COG112, an apoE-mimetic peptide closely related to COG1410, inhibited NFκB activation through inhibition of IκK signaling [48, 49]. Finally, evidence suggests that both the intact apoE holoprotein and peptides derived from the receptor-binding region reduce glial activation and blunt neuroinflammatory responses in vivo [15]. Cumulatively, these results suggest that apoE-mimetic peptides represent a promising, novel therapeutic strategy for the potential treatment of acute neurological diseases associated with glial activation and neuroinflammation, including ICH [20, 22, 50, 51].
In summary, we provide data demonstrating that administration of COG1410 (2 mg/kg) reduces pro-inflammatory events in the CNS following induction of ICH. This was associated with a reduction in microgliosis, cerebral edema, and improved functional outcome. Moreover, administration of COG1410 was not associated with any overt toxicity, and the beneficial effects were retained when administered up to 2 h after injury, suggesting a reasonable potential for clinical translation into a pilot trial for patients with ICH.
References
Broderick JP, Adams HP Jr, Barsan W, Feinberg W, Feldmann E, Grotta J, Kase C, Krieger D, Mayberg M, Tilley B, Zabramski JM, Zuccarello M. Guidelines for the management of spontaneous intracerebral hemorrhage: a statement for healthcare professionals from a special writing group of the stroke council, American heart association. Stroke. 1999;30:905–15.
James ML, Sullivan PM, Lascola CD, Vitek MP, Laskowitz DT. Pharmacogenomic effects of apolipoprotein e on intracerebral hemorrhage. Stroke. 2009;40:632–9.
James ML, Wang H, Venkatraman T, Song P, Lascola CD, Laskowitz DT. Brain natriuretic peptide improves long-term functional recovery after acute CNS injury in mice. J Neurotrauma. 2009;27:217–28.
James ML, Blessing R, Bennett E, Laskowitz DT. Apolipoprotein E modifies neurological outcome by affecting cerebral edema but not hematoma size after intracerebral hemorrhage in humans. J Stroke Cerebrovasc Dis. 2009;18:144–9.
James ML, Warner DS, Laskowitz DT. Preclinical models of intracerebral hemorrhage: a translational perspective. Neurocrit Care. 2007;9(1):135–52.
Mendelow AD, Gregson BA, Fernandes HM, Murray GD, Teasdale GM, Hope DT, Karimi A, Shaw MD, Barer DH. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the international surgical trial in intracerebral haemorrhage (STICH): a randomised trial. Lancet. 2005;365:387–97.
Aono M, Bennett ER, Kim KS, Lynch JR, Myers J, Pearlstein RD, Warner DS, Laskowitz DT. Protective effect of apolipoprotein E-mimetic peptides on N-methyl-d-aspartate excitotoxicity in primary rat neuronal-glial cell cultures. Neuroscience. 2003;116:437–45.
Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30:1350–60.
Aono M, Lee Y, Grant ER, Zivin RA, Pearlstein RD, Warner DS, Bennett ER, Laskowitz DT. Apolipoprotein E protects against NMDA excitotoxicity. Neurobiol Dis. 2002;11:214–20.
Colton CA, Brown CM, Czapiga M, Vitek MP. Apolipoprotein-E allele-specific regulation of nitric oxide production. Ann N Y Acad Sci. 2002;962:212–25.
Laskowitz DT, Goel S, Bennett ER, Matthew WD. Apolipoprotein E suppresses glial cell secretion of TNF alpha. J Neuroimmunol. 1997;76:70–4.
Laskowitz DT, Thekdi AD, Thekdi SD, Han SK, Myers JK, Pizzo SV, Bennett ER. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp Neurol. 2001;167:74–85.
Lomnitski L, Oron L, Sklan D, Michaelson DM. Distinct alterations in phospholipid metabolism in brains of apolipoprotein E-deficient mice. J Neurosci Res. 1999;58:586–92.
Lynch JR, Morgan D, Mance J, Matthew WD, Laskowitz DT. Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J Neuroimmunol. 2001;114:107–13.
Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, Warner DS, Laskowitz DT. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. 2003;278:48529–33.
Miyata M, Smith JD. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat Genet. 1996;14:55–61.
Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264:850–2.
Hoane MR, Kaufman N, Vitek MP, McKenna SE. COG1410 improves cognitive performance and reduces cortical neuronal loss in the traumatically injured brain. J Neurotrauma. 2009;26:121–9.
Laskowitz DT, Vitek MP. Apolipoprotein E and neurological disease: therapeutic potential and pharmacogenomic interactions. Pharmacogenomics. 2007;8:959–69.
Lynch JR, Wang H, Mace B, Leinenweber S, Warner DS, Bennett ER, Vitek MP, McKenna S, Laskowitz DT. A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Exp Neurol. 2005;192:109–16.
Tukhovskaya EA, Yukin AY, Khokhlova ON, Murashev AN, Vitek MP. COG1410, a novel apolipoprotein-E mimetic, improves functional and morphological recovery in a rat model of focal brain ischemia. J Neurosci Res. 2009;87:677–82.
Gao J, Wang H, Sheng H, Lynch JR, Warner DS, Durham L, Vitek MP, Laskowitz DT. A novel apoE-derived therapeutic reduces vasospasm and improves outcome in a murine model of subarachnoid hemorrhage. Neurocrit Care. 2006;4:25–31.
Mesis RG, Wang H, Lombard FW, Yates R, Vitek MP, Borel CO, Warner DS, Laskowitz DT. Dissociation between vasospasm and functional improvement in a murine model of subarachnoid hemorrhage. Neurosurg Focus. 2006;21:E4.
Christensen DJ, Ohkubo N, Oddo J, Van Kanegan MJ, Neil J, Li F, Colton CA, Vitek MP. Apolipoprotein-E and peptide mimetics modulate inflammation by binding the SET protein and activating protein phosphatase 2A. J Immunol. 2011;186(4):2535–42.
Rosenberg GA, Estrada E, Wesley M, Kyner WT. Autoradiographic patterns of brain interstitial fluid flow after collagenase-induced haemorrhage in rat. Acta Neurochir Suppl (Wien). 1990;51:280–2.
Hamm RJ, Pike BR, O’Dell DM, Lyeth BG, Jenkins LW. The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J Neurotrauma. 1994;11:187–96.
Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats: statistical validation. Stroke J Cereb Circ. 1995;26:627–34. (discussion 35).
Song EC, Chu K, Jeong SW, Jung KH, Kim SH, Kim M, Yoon BW. Hyperglycemia exacerbates brain edema and perihematomal cell death after intracerebral hemorrhage. Stroke. 2003;34:2215–20.
Dawson HN, Cantillana V, Chen L, Vitek MP. The tau N279 K exon 10 splicing mutation recapitulates frontotemporal dementia and parkinsonism linked to chromosome 17 tauopathy in a mouse model. J Neurosci. 2007;27:9155–68.
Dawson HN, Cantillana V, Jansen M, Wang H, Vitek MP, Wilcock DM, Lynch JR, Laskowitz DT. Loss of tau elicits axonal degeneration in a mouse model of Alzheimer’s disease. Neuroscience. 2010;169:516–31.
West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–97.
Hoe HS, Pocivavsek A, Chakraborty G, Fu Z, Vicini S, Ehlers MD, Rebeck GW. Apolipoprotein E receptor 2 interactions with the N-methyl-d-aspartate receptor. J Biol Chem. 2006;281:3425–31.
Misra UK, Adlakha CL, Gawdi G, McMillian MK, Pizzo SV, Laskowitz DT. Apolipoprotein E and mimetic peptide initiate a calcium-dependent signaling response in macrophages. J Leukoc Biol. 2001;70:677–83.
Laskowitz DT, McKenna SE, Song P, Wang H, Durham L, Yeung N, Christensen D, Vitek MP. COG1410, a novel apolipoprotein E-based peptide, improves functional recovery in a murine model of traumatic brain injury. J Neurotrauma. 2007;24:1093–107.
MacLellan CL, Silasi G, Poon CC, Edmundson CL, Buist R, Peeling J, Colbourne F. Intracerebral hemorrhage models in rat: comparing collagenase to blood infusion. J Cereb Blood Flow Metab. 2008;28:516–25.
MacLellan CL, Silasi G, Auriat AM, Colbourne F. Rodent models of intracerebral hemorrhage. Stroke J Cereb Circ. 2010;41:S95–8.
Laskowitz DT, Fillit H, Yeung N, Toku K, Vitek MP. Apolipoprotein E-derived peptides reduce CNS inflammation: implications for therapy of neurological disease. Acta Neurol Scand Suppl. 2006;185:15–20.
Leira R, Davalos A, Silva Y, Gil-Peralta A, Tejada J, Garcia M, Castillo J. Early neurologic deterioration in intracerebral hemorrhage: predictors and associated factors. Neurology. 2004;63:461–7.
Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908.
Wasserman JK, Zhu X, Schlichter LC. Evolution of the inflammatory response in the brain following intracerebral hemorrhage and effects of delayed minocycline treatment. Brain Res. 2007;1180:140–54.
Fujimoto S, Katsuki H, Ohnishi M, Takagi M, Kume T, Akaike A. Thrombin induces striatal neurotoxicity depending on mitogen-activated protein kinase pathways in vivo. Neuroscience. 2007;144:694–701.
Ohnishi M, Katsuki H, Fujimoto S, Takagi M, Kume T, Akaike A. Involvement of thrombin and mitogen-activated protein kinase pathways in hemorrhagic brain injury. Exp Neurol. 2007;206:43–52.
Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem. 1996;271:11059–62.
Ivaska J, Nissinen L, Immonen N, Eriksson JE, Kahari VM, Heino J. Integrin alpha 2 beta 1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3 beta. Mol Cell Biol. 2002;22:1352–9.
Shanley TP, Vasi N, Denenberg A, Wong HR. The serine/threonine phosphatase, PP2A: endogenous regulator of inflammatory cell signaling. J Immunol. 2001;166:966–72.
Liu Q, Hofmann PA. Protein phosphatase 2A-mediated cross-talk between p38 MAPK and ERK in apoptosis of cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H2204–12.
Kray AE, Carter RS, Pennington KN, Gomez RJ, Sanders LE, Llanes JM, Khan WN, Ballard DW, Wadzinski BE. Positive regulation of IkappaB kinase signaling by protein serine/threonine phosphatase 2A. J Biol Chem. 2005;280:35974–82.
Singh K, Chaturvedi R, Asim M, Barry DP, Lewis ND, Vitek MP, Wilson KT. The apolipoprotein E-mimetic peptide COG112 inhibits the inflammatory response to citrobacter rodentium in colonic epithelial cells by preventing NF-kappa B activation. J Biol Chem. 2008;283:16752–61.
Singh K, Chaturvedi R, Barry DP, Coburn LA, Asim M, Lewis ND, Piazuelo MB, Washington MK, Vitek MP, Wilson KT. The apolipoprotein E-mimetic peptide COG112 inhibits NF-{kappa}B signaling, proinflammatory cytokine expression, and disease activity in murine models of colitis. J Biol Chem. 2011;286(5):3839–50.
Li FQ, Sempowski GD, McKenna SE, Laskowitz DT, Colton CA, Vitek MP. Apolipoprotein E-derived peptides ameliorate clinical disability and inflammatory infiltrates into the spinal cord in a murine model of multiple sclerosis. J Pharmacol Exp Ther. 2006;318:956–65.
McAdoo JD, Warner DS, Goldberg RN, Vitek MP, Pearlstein R, Laskowitz DT. Intrathecal administration of a novel apoE-derived therapeutic peptide improves outcome following perinatal hypoxic–ischemic injury. Neurosci Lett. 2005;381:305–8.
Acknowledgments
The authors would like to thank Jessica Oddo for technical assistance. This research was made possible through grants from R43NS064651-01A1 (DJC), the American Heart Association Scientist Development Grant (MLJ), and Foundation of Anesthesia Education and Research Mentored Research Training Grant (MLJ). COG1410 was provided by Cognosci, Inc. (Research Triangle Park, NC, USA).
Conflict of interest
Dr. Laskowitz served as a consultant for Cognosci, Inc., ending greater than 6 months ago. Dale Christensen served as a Research associate for Cognosci, Inc. Michael P. Vitek served as an Executive officer at Cognosci, Inc. BeiLei Lei, Hana N. Dawson, Haichen Wang, Steven Bellows, and Michael L. James declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Laskowitz, D.T., Lei, B., Dawson, H.N. et al. The apoE-mimetic Peptide, COG1410, Improves Functional Recovery in a Murine Model of Intracerebral Hemorrhage. Neurocrit Care 16, 316–326 (2012). https://doi.org/10.1007/s12028-011-9641-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-011-9641-5