
Abstract
Background
Release of cardiac biomarkers is reported in patients with subarachnoid hemorrhage (SAH). Data addressing the impact of cardiac injury on outcome in these patients is sparse. This study was conducted to ascertain the association of elevation of serum cardiac Troponin-I (cTnI) with mortality and neurological outcome in patients with SAH.
Methods
Medical records of all patients admitted with a diagnosis of SAH and at least one measured cTnI were reviewed. Demographic and clinical variables including admission neurological status were collected. Conservative and non-parametric statistics were used to assess association between cTnI and death or neurological outcome at discharge.
Results
The study group comprised of 83 patients with a mean age of 59 years. There was a female (60%) and African-American (60%) preponderance. At admission, the median Glasgow Coma Scale (GCS) was 9, and 47% had a severe Hunt–Hess grade (HHG) of ≥4. Elevation of cTnI was found in 31 (37%) patients and was associated with worse baseline Fisher grade (p=0.01) and neurological status: GCS score (p=0.006) and HHG (p=0.007). Patients with abnormal cTnI were more likely to die (55% vs.27%; odds ratio 1.3–8.4, p = 0.01) and had a worse GCS score (p = 0.008) and HHG (p = 0.004) on discharge. On multivariate analysis, peak cTnI (p = 0.04) and admission GCS score of <12 (p = 0.02) were independent predictors of death at discharge.
Conclusion
Patients with subarachnoid hemorrhage and elevated cTnI are found to have worse neurological status at admission. These patients have a worse neurological outcome and in-hospital mortality.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Subarachnoid hemorrhage (SAH) is a devastating condition with substantial morbidity and mortality despite current advances in therapy. It is associated with an estimated annual cost of $1.75 billion and has a 30-day mortality ranging from 30 to 52% [1]. Cardiac injury in patients with SAH has long been recognized. The clinical manifestation of cardiac injury is variable, ranging from an abnormal electrocardiogram [2, 3] and elevated cardiac biomarkers, to pulmonary edema [4] and cardiogenic shock [5]. The frequency of elevated serum troponin (cTnI) in SAH ranges from 20 to 40% (6). A previous study suggested that the etiology of cardiac injury in the majority of patients with SAH is due to reversible left ventricular systolic dysfunction rather than underlying obstructive coronary artery disease [7]. The mechanism of cardiac injury is thought to be secondary to a sudden surge in catecholamine levels [8–10] resulting in functional myocardial stunning [7] and focal contraction band necrosis. Therefore, neurocardiac injury is likely directly responsible for left ventricular dysfunction and elevated cardiac biomarkers [11].
Adverse cardiac and pulmonary outcomes in patients with SAH and an elevated cTnI have been previously reported [5]. However, the prognostic importance of elevated cTnI on neurological outcome and death in SAH has not been well studied. The current investigation was conducted to establish the frequency of cardiac injury and to study its association with neurological outcome and mortality in patients with SAH.
Methods
Study Population
A convenience cohort was randomly chosen from all patients admitted to our hospital between January 1999 and December 2003, with a diagnosis of SAH (ICD—430), established by CT scan, or by xanthochromia of the cerebrospinal fluid if the CT scan was non-diagnostic (n = 250). We reviewed the hospital records of all these patients and excluded from our study those patients with SAH due to trauma, arteriovenous malformation or other secondary causes, and those patients with no cTnI measurement(s) (n = 158). Also excluded were 8 outside referral patients whose computed tomography scans were not available, and 1 patient whose follow-up information was missing. The remaining 83 patients formed the study group. The study was approved by the University Institutional Review Board.
Study Parameters
Demographic information collected on the patients included age, sex, and race. Cardiac measures including cTnI, 12 lead electrocardiogram, transthoracic 2-dimensional echocardiogram, chest radiographs confirming the presence or absence of pulmonary edema, and blood pressure measurements were also collected. Left ventricular dysfunction was defined as an ejection fraction ≤50% and the echocardiograms were independently analyzed by a cardiologist naïve to clinical status. Hemodynamic instability was defined as any systolic pressure <90 mm Hg, a mean pressure of <65 and/or use of any ionotropic support during the course of hospitalization.
We obtained clinical and neurological status of the patients from the admission notes. Neurological measures included Glasgow Coma Scale (GCS), Hunt & Hess Grade (HHG), and Fischer grade on head CT scan. A neurologist blinded to the clinical status evaluated the head CT scan done at admission for presence of SAH, intra-cerebral and intra-ventricular hemorrhage, hydrocephalus and midline shift. The degree and severity of SAH was defined using the Fischer grade [12]. Data on hospital course, medical management, and any surgical procedures such as clipping or coiling were also collected. Clinical outcome and follow-up information were obtained from hospital records which included documented neurological assessments of GCS, HHG and presence or absence of stroke.
Statistical Analysis
Any value of cTnI ≥2.0 ng/ml was considered abnormal, based on the reference level provided by our laboratory. The majority of the patients had more than one cTnI measured during the course of their hospitalization. A patient was considered to have elevated cTnI if any of their values was ≥2 ng/ml. Comparison of positive and negative groups was done using chi-square statistics for the categorical variables and Wilcoxon rank sum statistics for the continuous variables. Predictions of death and stroke were assessed through logistic regression models. Elevated patients were divided into two groups based on the median maximum cTnI value, and rates of mortality were determined for these two groups as well as for the group having no elevated cTnI. SAS© software (version 9.1) was used.
Results
Patient Characteristics
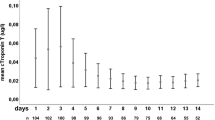
Our study population consisted of a total of 83 patients who were admitted with SAH and had at least one measured cTnI. Thirty-one patients (37%) had at least one elevated cTnI value while 52 patients (63%) had no elevated cTnI. Of the positive group, 18 patients had an abnormal value on the initial measurement, 12 from the second measurement and 1 from the third. Four patients (13%) had persistent elevation of cTnI. Troponin I was typically elevated early in the clinical course with median interval between SAH onset and peak cTnI level was 0 days. The mean maximum cTnI level in the group with SAH alone was 6.0 ng/ml and was somewhat higher at 9.0 ng/ml in patients with associated intra-ventricular hemorrhage or hydrocephalus. Of the 52 patients with a normal cTnI level, 24 (46%) had only a single measured value. Seven patients died before a second cTnI level was obtained. The time between the first cTnI measurement and death were: 0 days (2 patients); 1 day (3 patients); 2 days (1 patient); and 3 days (1 patient).
Table 1 provides characteristics of the total study population as well as the two subpopulations [patients with an elevated cTnI (Group A) and patients with measured but normal cTnI (Group B)]. The majority of the patients were African American (60%), women (60%) with a mean age of 58.7 years. Fifty-eight patients (69%) had a history of hypertension, 14% had documented underlying coronary artery disease (CAD) and 51% were smokers. The group was predominantly comprised of patients with a poor baseline neurological status as represented by the mean HHG of 3.2 and 47% had a very severe score of 4 or 5. Also, 23 and 22% of the patients had GCS score of 3 and 15 respectively; and the rest had scores between these two extremes.
Group A and Group B did not differ significantly by age, sex, or history of hypertension. Patients in Group A were mostly Caucasians without diabetes, and had a worse Fischer grade on head CT (p = 0.01). A higher HHG grade of >3 was also more prevalent in Group A than Group B (90% vs. 60%). There was no significant difference in prevalence of vasospasm on the trans-cranial Doppler study (42% vs. 50%; p = 0.47) between the two groups. Equal number of patients in both groups underwent either clipping or coiling.
An admission ECG was available in 77 patients and was abnormal in 71 (92%). The most common abnormality was QTc prolongation, occurring in 60% of the patients, which was not different between the two groups. However, abnormal ST depression was observed predominantly in Group A (29% vs. 6%; p = 0.016). Pulmonary edema on chest radiograph was also more prevalent in Group A (74% vs. 25%; p < 0.0001). The occurrence of left ventricular dysfunction tended to be more frequent in Group A and was found to be statistically significant. {7 patients (64%) vs. 2 patients (33%); p = 0.01}. Out of the 9 patients with LV dysfunction, 3 died.
Outcomes
The overall in-hospital mortality was 37%. Mortality was significantly higher in patients with abnormal cTnI (Group A 55% vs. Group B 27%; p = 0.01). Prediction of death was assessed through logistic regression models. Results from the univariate logistic regression models are given in Table 2. On univariate analysis, abnormal cTnI was a strong predictor of death during hospitalization (p = 0.005), as were admission GCS (p = 0.0003), HHG (p < 0.0001), and Fischer grade score on head CT (p = 0.0147). Patients with ST depression appeared more likely to die, but this trend did not reach statistical significance (p = 0.06). Results from the multiple logistic regression analysis are depicted in Table 3 which shows that after adjusting for the other covariates in the model, cTnI (odds ratio = 1.055; 95% CI 1.016–1.095; p = 0.0051) and admission GCS of <12 (Odds ratio 9.2; 95% CI 1.38–61.4; p = 0.02) remained as independent predictors of mortality. Odds ratio of 1.055 indicates that for each one-unit increase in maximum cTnI value, the likelihood of death increased by 5%. On further analysis, when patients were divided into tertiles of cTnI (Fig. 1), a direct, quantitative relationship between the degree of cTnI elevation and mortality could be observed. An increase in percentage of deaths was seen with increasing values of maximum cTnI.

Percentage of people who died based on tertile levels of Troponin
The overall stroke incidence was 23% with no significant difference between Groups A and B. Prediction of stroke was assessed through logistic regression models. Results from the univariate logistic regression models are given in Table 2 where it can be seen that having an admission HHG of 4 or 5 is significantly associated with increased likelihood of stroke. In the multiple logistic regression model (Table 3), both admission GCS and HHG showed statistical significance (p-values 0.002 and 0.04, respectively).
Discussion
The key, novel finding of the present study is that elevated cTnI in the setting of subarachnoid hemorrhage is a predictor of mortality during the initial hospitalization. Also, admission GCS score of <12 is an independent predictor of both in-hospital death and stroke at discharge. Furthermore, there is a direct, quantitative relationship between degree of cTnI elevation and mortality rate as seen by the astoundingly high mortality rate of 73% among patients in the highest tertile. Furthermore, this mortality correlation is independent of that provided by other traditional cardiac investigations.
Release of cardiac biomarkers in primarily non-cardiac conditions is usually due to loss of membrane integrity of cardiac myocytes not associated with fixed, flow-limiting coronary stenosis. It may reflect necrosis or apoptosis of myocytes due to oxygen supply/demand mismatch or oxidative stress. Circulating cytokines in the setting of septic shock are thought to mediate the cardiac injury. SAH produces an intense sympathetic discharge [13] that may produce myocardial necrosis and cTnI elevation. In animal models of SAH, myocardial injury with resultant global myocardial dysfunction in absence of coronary occlusion has been well described [14, 15]. Also, elevation of cardiac biomarkers has been reported in other intra-cranial conditions including ischemic stroke and is associated with poor prognosis [16].
There is limited information about the association and pattern of cardiac biomarker release and prognosis in patients with SAH. The 37% frequency of cTnI release in our SAH patients is comparable to the range of 20–40% reported in previous studies [6]. In a recent substudy of the Columbia University SAH Outcomes Project, Naidech et al. reported a 42% incidence of cTnI release in SAH [17]. The median interval between SAH onset and peak cTnI in our study is 0 days, and is different from that reported by Naidech et al. [17] where peak cTnI was detected later. This pattern of cTnI release is clearly different from that observed in acute coronary syndrome, thereby suggesting mechanisms other than coronary occlusion leading to this injury. Our findings confirm the previous observation [17] that these patients with SAH and cTnI release tend to have higher Fischer grade on head CT; thus they are likely to have intra-ventricular hemorrhage and hydrocephalus.
We observed a strong relationship between peak cTnI levels and mortality, with each one-unit increase in maximum cTnI value being associated with a 5% increased likelihood of in-hospital death. Indeed, a peak cTnI level of ≥9.5 ng/ml conferred an alarmingly high in-hospital mortality of 73%. Patients with an elevated cTnI were also more likely to have a worse neurological outcome. This finding is similar to that of Naidech et al., where cTnI elevation after SAH was associated with an increased risk of cardiopulmonary complications, delayed cerebral ischemia, and death or poor functional outcome at discharge.
Data addressing the role of other diagnostic cardiac testing in patients with SAH and prognosis is indiscriminate. Previous studies have documented the failure of specific ECG changes in predicting outcome [2, 3, 18]. Similar results were obtained in our study, where ST depression had a statistically insignificant trend in predicting mortality. Based on our results, we propose that evaluation of cardiac injury using serum biomarkers should be pursued irrespective of ECG changes. Determination of left ventricular ejection fraction with an echocardiogram in patients with SAH may provide useful clinical information but appears to be of limited short-term prognostic value. Left ventricular dysfunction in SAH is usually transient and associated with cTnI release [5, 6, 19, 20]. In our study, left ventricular dysfunction was present in more than half (52%) of the patients, but did not correlate with the extent of cTnI release. In addition, the degree of left ventricular dysfunction also did not predict mortality.
The fundamental limitation of the study is the selection bias. Of 250 patients with SAH during the study period, only 92 had cTnI measured. The clinical reasons prompting cTnI measurement in this group of patients could not be determined due to the retrospective design of the study. Presumably, there was no clear indication for ordering routine cTnI measurement, and it was performed only when a reasonable suspicion of cardiac dysfunction was present (e.g., abnormal ECG in 92% of patients), and/or in patients with poor neurological grade which is supported by the poor median clinical grade in our study population. Thus, this study is likely to over-represent the incidence of elevated cTnI. In addition, the effect of lack of repeat cTnI determination in those few patients, who succumbed to the illness and had an initial normal level, cannot be determined. Furthermore, the study population comprised mostly patients with a poor clinical grade: mean H&H of 3.4 and 47% of patients with H&H grades 4–5. Due to the retrospective nature of the study, it is possible that those with good neurological grades were excluded due to no cTnI measurements. In that scenario our study probably over represents the high incidence of elevated cTnI in high-grade patients.
Conclusion
In the setting of SAH, elevation of cTnI is common. Substantial elevation of cTnI carries an ominous prognosis, and cTnI is more informative about prognosis than other cardiac measures including electrocardiogram and echocardiography. It does not usually reflect an acute coronary thrombosis and myocardial infarction requiring coronary intervention, but is a sequel of the subarachnoid hemorrhage.
References
Marti-Fabregas J, Belvis R, Guardia E, Cocho D, Munoz J, Marruecos L, Marti-Vilalta JL. Prognostic value of Pulsatility Index in acute intracerebral hemorrhage. Neurology 2003;61(8):1051–6.
Zaroff JG, Rordorf GA, Newell JB, Ogilvy CS, Levinson JR. Cardiac outcome in patients with subarachnoid hemorrhage and electrocardiographic abnormalities. Neurosurgery 1999;44(1):34–9, discussion 39–40.
Sommargren CE, Zaroff JG, Banki N, Drew BJ. Electrocardiographic repolarization abnormalities in subarachnoid hemorrhage. J Electrocardiol 2002;35 Suppl:257–62.
Mayer SA, Fink ME, Homma S, Sherman D, Limandri G, Lennihan L, Solomon RA, Klebanoff LM, Beckford A, Raps EC. Cardiac injury associated with neurogenic pulmonary edema following subarachnoid hemorrhage. Neurology 1994;44(5):815–20.
Parekh N, Venkatesh B, Cross D, Leditschke A, Atherton J, Miles W, Winning A, Clague A, Rickard C. Cardiac troponin I predicts myocardial dysfunction in aneurysmal subarachnoid hemorrhage. J Am Coll Cardiol 2000;36(4):1328–35.
Deibert E, Aiyagari V, Diringer MN. Reversible left ventricular dysfunction associated with raised troponin I after subarachnoid haemorrhage does not preclude successful heart transplantation. Heart 2000;84(2):205–7.
Kono T, Morita H, Kuroiwa T, Onaka H, Takatsuka H, Fujiwara A. Left ventricular wall motion abnormalities in patients with subarachnoid hemorrhage: neurogenic stunned myocardium. J Am Coll Cardiol 1994;24(3):636–40.
Masuda T, Sato K, Yamamoto S, Matsuyama N, Shimohama T, Matsunaga A, Obuchi S, Shiba Y, Shimizu S, Izumi T. Sympathetic nervous activity and myocardial damage immediately after subarachnoid hemorrhage in a unique animal model. Stroke 2002;33(6):1671–6.
Zaroff JG, Rordorf GA, Ogilvy CS, Picard MH. Regional patterns of left ventricular systolic dysfunction after subarachnoid hemorrhage: evidence for neurally mediated cardiac injury. J Am Soc Echocardiogr 2000;13(8):774–9.
Zaroff JG, Rordorf GA, Titus JS, Newell JB, Nowak NJ, Torchiana DF, Aretz HT, Picard MH, Macdonald RL. Regional myocardial perfusion after experimental subarachnoid hemorrhage. Stroke 2000;31(5):1136–43.
Zaroff JG, Pawlikowska L, Miss JC, Yarlagadda S, Ha C, Achrol A, Kwok PY, McCulloch CE, Lawton MT, Ko N, Smith W, Young WL. Adrenoceptor polymorphisms and the risk of cardiac injury and dysfunction after subarachnoid hemorrhage. Stroke 2006;37(7):1680–5.
Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980;6(1):1–9.
Banki NM, Kopelnik A, Dae MW, Miss J, Tung P, Lawton MT, Drew BJ, Foster E, Smith W, Parmley WW, Zaroff JG. Acute neurocardiogenic injury after subarachnoid hemorrhage. Circulation 2005;112(21):3314–9.
Elrifai AM, Bailes JE, Shih SR, Dianzumba S, Brillman J. Characterization of the cardiac effects of acute subarachnoid hemorrhage in dogs. Stroke 1996;27(4):737–41, discussion 741–2.
Sato K, Masuda T, Izumi T. Subarachnoid hemorrhage and myocardial damage clinical and experimental studies. Jpn Heart J 1999;40(6):683–701 .
Di Angelantonio E, Fiorelli M, Toni D, Sacchetti ML, Lorenzano S, Falcou A, Ciarla MV, Suppa M, Bonanni L, Bertazzoni G, Aguglia F, Argentino C. Prognostic significance of admission levels of troponin I in patients with acute ischaemic stroke. J Neurol Neurosurg Psychiatry 2005;76(1):76–81.
Naidech AM, Kreiter KT, Janjua N, Ostapkovich ND, Parra A, Commichau C, Fitzsimmons BF, Connolly CS, Mayer SA. Cardiac troponin elevation, cardiovascular morbidity, and outcome after subarachnoid hemorrhage. Circulation 2005;112(18):2851–6.
Sakr YL, Ghosn I, Vincent JL. Cardiac manifestations after subarachnoid hemorrhage: a systematic review of the literature. Prog Cardiovasc Dis 2002;45(1):67–80.
Birns J, Sabharwal N, Senior R. Diagnosis of subarachnoid hemorrhage indicated by transthoracic echocardiography. J Am Soc Echocardiogr 2003;16(9):995–8.
Inoue F, Tsuzuki T, Thoma Y, Shiono S, Tabuse H, Hoshida T, Saito Y. Subarachnoid hemorrhage complicated with different manifestations of transient abnormal left ventricular wall motion: two case reports. J Cardiol 2006;47(5):245–54.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ramappa, P., Thatai, D., Coplin, W. et al. Cardiac Troponin-I: A Predictor of Prognosis in Subarachnoid Hemorrhage. Neurocrit Care 8, 398–403 (2008). https://doi.org/10.1007/s12028-007-9038-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-007-9038-7