
Abstract
As our understanding of the pathogenesis of autoimmune diseases is growing, new therapies are being developed to target disease-specific pathways. Since the introduction of etanercept in 1998, several biotechnological agents have been developed, most of them indicated in the treatment of rheumatoid arthritis, but also psoriatic arthritis. Most currently available molecules target TNF-alfa with different strategies (i.e., etanercept, infliximab, adalimumab, golimumab, and certolizumab pegol), IL-6 (tocilizumab), CTLA-4 (abatacept), and B cells (rituximab, belimumab) as they are key mediators in the cascade of inflammation. Further, small molecules have been recently developed to target intracellular signaling, such as Janus Kinases for tofacitinib, the first FDA-approved small molecule for rheumatoid arthritis. Most novel treatments are being developed for arthritis with specific differences between rheumatoid and psoriatic arthritis, as well as for systemic lupus erythematosus, following the approval of belimumab. Finally, biologic therapies are effective also in gout, mainly targeting interleukin-1 to block the inflammasome. This review article describes the new and upcoming treatment options for rheumatoid arthritis, psoriatic arthritis, systemic lupus erythematosus, and gout to dissect what we should be aware of when discussing these new and promising molecules.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The similarities across autoimmune diseases outnumber differences, particularly when treatment options are compared. Indeed, the therapeutics of autoimmune diseases are based on a growing armamentarium that includes monoclonal antibodies against cytokines or other mediators and now novel small molecules which act by targeting intracellular mediators with high specificity [1]. Nevertheless, a significant number of patients with paradigmatic inflammatory rheumatologic conditions, such as rheumatoid arthritis (RA), psoriatic arthritis (PsA), systemic lupus erythematosus (SLE), and gout arthritis, manifest an active disease with an incomplete response to the treatment, despite being treated with the currently available molecules. Further, important concerns remain about safety profiles of targeting pro-inflammatory mediators, while less obvious targets may hold important beneficial implications in the management of metabolic or inflammatory comorbidities.
Indeed, chronic inflammation is the main common trait of rheumatologic diseases, independent of the autoimmune (as in RA and SLE), chronic inflammatory (PsA), or metabolic (gout) bases. We are particularly intrigued by new treatment options that will be critically discussed in the present article. Furthermore, treatments may prove beneficial in different conditions but will be discussed in a disease-specific classification. This is indeed an ambitious goal due to the large number of molecules, the rapidly changing scenarios, the often partial reports at meetings rather than full articles, and the industrial need for secrecy.
The clinical epidemiology of rheumatic diseases
Before discussing the new treatment options for rheumatic diseases, their epidemiology, pathogenesis, and clinical features should be briefly reviewed.
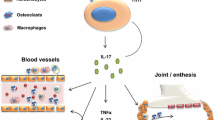
First, RA is an autoimmune systemic disease affecting approximately 0.5–1 % of the general population, with a striking predominance of women in their fourth–fifth decade of life. RA is a paradigmatic multifactorial disease, where genetics, risk factors (especially smoking), and autoimmunity contribute to the clinical manifestations of the disease. RA is characterized by symmetrical articular involvement, persistent synovitis, systemic inflammation, and specific serum autoantibodies (namely rheumatoid factor and antibodies against citrullinated peptides) [2]. RA can lead to joint deformity, global disability, and low quality of life, while significantly increasing the atherosclerosis risk, leading to cardiovascular events being the main cause of death in these patients [3, 4]. In 2010, the American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR) reported new classification criteria for early rheumatoid arthritis [5], which assess joint involvement, autoantibody status, and acute-phase response and symptoms duration. The pathogenesis of RA is based on chronic inflammation, that is initiated when a T cell is activated by an endogenous trigger, which, in turn, activates macrophages and fibroblasts to produce cytokines (particularly, but not limited to TNF-alfa, IL-6, IL-17, IL-1, IL-23), and then angiogenesis ensues and inflammatory cells concentrate in the inflammatory area, making inflammation permanent. Both B and T lymphocytes are involved in RA pathogenesis, and chronic inflammation ultimately leads to tissue remodeling and joint damage [2].
Second, PsA is a chronic inflammatory, seronegative oligoarthritis, which may also include an axial involvement, with inflammatory back pain. It affects nearly 40 % of patients with skin psoriasis, and it is more common in people carrying the HLA-B27 allele [6], while equally affecting men and women [7]. PsA is clinically characterized by an asymmetrical oligoarthritis and, differently from RA, dactylitis, enthesitis, and in some cases inflammation of the sacroiliac joints [8]. The case identification is based on the CASPAR classification criteria [9]. In PsA, the immune response is mediated by T and B lymphocytes that are present both in the enthesis and in the skin. In the last years, innate immunity has also developed a role in PsA pathogenesis, and NK cells may be a trigger that drives toward the immune-mediated disease [10–12]. Several cytokines are involved in PsA development: First, PsA was thought to be only a Th1-mediated disease, but the discovery of IL-17 and Th17 cells changed this view about disease pathogenesis [13] and IL-23 is now considered as important in enthesis inflammation, bone destruction, and ultimately joint damage [14].
Third, SLE is a prototypical chronic, multiorgan, autoimmune disease characterized by a wide range of clinical manifestations: from skin rash to anemia and lymphopenia, arthritis, nephritis, serositis, and central nervous system involvement. It predominantly affects women between 15 and 50 years of age [15]. Serum autoantibodies can be detected, particularly anti-double-stranded DNA (anti-dsDNA), which titer, differently from other serum autoantibodies, mirror disease activity [16]. T cells also play a role in SLE pathogenesis leading to B cell stimulation, when an antigen-presenting cell (APC) is activated by a trigger. Additionally, co-stimulatory molecules interact in the APC–T cell stimulation process; these include CD40–CD40 ligand and CD28–B7 which allow the T cell to activate, or to suppress activation. T cells produce cytokines (mainly TNF-alfa, IL-10, IFN-gamma) that stimulate autoantibodies production by B cells [17], but TNF-alfa may have a protective role toward SLE, based on controversial evidence. Among other cytokines, interleukin 10 (IL-10) and interferon-alfa levels are elevated in patients with SLE and correlate with disease activity [18]. B lymphocyte stimulator (BlyS) is a part of the TNF-ligand family and promotes the activation and proliferation of B cells, being present at high levels in the sera of patients with active SLE, but also in other rheumatic conditions, such as Sjogren’s syndrome and RA [19–23]. BLyS levels also correlate with disease activity and anti-dsDNA levels [24], these observations cumulatively led to the development of belimumab, an anti-BLyS monoclonal antibody, and its approval for SLE treatment.
Finally, gout is primarily a metabolic disease characterized by recurrent acute arthritis caused by urate crystals deposition in the joints. The disease affects up to 4 % of the population of the USA [25], more commonly men about 50 years of age and women exclusively after menopause. Hyperuricemia is an asymptomatic chronic condition that predisposes to gout, particularly with serum uric acid levels higher than 7 mg/dL (380 mmol/l), but during acute arthritis these may be significantly lower. The most commonly involved joint is usually the first metatarsophalangeal (MTP) joint. The definitive diagnosis is made with the aspiration of the joint fluid and the microscopic analysis showing urate crystals [26]. Current treatments are directed at solving the acute arthritis attack and to lower uric acid levels with different approaches.
Current and upcoming treatments for RA
RA treatment options have dramatically changed since the introduction of biologic agents over a decade ago. Currently, nine biologic agents are approved for RA treatment: abatacept, adalimumab, anakinra, certolizumab pegol, etanercept, golimumab, infliximab, rituximab, and tocilizumab. Most recently, the US Food and Drugs administration approved also the first small molecule, tofacitinib, for the treatment of RA. The main targets in RA treatment include TNF-alfa, but also B cells, IL-6, IL-1, and CD80/86. Traditional disease modifying anti-rheumatic drugs (DMARDs) and corticosteroids still remains crucial in the management of RA [27]. Another important aspect is the biologic agents expiring patents that will lead to biosimilars, drugs similar to the originator biological molecule, such as CTP13, to enter the market [28]. Novel treatment targets for RA include IL-17, IL-12/23, IL-15, oncostatin, and chemokines, while new molecules target both current targets such as B cells, IL-6, IL-1, but also protein kinases [29] (Tables 1, 2, 3).
Among molecular targets, the Th17 pathway in general and IL-17 in particular are involved in RA-associated bone destruction and joint damage [13]. IL-17 is the target of secukinumab, ixekizumab, and brodalumab. Secukinumab is a fully human IgG1 k anti-IL-17A monoclonal antibody, effective in the induction and maintenance therapy of moderate-to-severe plaque psoriasis (as will be discussed below). On the other hand, secukinumab failed a randomized controlled trial in RA, leading to rates of American College of Rheumatology 20 % (ACR20) response at week 16 not significantly different from placebo at monthly doses of 25, 75, 150, or 300 mg. Nonetheless, the study showed a clinically relevant decrease in the Disease Activity Score (DAS28), C-reactive protein (CRP), and a significant reduction in high-sensitivity CRP levels [30]. More recently, a 52-week study assessed secukinumab longer-term safety and efficacy and the authors reported that patients with active RA, who failed to respond to DMARDs and other biologic agents, had an improvement in RA symptoms after long-term treatment with secukinumab 150 mg/month. The incidence of adverse events (AE) was stable throughout the duration of the study [31]. Ixekizumab is a humanized anti-IL-17A IgG4 monoclonal antibody, firstly evaluated on RA patients on a stable dose of DMARDs. Ixekizumab has showed efficacy in all treatment groups compared with placebo [32]. A subsequent follow-up trial showed significant dose-related improvement in RA symptoms both in biological DMARD-naıve patients and in anti-TNF-alfa inadequate responders [33]. Ixekizumab is now undergoing a Phase III trial in RA, but, similar to secukinumab, its development is more focused on psoriasis and psoriatic arthritis. Brodalumab (AMG-827) is a fully human anti-IL-17RA IgG2 monoclonal antibody that binds to IL-17RA on circulating leukocytes and inhibits Il-17-mediated signaling. The safety profile of brodalumab in RA was recently reported with AE in 77 % of patients treated with brodalumab, and 3 serious AE [34]. In the same study, even though effectiveness was not the endpoint, brodalumab did not show any clinical improvement when compared with placebo. As for secukinumab and ixekizumab, the development plan for brodalumab is more focused on psoriasis and psoriatic arthritis. Among other Th17-related therapeutic targets, apilimod mesylate is an IL-12/IL-23 inhibitor, which is prepared as a daily oral drug. In an intermediate report of a Phase II trial, it failed to induce a meaningful clinical improvement in RA [35].
As studies and the efficacy of rituximab have shown, B cells play also a central role in RA pathogenesis [36, 37] and new molecules are being tested or have already completed trials to assess their efficacy in RA. Ocrelizumab is a humanized anti-CD20 monoclonal antibody [38] that underwent several trials to evaluate the efficacy and safety in a broad spectrum of patients, with different diseases. Higher AE rates have been shown in the highest dosage in combination with methotrexate, but the lowest dosage group did not show any significant benefit [39–43], so the development of ocrelizumab was terminated in 2010. Ofatumumab is a humanized anti-CD-20 monoclonal antibody that recognizes a unique membrane proximal epitope, different from the epitope recognized by rituximab and ocrelizumab. This may account for higher efficacy compared with other CD-20 blocking molecules. In the first clinical trials, iv and sc ofatumumab significantly improved all clinical outcomes in RA without determining immunogenicity at week 24 nor unexpected safety issues [44–46]. Ofatumumab is currently undergoing a Phase III trial. Belimumab, the anti-BLyS monoclonal antibody recently approved for SLE treatment, has completed a Phase II trial to evaluate efficacy, safety, and tolerability in RA. The results showed efficacy and safety in RA patients who previously failed DMARDs therapies [47]; indeed, belimumab may hold promises for the future of RA treatment. Atacicept is a human recombinant fusion protein that inhibits the B cell-stimulating factors: proliferation-inducing ligand (APRIL) and BLyS. Despite promising results in preclinical and Phase I studies [48], the results from two randomized, placebo-controlled, phase II trials suggest that atacicept is not superior to placebo. Nonetheless, atacicept showed a clear biological effect: It reduced Ig and rheumatoid factor but not anti-citrullinated peptide antibodies levels in a dose-dependent manner [49, 50]. Comparable results were obtained with atacicept in MTX inadequate responders. Tabalumab is a fully human IgG4 monoclonal antibody neutralizing soluble and membrane-bound BAFF. A Phase II trial showed that tabalumab significantly reduces RA signs and symptoms and also has a similar safety profile compared with placebo [51–53], and the development of tabalumab in RA was discontinued.
IL-6 is a key mediator of the inflammation in RA, and tocilizumab, an humanized anti-IL-6 receptor, is the only IL-6 blocking therapy currently approved for RA [54]. Sarilumab is fully human anti-IL-6 receptor α (Rα) monoclonal antibody which completed a Phase III trial [55]. Early reports show that sarilumab is effective in moderate-to-severe RA, with a safety profile similar to the other IL-6 inhibitors. Sirukumab is a human anti-soluble IL-6 monoclonal antibody for which a Phase II trial showed improvement in signs and symptoms of RA with a safety profile similar to other IL-6 inhibitors [56].
IL-1 [57], IL-15 [58], and oncostatin [59] are additional candidates as new targets for RA treatment. IL-1 is a crucial molecule in the pathogenesis of RA [60]. Anakinra is an IL-1 receptor antagonist, which, according to a Cochrane review, is modestly effective and relatively safe, although injection site reactions are significantly increased compared with placebo [61]. As cardiovascular disease is the major cause of death in RA [3], inhibiting IL-1, which increases nitrooxidative stress, could help reducing RA mortality. But due to the lack of solid evidence about its efficacy and safety, anakinra was not included in the 2012 ACR recommendations [62], even though it appears to improve the cardiovascular function and metabolic panel in RA [63, 64]. Newer molecules are in development to target IL1, including canakinumab, a fully human anti-IL-1beta monoclonal antibody [65]. In a phase II dose-finding study investigating efficacy and safety in patients with active RA, despite ongoing therapy at stable doses of methotrexate [66], canakinumab improved the therapeutic responses among patients with active RA. HuMax-IL-15 is a human IgG1 anti-IL-15 monoclonal antibody, which, in vitro, suppresses the proliferation and induces apoptosis of an IL-15-dependent cell line [67]. In a Phase II trial, it was shown to be safe and well tolerated [68]. Finally, GSK315234A is a monoclonal anti-oncostatin-M (OSM) antibody, which does not bind to IL-6 [69]. In a Phase II trial, GSK315234A showed limited clinical efficacy, possibly due to low binding affinity dose, but pharmacological and biologic effects, but also a dose-dependent decrease in platelets, which was not considered to be clinically relevant nor a safety issue.
Chemokines have been indicated in the last years as potential treatment targets for different autoimmune diseases [70], but data in RA have been disappointing (Table 3), likely due to the redundancy between chemokines and their receptors and in most cases led to the termination of development programs.
Most recently, small molecules have been developed (Table 2) to target intracellular signaling. In general terms, these differ from monoclonal antibodies because they are administered orally and act intracellularly, usually on specific kinase signaling, such as the four members of the Janus Kinases (JAK) family [71]. Targeting one or more JAK, small molecules regulate the downstream shared functions of different intracellular structures such as surface receptors, signaling proteins, and transcription of nuclear proteins. Small molecules against the JAK/STAT signaling pathway are the most advanced in the development process [29] with tofacitinib, an inhibitor of JAK1/JAK3, and to a lesser extent JAK2, which has been FDA-approved to treat adults with moderately to severely active RA who are inadequate responders of intolerant to methotrexate. A recent systematic review and meta-analysis confirmed that tofacitinib induces a significant improvement of ACR 20/50/70 response criteria when compared with placebo after 12 weeks, superior to adalimumab, without additional safety concerns [72]. In the phase II, III, and LTE trials of tofacitinib, tuberculosis (TB) was reported in 12 cases (10 in endemic countries), and non-disseminated herpes zoster, anemia, thrombocytopenia and neutropenia, and hypercholesterolemia were also reported with increased frequency. There may be a possible increased cancer risk, because of the blockade of interferons and NK cells. Lymphomas or other lymphoproliferative disorders had similar rate compared with other biologic agents and the general RA population [29]. Baricitinib is an oral JAK1/JAK2 inhibitor, currently undergoing Phase III trials in RA, following a Phase IIb trial that showed statistically significant improvements in RA signs and symptoms compared with placebo, and also a prolonged response rate during 12 additional weeks of blind treatment. More importantly, the clinical improvements were sustained throughout 52 weeks. The safety profile showed no opportunistic infections, no tuberculosis reactivation, or lymphomas, but one death attributed to presumed myocardial infarction was observed [73]. Ruxolitinib is a JAK1/JAK2 inhibitor currently in Phase II for RA, originally developed for myeloproliferative disorders [74]. VX-509 inhibits JAK3, and GLPG0634 inhibits JAK1; both molecules are under investigation in Phase II trials.
Additional small molecules that are being investigated are directed against the fourth JAK (Syk) in the case of fostamatinib, phosphodiesterase-4 (PDE4, Apremilast), and MAPK p38 (VX-702, SCIO-467, BMS-582949, PH-797804). Apremilast has been recently approved by the FDA for the treatment of psoriatic disease [75–79], as will be discussed below. It has shown potent anti-inflammatory action in several animal models of inflammation [80], as PDE4 is one of the major phosphodiesterases expressed in leukocytes. In vitro, apremilast inhibits the production of inflammatory mediators such as TNF-alpha, IL-12, IL-2, IFN-gamma, and IL-5. Whether apremilast may prove beneficial in RA remains to be determined.
Finally, fibroblasts are the latest treatment target in RA, based on their role in the synovial tissue, leading to bone erosion and cartilage destruction mediated by the secretion of RANKL [81]. Fibroblasts may be more specific targets, potentially with less systemic AE, due to the lack on immunosuppression. Novel therapies will likely target fibroblast survival, as studies have shown that cyclin-dependent kinases (CDK) control cellular proliferation and that CDK levels, especially p21, are low in RA fibroblasts [82]. Adenovirus-mediated p21 gene transferring is being evaluated in early trials in mice and rats, and preliminary data show that inflammatory cytokines in the synovial tissue were inhibited [83]. Second, cadherin-11 might become a potential target; it mediates adhesion between lining layer fibroblasts. Its levels are elevated in RA tissues, and as Kiener et al. [84] have shown, it promotes cartilage invasion. Third, fibroblast activation protein (FAP) is associated with RA [85]. FAP is an enzyme with dipeptidylpeptidase 4 activity. In a trial in mice through FAP inhibition increased cartilage invasion [86]. Fourth, mesenchymal stem cells (MSC) and fibroblast-like synoviocytes (FLS) normally control immune responses, but in RA this function is interrupted. Further investigations are needed to assess if MSC are a valid therapeutic target in RA [87].
Current and upcoming treatments for PsA
The treatment of psoriasis and PsA has changed dramatically in the recent years, despite the largely incomplete understanding of the disease pathogenesis and etiology [88]. Indeed, the 2012 EULAR Psoriatic Arthritis Management Recommendations [89] support the use of biologic agents if a traditional DMARD has failed or is not well tolerated, or as first line in the presence of dactylitis, enthesitis, or axial involvement. The main therapeutic target in PsA is TNF-alpha: Anti-TNF-alpha inhibitors (adalimumab, certolizumab pegol, etanercept, golimumab, and infliximab) have demonstrated efficacy for both skin and joint manifestations [90], but a subgroup of patients continues to manifest a largely incomplete response, thus justifying the need for new treatments.
New therapeutic targets in PsA include B cells, T cells, IL-12/23, IL-17, PDE4, and JAK (Table 4). Even though PsA is not a B cell-mediated disease, CD19 + cells are present in the affected synovia. As such, rituximab has been used to treat refractory PsA, but there are no Phase III data to support its use [91]. Similarly, abatacept inhibits the co-stimulation of T cells by binding to CTLA4, and additionally case reports suggest that abatacept induces clinical improvement in PsA [92–95], along with one phase II RCT [96]. Ustekinumab is a human monoclonal antibody that binds the p40 subunit shared by IL-12 and IL-23. These cytokines appear central to the disease effector mechanisms [97, 98] and genetic susceptibility [99, 100]. It is currently approved treating skin psoriasis, based on a significant and rapid effectiveness, and data from the PSUMMIT 1 and 2 studies are very encouraging also for PsA [101]. More importantly, the Phase III trial dalimiteeta showed that ustekinumab reduces joint radiological progression and improves the signs and symptoms of PsA also in patients previously treated with TNFalpha inhibitors [102, 103]. Downstream to IL-12 and IL-23, IL-17 is family of pro-inflammatory cytokines of which IL-17A appears most involved in psoriasis and PsA pathogenesis [13, 104]. Secukinumab is a fully human anti-IL-17A monoclonal antibody with significant effectiveness in Phase II studies on psoriasis. In the case of PsA, secukinumab did not achieve the primary endpoint in a phase II trial, but the rapid clinical, serological, and quality of life improvements led to the ongoing Phase III trials in both conditions [105]. The results of two Phase III trials on psoriasis were recently published and showed that secukinumab is effective in the treatment of psoriasis [106]. Ixekizumab is a humanized anti-IL-17A monoclonal antibody, for which there are no data in PsA, but Phase II trials in psoriasis reported improved symptoms, and a Phase III trial is ongoing. Brodalumab is a fully human anti-IL-17RA monoclonal antibody, which demonstrated a good response in PsA compared with placebo at two different dosages (140 and 280 mg) subcutaneously every 2 weeks [107].
Apremilast is an oral PDE4 inhibitor [79], which has been recently approved by the FDA for PsA following a phase III trial with significant improvements in the disease clinical features and the related functional ability [76]. The Psoriatic Arthritis Long-Term Assessment of Clinical Efficacy (PALACE) program is studying the effect of apremilast on sign and symptoms of PsA in 4 trials, and preliminary results have confirmed the clinical efficacy and safety of apremilast [76]. Of note, apremilast is being investigated in ankylosing spondylitis, Behçet disease, and RA.
JAK inhibitors may be promising that also in PsA [75], a Phase III study of tofacitinib is ongoing for moderate-to-severe plaque psoriasis.
Current and upcoming treatments for SLE
Current SLE treatment options include mostly corticosteroids, hydroxychloroquine, and immunosuppressive drugs, such as cyclophosphamide, azathioprine, and mycophenolate mofetil. Belimumab is the first and only biologic agent approved for SLE, and it acts inhibiting the B lymphocyte stimulator (BLyS). Belimumab increases complement levels, while reducing anti-dsDNA antibody titers and activated B cells [108]. Based on these data, old and new biologic agents are being investigated to target B cells, co-stimulatory molecules, and IL-6 (Table 5). Rituximab a chimeric anti-CD-20 monoclonal antibody failed the primary endpoints in clinical trials [109]. Albeit off-label, rituximab remains recommended by EULAR for refractory SLE nephritis [110]. Ocrelizumab is a humanized anti-CD20 monoclonal antibody, which was evaluated in two phase III RCT, and achieved similar results to rituximab, but was stopped due to the incidence of infectious AE [111]. Epratuzumab is a humanized monoclonal antibody that activates the CD22 regulatory pathway on B cells [112]. Trials were terminated because of drug supply issues although the drug was found effective in improving the British Isles Lupus Assessment Group (BILAG) activity scores and reducing the need for steroids. Atacicept is a soluble fully human recombinant anti-APRIL fusion protein, which reduces B cells and Ig levels [113]. In a 76-week trial, patients with SLE were randomized to receive atacicept 75 mg or 150 mg subcutaneously or placebo twice weekly for 4 weeks, then weekly for 48 weeks. The enrollment in the atacicept 150 mg group was terminated because of two deaths, while there were no differences between atacicept 75 mg and placebo [114]. Abatacept [115], a fusion protein that inhibits the co-stimulation of T cells by binding to CTLA4, was tested in a phase IIb trial with patients with non-life-threatening manifestations randomized to receive abatacept 10 mg/kg or placebo plus prednisone 30 mg/day for 1 month. The primary and secondary endpoints were not met, along with adverse events occurring with similar frequency in both treatment groups [116]. Tocilizumab is an anti-IL-6R monoclonal antibody, which is highly effective in RA as monotherapy. In an open-label phase I trial, patients randomized to receive tocilizumab at different dosages had a lower disease activity, but the highest dosage (8 mg/kg) was associated with neutropenia [117]. Another IL-6 inhibitor, sirukumab, has been investigated to assess safety profile and pharmacokinetic in healthy subjects [118]. Small molecules, such as JAK inhibitors, may have a role in SLE, according to preclinical studies [119]. IL-12/IL-23 (ustekinumab) and IL-17 (secukinumab, brodalumab, ixekizumab) may be promising targets also in SLE, since increased levels of IL-17 have been observed in neutrophils from patients with SLE [104], but no data are now available. Peptide therapies may prove beneficial based on the use of tolerogenic T cells epitopes, which modulate only the cell that are involved in SLE pathogenesis, without interfering with other cell types. Two peptides, P140 and hCDR1, were tested in clinical trials and demonstrated a good safety profile and tolerability [120] and are currently being investigated.
Current and upcoming treatments for gout
Biotechnological therapies are being developed also for gout, mainly because of better understanding of the disease effector mechanisms. The current treatment is based on lowering uric acid levels and reducing the pain and inflammation during the acute arthritis attacks mostly by colchicine, NSAIDs, and steroids [121]. Gout is now considered as an adult autoinflammatory disease based on the involvement of the inflammasome and IL-1, which interacts with monosodium urate crystals. Anakinra, canakinumab and rilonacept target IL-1beta and have been developed mostly for pediatric forms of autoinflammation (Table 6). One small pilot study with anakinra showed that it might be effective in gout and it is well tolerated [122], but evidences are lacking. On the other hand, canakinumab has demonstrated promising results in acute gout arthritis trials in patients refractory or in which NSAIDs or steroids were contraindicated [123, 124]. It may hold promises in a niche of patients with uncontrolled disease and recurrent arthritis attacks. In one of the first clinical RCT, double-blind trials, canakinumab was given at different dosages, and compared with triamcinolone acetonide, the results showed that canakinumab 150 mg provides sustained pain relief in acute gout arthritis and reduces the risk of attacks recurrence [125, 126]. Rilonacept is a fusion protein designed to bind IL-1 before it interacts with the cell surface and initiates inflammation. A phase III trial showed significant reductions in acute gout arthritis when it was started associated with allopurinol [127]. The results of a Phase III safety trial have recently been published and show that rilonacept 160 mg sc weekly has a safe profile [128]. An additional new anti-IL-1 beta inhibitor is being investigated, but data are awaited [129].
Final thoughts on an exciting scenario
A large number of new therapies are being investigated in different inflammatory rheumatologic diseases. In some cases, molecules are being tested across indications, while in some others new pathways are being addressed and appear to be specific to each condition (as for IL-12/IL-23/IL-17 in PsA, IL-1beta in gout). While safety issues remain a concern, we are particularly eager to identify which molecules will allow a sustainable and effective treatment of these conditions with their growing social costs. Finally, we are convinced that, as illustrated for the JAK inhibitors, a better understanding of the disease pathogenesis is a necessary step to develop new effective and safety treatments.
References
Choy EH, Kavanaugh AF, Jones SA. The problem of choice: current biologic agents and future prospects in RA. Nat Rev Rheumatol. 2013;9(3):154–63. doi:10.1038/nrrheum.2013.8.
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–19. doi:10.1056/NEJMra1004965. doi:10.7748/phc2011.11.21.9.29.c8797.
Meune C, Touze E, Trinquart L, Allanore Y. Trends in cardiovascular mortality in patients with rheumatoid arthritis over 50 years: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford, England). 2009;48(10):1309–13. doi:10.1093/rheumatology/kep252.
Gabriel SE. Heart disease and rheumatoid arthritis: understanding the risks. Ann Rheum Dis. 2010;69(Suppl 1):i61–4. doi:10.1136/ard.2009.119404.
Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569–81. doi:10.1002/art.27584.
Gladman DD. Psoriatic arthritis. Dermatol Ther. 2009;22(1):40–55. doi:10.1111/j.1529-8019.2008.01215.x.
Gabriel SE, Michaud K. Epidemiological studies in incidence, prevalence, mortality, and comorbidity of the rheumatic diseases. Arthritis Res Ther. 2009;11(3):229. doi:10.1186/ar2669.
Duarte GV, Faillace C, Freire de Carvalho J. Psoriatic arthritis. Best Pract Res Clin Rheumatol. 2012;26(1):147–56. doi:10.1016/j.berh.2012.01.003.
Taylor WJ, Helliwell PS. Development of diagnostic criteria for psoriatic arthritis: methods and process. Curr Rheumatol Rep. 2004;6(4):299–305.
Chimenti MS, Ballanti E, Perricone C, Cipriani P, Giacomelli R, Perricone R. Immunomodulation in psoriatic arthritis: focus on cellular and molecular pathways. Autoimmun Rev. 2013;12(5):599–606. doi:10.1016/j.autrev.2012.10.002.
Conigliaro P, Scrivo R, Valesini G, Perricone R. Emerging role for NK cells in the pathogenesis of inflammatory arthropathies. Autoimmun Rev. 2011;10(10):577–81. doi:10.1016/j.autrev.2011.04.017.
Lories RJ, de Vlam K. Is psoriatic arthritis a result of abnormalities in acquired or innate immunity? Curr Rheumatol Rep. 2012;14(4):375–82. doi:10.1007/s11926-012-0257-3.
Kirkham BW, Kavanaugh A, Reich K. Interleukin-17A: a unique pathway in immune-mediated diseases: psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology. 2014;141(2):133–42. doi:10.1111/imm.12142.
Scarpa R. New insights into the concept of psoriatic disease. J Rheumatol Suppl. 2012;89:4–6. doi:10.3899/jrheum.120231.
Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet. 2014;. doi:10.1016/s0140-6736(14)60128-8.
Cozzani E, Drosera M, Gasparini G. Serology of lupus erythematosus: correlation between immunopathological features and clinical aspects. Autoimmun Dis. 2014;2014:321359. doi:10.1155/2014/321359.
Mak A, Kow NY. The pathology of T cells in systemic lupus erythematosus. J Immunol Res. 2014;2014:419029. doi:10.1155/2014/419029.
Peng H, Wang W, Zhou M, Li R, Pan HF, Ye DQ. Role of interleukin-10 and interleukin-10 receptor in systemic lupus erythematosus. Clin Rheumatol. 2013;32(9):1255–66. doi:10.1007/s10067-013-2294-3.
Candon S, Gottenberg JE, Bengoufa D, Chatenoud L, Mariette X. Quantitative assessment of antibodies to ribonucleoproteins in primary Sjogren syndrome: correlation with B-cell biomarkers and disease activity. Ann Rheum Dis. 2009;68(7):1208–12. doi:10.1136/ard.2008.095257.
Pers JO, Youinou P. Are the B cells cast with the leading part in the Sjogren’s syndrome scenario? Oral Dis. 2013;. doi:10.1111/odi.12153.
Voulgarelis M, Tzioufas AG. Current aspects of pathogenesis in Sjogren’s Syndrome. Ther Adv Musculoskelet Dis. 2010;2(6):325–34. doi:10.1177/1759720x10381431.
Leandro MJ, Cambridge G. Expression of B cell activating factor (BAFF) and BAFF-binding receptors in rheumatoid arthritis. J Rheumatol. 2013;40(8):1247–50. doi:10.3899/jrheum.130677.
Moura RA, Canhao H, Polido-Pereira J, Rodrigues AM, Navalho M, Mourao AF, et al. BAFF and TACI gene expression are increased in patients with untreated very early rheumatoid arthritis. J Rheumatol. 2013;40(8):1293–302. doi:10.3899/jrheum.121110.
Scholz JL, Oropallo MA, Sindhava V, Goenka R, Cancro MP. The role of B lymphocyte stimulator in B cell biology: implications for the treatment of lupus. Lupus. 2013;22(4):350–60. doi:10.1177/0961203312469453.
Juraschek SP, Miller ER 3rd, Gelber AC. Body mass index, obesity, and prevalent gout in the United States in 1988–1994 and 2007–2010. Arthritis Care Res. 2013;65(1):127–32. doi:10.1002/acr.21791.
Roddy E, Mallen CD, Doherty M. Gout. BMJ (Clinical Research ed). 2013;347:f5648. doi:10.1136/bmj.f5648.
Horton SC, Emery P. Biological therapy for rheumatoid arthritis: where are we now? British J Hosp Med (Lond Engl: 2005). 2012;73(1):12–8.
Dorner T, Strand V, Castaneda-Hernandez G, Ferraccioli G, Isaacs JD, Kvien TK, et al. The role of biosimilars in the treatment of rheumatic diseases. Ann Rheum Dis. 2013;72(3):322–8. doi:10.1136/annrheumdis-2012-202715.
Fleischmann R. Novel small-molecular therapeutics for rheumatoid arthritis. Curr Opin Rheumatol. 2012;24(3):335–41. doi:10.1097/BOR.0b013e32835190ef.
Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Mazurov V, et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann Rheum Dis. 2013;72(6):863–9. doi:10.1136/annrheumdis-2012-201601.
Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Aelion JA, et al. One-year efficacy and safety results of secukinumab in patients with rheumatoid arthritis: phase II, dose-finding, double-blind, randomized, placebo-controlled study. J Rheumatol. 2014;41(3):414–21. doi:10.3899/jrheum.130637.
Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, et al. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. 2010;62(4):929–39. doi:10.1002/art.27334.
Genovese MC, Greenwald M, Cho CS, Berman A, Jin L, Cameron GS et al. A Phase II Randomized Study of Subcutaneous Ixekizumab, an Anti-Interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumat (Hoboken, NJ). 2014;66(7):1693–704. doi:10.1002/art.38617.
Martin DA, Churchill M, Flores-Suarez L, Cardiel MH, Wallace D, Martin R, et al. A phase Ib multiple ascending dose study evaluating safety, pharmacokinetics, and early clinical response of brodalumab, a human anti-IL-17R antibody, in methotrexate-resistant rheumatoid arthritis. Arthritis Res Ther. 2013;15(5):R164. doi:10.1186/ar4347.
Krausz S, Boumans MJ, Gerlag DM, Lufkin J, van Kuijk AW, Bakker A, et al. Brief report: a phase IIa, randomized, double-blind, placebo-controlled trial of apilimod mesylate, an interleukin-12/interleukin-23 inhibitor, in patients with rheumatoid arthritis. Arthritis Rheum. 2012;64(6):1750–5. doi:10.1002/art.34339.
Bugatti S, Vitolo B, Caporali R, Montecucco C, Manzo A. B cells in rheumatoid arthritis: from pathogenic players to disease biomarkers. BioMed Res Int. 2014;2014:681678. doi:10.1155/2014/681678.
Mei HE, Schmidt S, Dorner T. Rationale of anti-CD19 immunotherapy: an option to target autoreactive plasma cells in autoimmunity. Arthritis Res Ther. 2012;14(Suppl 5):S1. doi:10.1186/ar3909.
Genovese MC, Kaine JL, Lowenstein MB, Del Giudice J, Baldassare A, Schechtman J, et al. Ocrelizumab, a humanized anti-CD20 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I/II randomized, blinded, placebo-controlled, dose-ranging study. Arthritis Rheum. 2008;58(9):2652–61. doi:10.1002/art.23732.
Emery P, Rigby W, Tak PP, Dorner T, Olech E, Martin C, et al. Safety with ocrelizumab in rheumatoid arthritis: results from the ocrelizumab phase III program. PLoS One. 2014;9(2):e87379. doi:10.1371/journal.pone.0087379.
Harigai M, Tanaka Y, Maisawa S. Safety and efficacy of various dosages of ocrelizumab in Japanese patients with rheumatoid arthritis with an inadequate response to methotrexate therapy: a placebo-controlled double-blind parallel-group study. J Rheumatol. 2012;39(3):486–95. doi:10.3899/jrheum.110994.
Rigby W, Tony HP, Oelke K, Combe B, Laster A, von Muhlen CA, et al. Safety and efficacy of ocrelizumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a forty-eight-week randomized, double-blind, placebo-controlled, parallel-group phase III trial. Arthritis Rheum. 2012;64(2):350–9. doi:10.1002/art.33317.
Stohl W, Gomez-Reino J, Olech E, Dudler J, Fleischmann RM, Zerbini CA, et al. Safety and efficacy of ocrelizumab in combination with methotrexate in MTX-naive subjects with rheumatoid arthritis: the phase III FILM trial. Ann Rheum Dis. 2012;71(8):1289–96. doi:10.1136/annrheumdis-2011-200706.
Tak PP, Mease PJ, Genovese MC, Kremer J, Haraoui B, Tanaka Y, et al. Safety and efficacy of ocrelizumab in patients with rheumatoid arthritis and an inadequate response to at least one tumor necrosis factor inhibitor: results of a forty-eight-week randomized, double-blind, placebo-controlled, parallel-group phase III trial. Arthritis Rheum. 2012;64(2):360–70.
Kurrasch R, Brown JC, Chu M, Craigen J, Overend P, Patel B, et al. Subcutaneously administered ofatumumab in rheumatoid arthritis: a phase I/II study of safety, tolerability, pharmacokinetics, and pharmacodynamics. J Rheumatol. 2013;40(7):1089–96. doi:10.3899/jrheum.121118.
Ostergaard M, Baslund B, Rigby W, Rojkovich B, Jorgensen C, Dawes PT, et al. Ofatumumab, a human anti-CD20 monoclonal antibody, for treatment of rheumatoid arthritis with an inadequate response to one or more disease-modifying antirheumatic drugs: results of a randomized, double-blind, placebo-controlled, phase I/II study. Arthritis Rheum. 2010;62(8):2227–38. doi:10.1002/art.27524.
Taylor PC, Quattrocchi E, Mallett S, Kurrasch R, Petersen J, Chang DJ. Ofatumumab, a fully human anti-CD20 monoclonal antibody, in biological-naive, rheumatoid arthritis patients with an inadequate response to methotrexate: a randomised, double-blind, placebo-controlled clinical trial. Ann Rheum Dis. 2011;70(12):2119–25. doi:10.1136/ard.2011.151522.
Stohl W, Merrill JT, McKay JD, Lisse JR, Zhong ZJ, Freimuth WW, et al. Efficacy and safety of belimumab in patients with rheumatoid arthritis: a phase II, randomized, double-blind, placebo-controlled, dose-ranging Study. J Rheumatol. 2013;40(5):579–89. doi:10.3899/jrheum.120886.
Tak PP, Thurlings RM, Rossier C, Nestorov I, Dimic A, Mircetic V, et al. Atacicept in patients with rheumatoid arthritis: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating, single- and repeated-dose study. Arthritis Rheum. 2008;58(1):61–72. doi:10.1002/art.23178.
Genovese MC, Kinnman N, de La Bourdonnaye G, Pena Rossi C, Tak PP. Atacicept in patients with rheumatoid arthritis and an inadequate response to tumor necrosis factor antagonist therapy: results of a phase II, randomized, placebo-controlled, dose-finding trial. Arthritis Rheum. 2011;63(7):1793–803. doi:10.1002/art.30373.
van Vollenhoven RF, Kinnman N, Vincent E, Wax S, Bathon J. Atacicept in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase II, randomized, placebo-controlled trial. Arthritis Rheum. 2011;63(7):1782–92. doi:10.1002/art.30372.
Genovese MC, Bojin S, Biagini IM, Mociran E, Cristei D, Mirea G, et al. Tabalumab in rheumatoid arthritis patients with an inadequate response to methotrexate and naive to biologic therapy: a phase II, randomized, placebo-controlled trial. Arthritis Rheum. 2013;65(4):880–9. doi:10.1002/art.37820.
Genovese MC, Fleischmann RM, Greenwald M, Satterwhite J, Veenhuizen M, Xie L, et al. Tabalumab, an anti-BAFF monoclonal antibody, in patients with active rheumatoid arthritis with an inadequate response to TNF inhibitors. Ann Rheum Dis. 2013;72(9):1461–8. doi:10.1136/annrheumdis-2012-202775.
Genovese MC, Lee E, Satterwhite J, Veenhuizen M, Disch D, Berclaz PY, et al. A phase 2 dose-ranging study of subcutaneous tabalumab for the treatment of patients with active rheumatoid arthritis and an inadequate response to methotrexate. Ann Rheum Dis. 2013;72(9):1453–60. doi:10.1136/annrheumdis-2012-202864.
Woodrick RS, Ruderman EM. IL-6 inhibition for the treatment of rheumatoid arthritis and other conditions. Bull NYU Hosp Joint Dis. 2012;70(3):195–9.
Huizinga TW, Fleischmann RM, Jasson M, Radin AR, van Adelsberg J, Fiore S, et al. Sarilumab, a fully human monoclonal antibody against IL-6Ralpha in patients with rheumatoid arthritis and an inadequate response to methotrexate: efficacy and safety results from the randomised SARIL-RA-MOBILITY Part A trial. Ann Rheum Dis. 2013;. doi:10.1136/annrheumdis-2013-204405.
Smolen JS, Weinblatt ME, Sheng S, Zhuang Y, Hsu B. Sirukumab, a human anti-interleukin-6 monoclonal antibody: a randomised, 2-part (proof-of-concept and dose-finding), phase II study in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2014;. doi:10.1136/annrheumdis-2013-205137.
Geyer M, Muller-Ladner U. Actual status of antiinterleukin-1 therapies in rheumatic diseases. Curr Opin Rheumatol. 2010;22(3):246–51. doi:10.1097/BOR.0b013e3283373fa0.
Ruckert R, Brandt K, Ernst M, Marienfeld K, Csernok E, Metzler C, et al. Interleukin-15 stimulates macrophages to activate CD4 + T cells: a role in the pathogenesis of rheumatoid arthritis? Immunology. 2009;126(1):63–73. doi:10.1111/j.1365-2567.2008.02878.x.
Hintzen C, Quaiser S, Pap T, Heinrich PC, Hermanns HM. Induction of CCL13 expression in synovial fibroblasts highlights a significant role of oncostatin M in rheumatoid arthritis. Arthritis Rheum. 2009;60(7):1932–43. doi:10.1002/art.24602.
Kay J, Calabrese L. The role of interleukin-1 in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford, England). 2004;43(Suppl 3):iii2–9. doi:10.1093/rheumatology/keh201.
Mertens M, Singh JA. Anakinra for rheumatoid arthritis. Cochrane Database of Syst Rev. 2009(1):Cd005121. doi:10.1002/14651858.CD005121.pub3.
Singh JA, Furst DE, Bharat A, Curtis JR, Kavanaugh AF, Kremer JM, et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res. 2012;64(5):625–39. doi:10.1002/acr.21641.
Ikonomidis I, Lekakis JP, Nikolaou M, Paraskevaidis I, Andreadou I, Kaplanoglou T, et al. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. 2008;117(20):2662–9. doi:10.1161/circulationaha.107.731877.
Ljung L, Olsson T, Engstrand S, Wallberg-Jonsson S, Soderberg S, Rantapaa-Dahlqvist S. Interleukin-1 receptor antagonist is associated with both lipid metabolism and inflammation in rheumatoid arthritis. Clin Exp Rheumatol. 2007;25(4):617–20.
Chakraborty A, Tannenbaum S, Rordorf C, Lowe PJ, Floch D, Gram H, et al. Pharmacokinetic and pharmacodynamic properties of canakinumab, a human anti-interleukin-1beta monoclonal antibody. Clin Pharmacokinet. 2012;51(6):e1–18. doi:10.2165/11599820-000000000-00000.
Alten R, Gomez-Reino J, Durez P, Beaulieu A, Sebba A, Krammer G, et al. Efficacy and safety of the human anti-IL-1beta monoclonal antibody canakinumab in rheumatoid arthritis: results of a 12-week, Phase II, dose-finding study. BMC Musculoskelet Disord. 2011;12:153. doi:10.1186/1471-2474-12-153.
Masuko-Hongo K, Kurokawa M, Kobata T, Nishioka K, Kato T. Effect of IL15 on T cell clonality in vitro and in the synovial fluid of patients with rheumatoid arthritis. Ann Rheum Dis. 2000;59(9):688–94.
Baslund B, Tvede N, Danneskiold-Samsoe B, Larsson P, Panayi G, Petersen J, et al. Targeting interleukin-15 in patients with rheumatoid arthritis: a proof-of-concept study. Arthritis Rheum. 2005;52(9):2686–92. doi:10.1002/art.21249.
Choy EH, Bendit M, McAleer D, Liu F, Feeney M, Brett S, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of an anti- oncostatin M monoclonal antibody in rheumatoid arthritis: results from phase II randomized, placebo-controlled trials. Arthritis Res Ther. 2013;15(5):R132. doi:10.1186/ar4312.
Keystone E, Cohen MD. Cell-signaling therapy in rheumatoid arthritis. Curr Rheumatol Rep. 2013;15(10):368. doi:10.1007/s11926-013-0368-5.
Migita K, Izumi Y, Torigoshi T, Satomura K, Izumi M, Nishino Y, et al. Inhibition of Janus kinase/signal transducer and activator of transcription (JAK/STAT) signalling pathway in rheumatoid synovial fibroblasts using small molecule compounds. Clin Exp Immunol. 2013;174(3):356–63. doi:10.1111/cei.12190.
Kawalec P, Mikrut A, Wisniewska N, Pilc A. The effectiveness of tofacitinib, a novel Janus kinase inhibitor, in the treatment of rheumatoid arthritis: a systematic review and meta-analysis. Clin Rheumatol. 2013;32(10):1415–24. doi:10.1007/s10067-013-2329-9.
Shi JG, Chen X, Lee F, Emm T, Scherle PA, Lo Y, et al. The pharmacokinetics, pharmacodynamics and safety of Baricitinib, an oral JAK 1/2 Inhibitor, in Healthy Volunteers. J Clin Pharmacol. 2014;. doi:10.1002/jcph.354.
Mesa RA. Ruxolitinib, a selective JAK1 and JAK2 inhibitor for the treatment of myeloproliferative neoplasms and psoriasis. IDrugs Investig Drugs J. 2010;13(6):394–403.
Gan EY, Chong WS, Tey HL. Therapeutic strategies in psoriasis patients with psoriatic arthritis: focus on new agents. BioDrugs Clin Immunother Biopharm Gene Ther. 2013;27(4):359–73. doi:10.1007/s40259-013-0025-6.
Kavanaugh A, Mease PJ, Gomez-Reino JJ, Adebajo AO, Wollenhaupt J, Gladman DD, et al. Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis. 2014;73(6):1020–6. doi:10.1136/annrheumdis-2013-205056.
Palfreeman AC, McNamee KE, McCann FE. New developments in the management of psoriasis and psoriatic arthritis: a focus on apremilast. Drug Des Dev Ther. 2013;7:201–10. doi:10.2147/dddt.s32713.
Schett G, Wollenhaupt J, Papp K, Joos R, Rodrigues JF, Vessey AR, et al. Oral apremilast in the treatment of active psoriatic arthritis: results of a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2012;64(10):3156–67. doi:10.1002/art.34627.
Wittmann M, Helliwell PS. Phosphodiesterase 4 inhibition in the treatment of psoriasis, psoriatic arthritis and other chronic inflammatory diseases. Dermatol Ther. 2013;3(1):1–15. doi:10.1007/s13555-013-0023-0.
Schett G, Sloan VS, Stevens RM, Schafer P. Apremilast: a novel PDE4 inhibitor in the treatment of autoimmune and inflammatory diseases. Ther Adv Musculoskelet Dis. 2010;2(5):271–8. doi:10.1177/1759720x10381432.
Kim HR, Kim KW, Kim BM, Jung HG, Cho ML, Lee SH. Reciprocal activation of CD4+ T cells and synovial fibroblasts by stromal cell-derived factor 1 promotes RANKL expression and osteoclastogenesis in rheumatoid arthritis. Arthritis Rheumatol (Hoboken, NJ). 2014;66(3):538–48. doi:10.1002/art.38286.
Perlman H, Bradley K, Liu H, Cole S, Shamiyeh E, Smith RC, et al. IL-6 and matrix metalloproteinase-1 are regulated by the cyclin-dependent kinase inhibitor p21 in synovial fibroblasts. J Immunol. 2003;170(2):838–45.
Filer A. The fibroblast as a therapeutic target in rheumatoid arthritis. Curr Opin Pharmacol. 2013;13(3):413–9. doi:10.1016/j.coph.2013.02.006.
Kiener HP, Niederreiter B, Lee DM, Jimenez-Boj E, Smolen JS, Brenner MB. Cadherin 11 promotes invasive behavior of fibroblast-like synoviocytes. Arthritis Rheum. 2009;60(5):1305–10. doi:10.1002/art.24453.
Bauer S, Jendro MC, Wadle A, Kleber S, Stenner F, Dinser R, et al. Fibroblast activation protein is expressed by rheumatoid myofibroblast-like synoviocytes. Arthritis Res Ther. 2006;8(6):R171. doi:10.1186/ar2080.
Ospelt C, Mertens JC, Jungel A, Brentano F, Maciejewska-Rodriguez H, Huber LC, et al. Inhibition of fibroblast activation protein and dipeptidylpeptidase 4 increases cartilage invasion by rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2010;62(5):1224–35. doi:10.1002/art.27395.
El-Jawhari JJ, El-Sherbiny YM, Jones EA, McGonagle D. Mesenchymal stem cells, autoimmunity and rheumatoid arthritis. QJM Mon J Assoc Physicians. 2014;107(7):505–14. doi:10.1093/qjmed/hcu033.
Huynh D, Kavanaugh A. Psoriatic arthritis: current therapy and future directions. Expert Opin Pharmacother. 2013;14(13):1755–64. doi:10.1517/14656566.2013.810208.
Gossec L, Smolen JS, Gaujoux-Viala C, Ash Z, Marzo-Ortega H, van der Heijde D, et al. European League Against Rheumatism recommendations for the management of psoriatic arthritis with pharmacological therapies. Ann Rheum Dis. 2012;71(1):4–12. doi:10.1136/annrheumdis-2011-200350.
Papoutsaki M, Costanzo A. Treatment of psoriasis and psoriatic arthritis. BioDrugs Clin Immunother Biopharm Gene Ther. 2013;27(Suppl 1):3–12. doi:10.1007/bf03325637.
Jimenez-Boj E, Stamm TA, Sadlonova M, Rovensky J, Raffayova H, Leeb B, et al. Rituximab in psoriatic arthritis: an exploratory evaluation. Ann Rheum Dis. 2012;71(11):1868–71. doi:10.1136/annrheumdis-2012-201897.
Altmeyer MD, Kerisit KG, Boh EE. Therapeutic hotline. Abatacept: our experience of use in two patients with refractory psoriasis and psoriatic arthritis. Dermatol Ther. 2011;24(2):287–90. doi:10.1111/j.1529-8019.2011.01405.x.
Rodrigues CE, Vieira FJ, Callado MR, Gomes KW, de Andrade JE, Vieira WP. Use of the abatacept in a patient with psoriatic arthritis. Revista Brasileira de Reumatologia. 2010;50(3):340–5.
Ursini F, Naty S, Russo E, Grembiale RD. Abatacept in psoriatic arthritis: case report and short review. J Pharmacol Pharmacother. 2013;4(Suppl 1):S29–32. doi:10.4103/0976-500x.120943.
Vieira FJ, Callado MR, Vieira WP. Abatacept as an option therapy in difficult to treat psoriatic arthritis. Rheumatol Int. 2010;30(6):849–50. doi:10.1007/s00296-009-1041-1.
Mease P, Genovese MC, Gladstein G, Kivitz AJ, Ritchlin C, Tak PP, et al. Abatacept in the treatment of patients with psoriatic arthritis: results of a six-month, multicenter, randomized, double-blind, placebo-controlled, phase II trial. Arthritis Rheum. 2011;63(4):939–48. doi:10.1002/art.30176.
Cauli A, Mathieu A. Th17 and interleukin 23 in the pathogenesis of psoriatic arthritis and spondyloarthritis. J Rheumatol Suppl. 2012;89:15–8. doi:10.3899/jrheum.120234.
Suzuki E, Mellins ED, Gershwin ME, Nestle FO, Adamopoulos IE. The IL-23/IL-17 axis in psoriatic arthritis. Autoimmun Rev. 2014;13(4–5):496–502. doi:10.1016/j.autrev.2014.01.050.
Zhu KJ, Zhu CY, Shi G, Fan YM. Meta-analysis of IL12B polymorphisms (rs3212227, rs6887695) with psoriasis and psoriatic arthritis. Rheumatol Int. 2013;33(7):1785–90. doi:10.1007/s00296-012-2637-4.
Catanoso MG, Boiardi L, Macchioni P, Garagnani P, Sazzini M, De Fanti S, et al. IL-23A, IL-23R, IL-17A and IL-17R polymorphisms in different psoriatic arthritis clinical manifestations in the northern Italian population. Rheumatol Int. 2013;33(5):1165–76. doi:10.1007/s00296-012-2501-6.
McInnes IB, Kavanaugh A, Gottlieb AB, Puig L, Rahman P, Ritchlin C, et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet. 2013;382(9894):780–9. doi:10.1016/s0140-6736(13)60594-2.
Kavanaugh A, Ritchlin C, Rahman P, Puig L, Gottlieb AB, Li S, et al. Ustekinumab, an anti-IL-12/23 p40 monoclonal antibody, inhibits radiographic progression in patients with active psoriatic arthritis: results of an integrated analysis of radiographic data from the phase 3, multicentre, randomised, double-blind, placebo-controlled PSUMMIT-1 and PSUMMIT-2 trials. Ann Rheum Dis. 2014;73(6):1000–6. doi:10.1136/annrheumdis-2013-204741.
Ritchlin C, Rahman P, Kavanaugh A, McInnes IB, Puig L, Li S, et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann Rheum Dis. 2014;73(6):990–9. doi:10.1136/annrheumdis-2013-204655.
Patel DD, Lee DM, Kolbinger F, Antoni C. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Ann Rheum Dis. 2013;72 (Suppl 2):ii116–23. doi:10.1136/annrheumdis-2012-202371.
McInnes IB, Sieper J, Braun J, Emery P, van der Heijde D, Isaacs JD, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis. 2014;73(2):349–56. doi:10.1136/annrheumdis-2012-202646.
Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371(4):326–38. doi:10.1056/NEJMoa1314258.
Mease PJ, Genovese MC, Greenwald MW, Ritchlin CT, Beaulieu AD, Deodhar A, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med. 2014;370(24):2295–306. doi:10.1056/NEJMoa1315231.
Zouali M, Uy EA. Belimumab therapy in systemic lupus erythematosus. BioDrugs Clin Immunother Biopharm Gene Ther. 2013;27(3):225–35. doi:10.1007/s40259-013-0031-8.
Cobo-Ibanez T, Loza-Santamaria E, Pego-Reigosa JM, Marques AO, Rua-Figueroa I, Fernandez-Nebro A, et al. Efficacy and safety of rituximab in the treatment of non-renal systemic lupus erythematosus: a systematic review. Semin Arthritis Rheum. 2014;. doi:10.1016/j.semarthrit.2014.04.002.
Bertsias GK, Tektonidou M, Amoura Z, Aringer M, Bajema I, Berden JH, et al. Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of adult and paediatric lupus nephritis. Ann Rheum Dis. 2012;71(11):1771–82. doi:10.1136/annrheumdis-2012-201940.
Reddy V, Jayne D, Close D, Isenberg D. B-cell depletion in SLE: clinical and trial experience with rituximab and ocrelizumab and implications for study design. Arthritis Res Ther. 2013;15(Suppl 1):S2. doi:10.1186/ar3910.
Daridon C, Blassfeld D, Reiter K, Mei HE, Giesecke C, Goldenberg DM, et al. Epratuzumab targeting of CD22 affects adhesion molecule expression and migration of B-cells in systemic lupus erythematosus. Arthritis Res Ther. 2010;12(6):R204. doi:10.1186/ar3179.
Pena-Rossi C, Nasonov E, Stanislav M, Yakusevich V, Ershova O, Lomareva N, et al. An exploratory dose-escalating study investigating the safety, tolerability, pharmacokinetics and pharmacodynamics of intravenous atacicept in patients with systemic lupus erythematosus. Lupus. 2009;18(6):547–55. doi:10.1177/0961203309102803.
Isenberg D, Gordon C, Licu D, Copt S, Rossi CP, Wofsy D. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann Rheum Dis. 2014;. doi:10.1136/annrheumdis-2013-205067.
Merrill JT. Co-stimulatory molecules as targets for treatment of lupus. Clin Immunol (Orlando, FL). 2013;148(3):369–75. doi:10.1016/j.clim.2013.04.012.
Merrill JT, Burgos-Vargas R, Westhovens R, Chalmers A, D’Cruz D, Wallace DJ, et al. The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010;62(10):3077–87. doi:10.1002/art.27601.
Illei GG, Shirota Y, Yarboro CH, Daruwalla J, Tackey E, Takada K, et al. Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. 2010;62(2):542–52. doi:10.1002/art.27221.
Xu Z, Bouman-Thio E, Comisar C, Frederick B, Van Hartingsveldt B, Marini JC, et al. Pharmacokinetics, pharmacodynamics and safety of a human anti-IL-6 monoclonal antibody (sirukumab) in healthy subjects in a first-in-human study. Br J Clin Pharmacol. 2011;72(2):270–81. doi:10.1111/j.1365-2125.2011.03964.x.
O’Shea JJ, Kontzias A, Yamaoka K, Tanaka Y, Laurence A. Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis. 2013;72(Suppl 2):ii111–5. doi:10.1136/annrheumdis-2012-202576.
Sthoeger Z, Sharabi A, Mozes E. Novel approaches to the development of targeted therapeutic agents for systemic lupus erythematosus. J Autoimmun. 2014;. doi:10.1016/j.jaut.2014.06.002.
Crittenden DB, Pillinger MH. New therapies for gout. Annu Rev Med. 2013;64:325–37. doi:10.1146/annurev-med-080911-105830.
So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9(2):R28. doi:10.1186/ar2143.
Schlesinger N, De Meulemeester M, Pikhlak A, Yucel AE, Richard D, Murphy V, et al. Canakinumab relieves symptoms of acute flares and improves health-related quality of life in patients with difficult-to-treat Gouty Arthritis by suppressing inflammation: results of a randomized, dose-ranging study. Arthritis Res Ther. 2011;13(2):R53. doi:10.1186/ar3297.
Schlesinger N, Alten RE, Bardin T, Schumacher HR, Bloch M, Gimona A, et al. Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Ann Rheum Dis. 2012;71(11):1839–48. doi:10.1136/annrheumdis-2011-200908.
So A, De Meulemeester M, Pikhlak A, Yucel AE, Richard D, Murphy V, et al. Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis: Results of a multicenter, phase II, dose-ranging study. Arthritis Rheum. 2010;62(10):3064–76. doi:10.1002/art.27600.
Schlesinger N, Mysler E, Lin HY, De Meulemeester M, Rovensky J, Arulmani U, et al. Canakinumab reduces the risk of acute gouty arthritis flares during initiation of allopurinol treatment: results of a double-blind, randomised study. Ann Rheum Dis. 2011;70(7):1264–71. doi:10.1136/ard.2010.144063.
Schumacher HR Jr, Evans RR, Saag KG, Clower J, Jennings W, Weinstein SP, et al. Rilonacept (interleukin-1 trap) for prevention of gout flares during initiation of uric acid-lowering therapy: results from a phase III randomized, double-blind, placebo-controlled, confirmatory efficacy study. Arthritis Care Res. 2012;64(10):1462–70. doi:10.1002/acr.21690.
Sundy JS, Schumacher HR, Kivitz A, Weinstein SP, Wu R, King-Davis S, et al. Rilonacept for gout flare prevention in patients receiving uric acid-lowering therapy: results of RESURGE, a Phase III, international safety study. J Rheumatol. 2014;. doi:10.3899/jrheum.131226.
Goh AX, Bertin-Maghit S, Ping Yeo S, Ho AW, Derks H, Mortellaro A et al. A novel human anti-interleukin-1beta neutralizing monoclonal antibody showing in vivo efficacy. mAbs. 2014;6(3):765–73. doi:10.4161/mabs.28614.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Selmi, C., Generali, E., Massarotti, M. et al. New treatments for inflammatory rheumatic disease. Immunol Res 60, 277–288 (2014). https://doi.org/10.1007/s12026-014-8565-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-014-8565-5