
Abstract
Type 1A diabetes (autoimmune) is now immunologically predictable in man, but preventable only in animal models. What triggers the development of autoimmunity in genetically susceptible individuals remains unknown. Studies of non-obese diabetic (NOD) mice reveal that interactions between T-cell receptors of diabetogenic T cell and an MHC class II loaded with an autoantigen are key determinates of the disease. With insulin as the primary target in the NOD mouse, likely man, and possibly the RT1-U rat models, therapeutic targeting of the components of these anti-insulin trimolecular complexes we believe provide a fulcrum for development of preventive therapy. In particular for the NOD mouse model, there is extensive evidence that the dominant insulin peptide driving disease initiation is insulin B chain amino acids 9-23 (SHLVEALYLVCGERG) recognized predominantly by germ-line sequences of a specific T-cell receptor Valpha (TRAV5D-4), and small molecules or monoclonal antibodies directed at this recognition complex can prevent diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 1A diabetes is a prototypic organ-specific autoimmune disorder that manifests itself through systemic insulin deficiency due to decreased numbers of insulin-secreting beta cells. Within the pancreas, it is only cells of the islets that are destroyed (acinar cell atrophy occurs apparently secondary to loss of trophic effects of insulin), and within the islets, it is currently thought that only beta cells are dramatically lost (usually cells producing glucagon, somatostatin, and pancreatic polypeptides are preserved). Islets lacking beta cells are termed pseudoatrophic, and the presence of pseudoatrophic islets is pathognomonic of the disease. Clinical data show slow, but predictable, progression from the initial activation of autoimmunity, as indicated by the development of multiple islet autoantibodies (e.g., reacting with insulin, GAD65, IA-2, and ZnT8), to the overt disease (Fig. 1). The best predictors for progression are the age of appearance of autoantibodies, the number of autoantigens targeted by these antibodies, the level of autoantibodies reacting with insulin, and the progressive loss of insulin secretion [1, 2].

Model of Type 1 diabetes divided into stages beginning with genetic susceptibility, followed by an unknown triggering event and the appearance of autoimmunity resulting in progressive beta cell destruction. We hypothesize that targeting of insulin/proinsulin by CD4+ T cells is a key event given the genetic predisposition primarily determined by MHC alleles (e.g., DQ8 in man and I-Ag7 in the NOD mouse) presenting a peptide of insulin (for NOD mouse B chain amino acids 9-23) to T-cell receptors whose germ-line sequences favor targeting of the MHC-insulin peptide complex (modified from [46])
The mechanism of beta cell destruction is not clear, and it may differ in mice and humans. Studies in non-obese diabetic (NOD) mice show that beta cells die by apoptosis, and not by necrosis [3, 4]. There is absence of similar data for humans due to lack of suitable samples and the general difficulties in finding apoptotic cells due to their prompt clearance in vivo. Results from NOD mice show clear involvement of multiple cell lineages in beta cell death, including CD4+ and CD8+ T cells [5–8], and macrophages [9]. However, it is unclear how these cells induce or contribute to apoptosis in beta cells. In their excellent review, Benoit and Mathis [10] proposed two possible mechanisms for induction of apoptosis in beta cells: (1) “recognition-linked”, whereupon the “killer” recognizes beta cell directly and utilizes either a perforin/granzyme or Fas/FasL pathways and (2) an “activation-linked”, where the beta cell is indirectly targeted by closely residing activated “killers” using variety of lethal weapons including FasL or a milieu of proinflammatory cytokines.
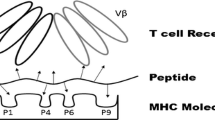
Regardless of the detailed mechanism involved, we believe that the fundamental cause of Type 1A diabetes is the recognition of key islet antigens presented by “diabetogenic” MHC class II alleles to “diabetogenic” CD4+ T cells in the background of a disregulated immune system. In particular, studies in mice implicate peptides derived from the beta chain of insulin (B:9-23), MHC class II molecule I-Ag7, with CD4+ T cells recognizing these MHC-peptide complexes [11–13], and a host of genes regulating immune cell activation and homeostasis [14]. Likewise, genetic studies in man implicate insulin/proinsulin as a major target of autoimmunity, predominantly MHC class II alleles such as HLA-DQ8 (highly homologous to I-Ag7), and genes involved in the regulation of the immune system [15, 16]. Interventions targeting this trimolecular complex composed of MHC class II, B:9-23 peptide, and specific T-cell receptor families might provide a way to prevent the disease. Therefore, it is of upmost importance to understand the details regarding formation of this complex.
Specific MHC class II molecule
Genetic studies in mice and man have identified the MHC locus as the major risk factor for the development of T1D. In mice, the presence of I-Ag7 allele is essential for disease development, while other alleles protect against the disease in a dominant fashion [12, 17]. A similar situation takes place in man: HLA alleles DRB1*0401, DRB1*0402, and DRB1*0405 together with DQA1*0301and DQB1*0302 (DQ8), as well as DRB1*0301 with DQA1*0501/DQB1*0201 (DQ2) predispose the carrier to the disease, while DRB1*1501 with DQB1*0602 alleles protect in a dominant fashion [18, 19]. Only two percent of newborns in Denver Colorado have the heterozygous DR3/4-DQ2/8 genotype but approximately 40 % of children developing Type 1 diabetes have this HLA genotype. Of note, the human DQ8 sequence is very similar to the NOD mouse I-Ag7 allele.
Primary antigen
Given that there are multiple autoantigens targeted by B and/or T lymphocytes, is there a primary antigenic target necessary for disease activation or progression? Our definition of a primary autoantigen implies the necessity of this antigen for disease initiation or progression (which can be tested in animal models) and precludes other secondary antigens that contribute to diabetes development, but in whose absence diabetes develops nonetheless. Clinical data documenting early development of anti-insulin antibodies in T1D patients, and the strong correlation between certain alleles of insulin gene and the risk for the development of the disease suggest that insulin is a primary autoantigen in humans [20].
Although T1D patients develop antibodies against multiple antigens, notably GAD65, IA-2, and ZnT8, it is only the levels of insulin autoantibodies that are positively correlated with the age of onset and the rate at which children progress to diabetes [1]. However, children with only insulin autoantibodies rarely progress to diabetes with some indication that such single insulin autoantibodies are different (e.g., lower affinity, less cross-reactivity with proinsulin) and may be biologic false positives [21]. The requirement for multiple antibody specificities might indicate that the initial response triggered by insulin must attain a certain magnitude to cause the disease and that targeting of other antigens is a byproduct of this damage. This scenario has been confirmed for islet-specific glucose-6-phosphatase–related protein (IGRP) that is targeted by T cells in NOD mice. Krishnamurthy et al. showed that even though both insulin and IGRP elicit strong T-cell responses in NOD mice, anergizing the anti-IGRP response had no effect on the disease, while anergizing the insulin-specific T cells prevented the disease and prevented the development of T-cell-targeting IGRP [22].
The importance of insulin for the development of diabetes is also underscored by the genetic data showing that a polymorphism (VNTR) in the 5′ region of the insulin gene is the second most important genetic determinant of the disease [23]. This region has been shown to regulate the expression of insulin in the thymus, thus influencing the central tolerance against the antigen. This conclusion is also supported by the finding that Type 1 diabetes develops in patients with compromised expression of insulin in the thymus due to defective function of AIRE gene [24].
Strong evidence for insulin being the primary autoantigen in the NOD mouse comes from experiments aimed at eliminating this protein from NOD mice. Mice have two insulin genes, pancreas-specific INS-1 and INS-2 expressed in both pancreas and the thymus. Elimination of INS-1 gene resulted in protection, whereas deletion of INS-2 accelerated the disease [25, 26]. These results fit with our hypothesis of insulin being a primary autoantigen: elimination of it from the thymus (INS-2 KO) would eliminate a central tolerance mechanism for this antigen. However, proving the primacy required a more complex experiment since insulin cannot be eliminated completely due to its essential function during embryonic development. Nakayama et al. [11] bypassed this requirement by generating NOD mice deficient in both insulin genes, but expressing a mutant form of insulin as a transgene. Previous experiments showed that changing a single tyrosine residue at position 16 of the insulin beta chain to an alanine impaired recognition of this antigen by the immune system [27]. Thus, B16:A insulin could “hide” from the immune system, but function normally at physiological level. The observation that NOD mice lacking both insulin genes with only B16:A insulin did not develop diabetes is consistent with the primary role insulin plays as a diabetes autoantigen.
In contrast to the effects of knocking out insulin genes, knockouts of multiple other islet expressed molecules to date have had no effect upon progression to diabetes of NOD mice, including GAD65 [28], IA-2 [29], IA-2beta [30], IAPP [31], and IGRP [32]. These knockouts neither accelerate nor inhibit progression to diabetes. There remain additional knockouts to test such as chromogranin and potentially yet to be discovered islet autoantigens.
Primary peptide
As mentioned above, knocking out both insulin genes, with transgenic replacement of insulin mutated to avoid activation of most T-cells-targeting insulin, prevents essentially all development of diabetes of the NOD mouse. This result not only proves the importance of insulin but also narrows it to the region of the beta chain that contains the mutation. Daniel and coworkers originally identified a short region of insulin B chain located between residues 9 and 23 (SHLVEALYLVCGERG) as an antigen targeted by more than 90 percent of anti-insulin CD4 T-cell clones isolated directly from pancreatic islets of NOD mice [33]. Studies from humans confirm the importance of this epitope [34]. This B:9-23 epitope contained the above mentioned B16:A mutation that prevents the development of diabetes in the doubly deficient NOD mice.
Primary register
Work by Stadinski et al. [35] brought a new focus to the details of antigen presentation in Type 1 diabetes. This study used a method of register trapping to determine which “face” of B:9-23 peptide is recognized by autoreactive T cells and which binds to I-Ag7. The register in which a peptide binds to MHC is defined as the specific linear orientation of the peptide within the MHC groove. As one moves a peptide one amino acid up or down the groove of the MHC, the peptide has to rotate for different side chains of the peptide to bind to the pockets of the MHC and by rotating change the side chains facing outward and interacting with T-cell receptors. By fixing the way the insulin B:9-23 peptide binds to I-Ag7, Stadinski and coworkers showed that B:9-23 stimulated a panel of insulin-specific CD4+ T-cell hybridomas only when bound in “register 3” where arginine of the native peptide is bound in pocket 9 of the I-Ag7 MHC. The mutated peptide, denoted B:9-23(RE), has mutated anchor amino acids corresponding to the p1 and p9 pockets of I-Ag7 with R replacing A at pocket 1 and E replacing R at pocket 9 (SHLVERLYLVCGEEG). This single register directly contradicted findings by Levisetti et al. [36] who proposed register 1 and 2 as being recognized by the two classes of T-cell hybridomas targeting insulin peptide B:9-23 in different registers. However, changes in T-cell responses could also be explained as altering peptide epitopes, while the peptides remained bound in register 3, which studies of Levisetti clones by Kappler’s group confirm.
Another paper from Kappler’s group showed that some truncations of the B:9-23 peptide might in fact increase T-cell reactivity. For example, C-terminal truncation to B21 eliminates the arginine residue that conflicts with the I-Ag7 p9-binding pocket when bound in register 3, and truncation to B20 eliminates glutamic acid that interferes with recognition by some T-cell clones [37]. Building on this knowledge, Kappler’s group generated a series of B:9-23/I-Ag7 tetramers that stain majority of insulin-specific CD4+ T-cell hybridomas.
These newly developed B:9-23 tetramers directly stain about 5 % of CD4+ T cells present in the pancreas of NOD mice. The authors also proposed a model explaining how the register 3 peptide might be recognized as a “de novo” antigen expressed specifically in the pancreas. According to this model, pancreatic beta cells generate truncated version(s) of the peptide that are not present in the thymus during the maturation of thymocytes. Absent during the negative selection, these peptides cannot delete autoreactive thymocytes recognizing insulin. Thus, this unique version of the peptide can be considered a “neo-antigen”. An ability of pancreatic beta cells to process insulin has been described previously [38]. This paper showed that a small percentage of beta cells had secretory granules containing B:9-23 peptide and that this peptide could be “picked up” by resident dendritic cells. Therefore, it is conceivable that this unique register 3 insulin peptide can be generated only in the pancreas.
In summary, identification of register 3 presentation of the insulin B:9-23 peptide to autoreactive T-cell receptors has opened new avenues of research. First, the panel of I-Ag7/B9-23 tetramers will enable in vivo studying of insulin-specific, I-Ag7-restricted CD4+ T cells in NOD mice. Second, the analogous approach might lead to the generation of similar reagents for human system. Finally, administration of an insulin peptide mimetope designed to bind in relevant register 3 can induce dominant tolerance that prevents NOD diabetes (see below) [39].
Specific T-cell receptor alpha chain sequences
The third component of the trimolecular recognition complex is the T-cell receptor which in the NOD mouse model has unusual features. Simone et al. studied thirteen B:9-23 specific T-cell clones isolated from pancreatic islets and reported that 10 of them expressed TCRs containing alpha chains derived from the TRAV5D-4 (formerly termed Vα 13S3) gene [40]. There was neither apparent restriction of CDR3 sequences nor restriction of the sequence of TCR beta chains. The residues of the CDR1 and CDR2 regions contributed to critical interactions for the binding of these TCRs to I-Ag7/B:9-23 complex. In a recent paper, Nakayama confirmed these finding and showed that germ-line-encoded TRAV5D-4 sequences in combination with many different CDR3 sequences induced anti-insulin autoimmunity in NOD mice [41]. We estimate that approximately one percent of NOD mouse TRAV5D-4-expressing T cells target the B:9-23 peptide [42]. To test if this finding is relevant to human disease, we identified TRAV13-1 as a human ortholog of the mouse TRAV-5D-4 and showed that CDR1 and CRD2 regions of this gene can cause diabetes in alpha chain retrogenic NOD mice when grafted onto mouse TCR alpha of two diabetogenic murine clones, 8–1.1 and 12–4.1 [41]. Of note, mouse and man share sequence identity for the mouse insulin 2 B:9-23 peptide.
Even though the specific autoantigenic targets have not been identified for the RT1-U-associated autoimmune diabetes rat models (e.g., BB rat and Lew1.WR1), a recent report indicates that disease is dependent upon germ-line-encoded sequences of TCR Vbeta15 [43]. This would suggest a specific target that can be recognized by genomic sequences of the CDR1 and CDR2 regions of the T-cell receptor. We speculate that the target antigen for these Vbeta15-expressing T cells is likely to be insulin.
Trimolecular complex: therapeutic interventions
As suggested in the introduction, clinical interventions targeting the trimolecular complex of MHC class II, B:9-23, and TCR might provide a generalizable way to prevent autoimmune diabetes. As shown in Fig. 2, one might envision interventions that alter trimolecular recognition complex components (e.g., eliminating T cells expressing specific T-cell receptor segments) block the interactions of the components (e.g., using antibodies or small molecules) or modulate the interactions to induce non-diabetogenic effects (e.g., induce insulin-specific regulatory T cells). In fact, such interventions have been tried, and they show promise in preventing the disease in autoimmune diabetes models (Fig. 2).

The trimolecular complex and four modes of interfering with it. MHC class II molecule binds insulin B chain peptide containing amino acids 9-23 (B:9-23), and this complex in then recognized by TCR. B:9-23 is depicted as not binding well at its C-terminus (right side in the picture) due to electrostatic hindrance existing between positively charged pocket 9 and an arginine of the peptide when bound in register 3. Figure shows three potential ways of interfering with the formation of the TCR:MHC/peptide complex: (1) antibody-mediated deletion of the specific T cells, (2) blockage of the TCR-binding “face” of the MHC/peptide complex with an antibody, and (3) blocking of the peptide from MHC (or changing its conformation) using a small molecule. In addition, one might use a small molecule to increase the strength of peptide binding in register 3 and thus by altering the function of the activated T cells from diabetogenic (disease-causing) to regulatory (disease-preventing)
Elimination of diabetogenic T cells in vivo has been studied in the RT1-U-associated autoimmune rat models [43]. As mentioned before, a recent paper reported that the diabetes susceptibility locus Iddm14 encodes the TCR Vbeta15 gene. To show the importance of the germ-line-encoded Vbeta15, the authors depleted T cells using specific anti-Vbeta15 antibody. The antibody prevented diabetes of genetically susceptible rats, whether the autoimmune diabetes was induced by poly I:C stimulation, viral challenge, or developed spontaneously. In every case, the antibody depleted T cells expressing Vbeta15 protein and at the same time prevented development of diabetes. Depletions of specific Vbeta T cells might not be as effective when the autoimmune response has had more time to spread to multiple antigens. It might also be possible that in human disease there is no single TCR germ-line sequence that is essential for autoimmune diabetes even for a specific MHC genotype.
Another studied intervention blocks the interaction between TCR and B:9-23/MHC complex using monoclonal antibodies. Li Zhang in our laboratory has immunized NOD mice with soluble I-Ag7 loaded with the register 3-binding B:9-23(RE) form of the peptide and showed protection from the disease that was associated with generation of specific anti- I-Ag7/B:9-23(RE) antibodies [44]. Moreover, Zhang went on to develop a monoclonal antibody that can inhibit formation of the relevant trimolecular complex and the in vitro activation of insulin-specific T-cell hybridoma (personal communication).
Blocking the interaction between TCR and B:9-23/MHC may also be accomplished by small molecules that can bind to one of the elements and thus interfere with the complex formation. Michels and colleagues used an in silico molecular docking algorithm to identify small molecules of a 140,000 compound NCI library that could bind to pockets of I-Ag7. The top 40 compounds for each pocket were screened for in vitro effects on B:9-23 peptide stimulation of insulin-specific T-cell hybridomas. The approach yielded several candidate molecules that could fulfill requirements for specificity and sensitivity [45]. For example, tetraazatricyclododecane (p6:4) was able to interfere with binding of the B:9-23 peptide to I-Ag7, and it also inhibited peptide-dependent stimulation of several insulin-specific T-cell hybridomas. The compound was specific since it did not interfere with stimulation by non-related MHC molecules or with stimulation of non-related TCRs. Moreover, the same compound was able to inhibit interaction between human DQ8 loaded with B:9-23 and a hybridoma expressing B:9-23-specific TCR cloned from T1D patient.
Unexpected was the finding that glyphosine, a small molecule predicted to bind in pocket 9 of I-Ag7, shifted the responses of insulin-specific T cells from IFN-γ to the protective cytokine IL-10 [45]. This was again true for both murine and human systems. The exciting consequence of this shift was inhibition of diabetes in NOD mice for as long as glyphosine was administered. One hypothesis is that glyphosine (highly negatively charged) promotes binding of B:9-23 in register 3 through altering of electrostatic charge of pocket 9. An “altered” register 3-bound peptide might stimulate T cells to produce IL-10 and thus induce regulatory T cells. If this is the case, one might speculate that induction of regulatory T cells by insulin peptide would not only transform diabetogenic T cells into regulatory T cells in the pancreas, but also would provide dominant protection against T cells with other autoantigen specificities.
That register 3-bound B:9-23 peptide can induce regulatory T cells was also reported by Daniel et al. [39] This group infused small amounts of B:9-23 (R22E) mimetope that was designed to bind in register 3 to I-Ag7 and showed that this regiment generated insulin-specific regulatory T cells. The most exciting result of this work was the generation of dominant tolerance to polyclonal responses to insulin. Again, the generation of dominant tolerance might be very important for treating diabetes that has progressed beyond recognition of insulin, where multiple autoantigens are targeted by diverse population of T cells.
Conclusion
Studies of Type 1A diabetes are leading to the conclusion that the disorder develops primarily as an accident in nature such that three interacting molecules create a recognition complex enhancing targeting of a major beta cell specific molecule. The MHC alleles of the complex for man, rat, and mouse are polymorphic and critical to the development of diabetes. Gene knockout studies strongly implicate targeting of insulin and not GAD, IA-2, IAPP, or IGRP, as essential for the development of NOD diabetes. Both rat and mouse have germ-line-encoded specific T-cell receptor segment sequences that influence progression to autoimmunity. Similar understanding of anti-islet molecular T-cell recognition of islet antigens are lacking for Type 1 diabetes of man.
Though components of the trimolecular recognition complex appear to be critical for the disease in analogy to the NOD mouse, they might not determine (if not polymorphic) which individual will develop disease beyond MHC risk. It is obvious that additional factors determine individual risk of diabetes, including unknown environmental factors (e.g., evidenced by increasing incidence of Type 1 diabetes), as well as multiple genetic factors predominantly influencing maintenance of tolerance.
Given ability to predict the Type 1 diabetes [1], the major task ahead is prevention. Therapeutic targeting of the trimolecular complex seems an obvious pathway to pursue though not yet successfully accomplished for any autoimmune disorder. The treatment of insulin-dependent diabetes is developing rapidly with availability of continuous glucose monitors that are able to turn off insulin pumps and prevent severe hypoglycemia already approved in Europe. This raises the bar for immunotherapy of diabetes such that most forms of immunosuppression will be excluded. Thus, antigen specific or trimolecular complex specific therapies will likely be required. Such novel therapies have their own theoretical risks, including disease induction with peptide immunization, inconvenience of monoclonal antibody therapeutics, potential creation of holes in immune repertoire, and potential induction of immune responses to neo-epitopes. Nevertheless, the logic of trimolecular complex targeting is very appealing, and perhaps in stages, development of disease specific therapies should be possible. If this is achieved for Type 1A diabetes, it will be a model for most autoimmune disorders, though one drug will not fit all, and drugs will need to be tailored to specific MHC alleles as well as disease-related peptides/registers (truly personalized medicine). The lessons from autoimmune diabetes are also likely to be of relevance to general immune function. Which epitope is targeted and the register in which it is recognized by CD4 T cells is potentially predetermined by germ-line sequences of the T-cell receptor repertoire.
References
Steck AK, Johnson K, Barriga KJ, Miao D, Yu L, Hutton JC, et al. Age of islet autoantibody appearance and mean levels of insulin, but not GAD or IA-2 autoantibodies, predict age of diagnosis of type 1 diabetes: diabetes autoimmunity study in the young. Diabetes Care. 2011;34(6):1397–9.
Mrena S, Virtanen SM, Laippala P, Kulmala P, Hannila ML, Akerblom HK, et al. Models for predicting type 1 diabetes in siblings of affected children. Diabetes Care. 2006;29(3):662–7.
Kurrer MO, Pakala SV, Hanson HL, Katz JD. Beta cell apoptosis in T cell-mediated autoimmune diabetes. PNAS USA. 1997;94(1):213–8.
O’Brien BA, Harmon BV, Cameron DP, Allan DJ. Apoptosis is the mode of β-cell death responsible for the development of IDDM in the nonobese diabetic (NOD) mouse. Diabetes. 1997;46:750–7.
Wegmann DR, Norbury-Glaser M, Daniel D. Insulin-specific T cells are a predominant component of islet infiltrates in pre-diabetic NOD mice. Eur J Immunol. 1994;24(8):1853–7.
DiLorenzo TP, Serreze DV. The good turned ugly: immunopathogenic basis for diabetogenic CD8+ T cells in NOD mice. Immunol Rev. 2005;204:250–63. (250–263).
Burton AR, Vincent E, Arnold PY, Lennon GP, Smeltzer M, Li CS, et al. On the pathogenicity of autoantigen-specific T-cell receptors. Diabetes. 2008;57(5):1321–30.
Roep BO, Peakman M. Diabetogenic T lymphocytes in human type 1 diabetes. Curr Opin Immunol. 2011;23(6):746–53.
Calderon B, Suri A, Unanue ER. In CD4+ T-cell-induced diabetes, macrophages are the final effector cells that mediate islet beta-cell killing: studies from an acute model. Am J Pathol. 2006;169(6):2137–47.
Mathis D, Vence L, Benoist C. Beta-cell death during progression to diabetes. Nature. 2001;414(6865):792–8.
Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;435(7039):220–3.
Wicker LS, Miller BJ, Coker LZ, McNally SE, Scott S, Mullen Y, et al. Genetic control of diabetes and insulitis in the nonobese diabetic (NOD) mouse. J Exp Med. 1987;165:1639–54.
Suri A, Katz JD. Dissecting the role of CD4+ T cells in autoimmune diabetes through the use of TCR transgenic mice. Immunol Rev. 1999;169:55–65.
Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol. 2011;33(1):67–87.
Noble JA, Valdes AM, Cook M, Klitz W, Thomson G, Erlich HA. The role of HLA class II genes in insulin-dependent diabetes mellitus: molecular analysis of 180 Caucasian, multiplex families. Am J Human Genet. 1996;59(5):1134–48.
Baschal EE, Eisenbarth GS. Extreme genetic risk for type 1A diabetes in the post-genome era. J Autoimmun. 2008;31(1):1–6.
Slattery RM, Kjer-Nielsen L, Allison J, Charlton B, Mandel TE, Miller JFAP. Prevention of diabetes in non-obese diabetic I-Ak transgenic mice. Nature. 1990;345(6277):724–6.
Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med. 2009;360(16):1646–54.
Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes. 2008;57:1084–92.
Burren OS, Adlem EC, Achuthan P, Christensen M, Coulson RM, Todd JA. T1DBase: update 2011, organization and presentation of large-scale data sets for type 1 diabetes research. Nucleic Acids Res. 2011;39:D997–1001.
Yu L, Miao D, Scrimgeour L, Johnson K, Rewers M, Eisenbarth GS. Distinguishing persistent insulin autoantibodies with differential risk: nonradioactive bivalent proinsulin/insulin autoantibody assay. Diabetes. 2012;61(1):179–86.
Krishnamurthy B, Dudek NL, McKenzie MD, Purcell AW, Brooks AG, Gellert S, et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest. 2006;116(12):3258–65.
Anjos S, Polychronakos C. Mechanisms of genetic susceptibility to type I diabetes: beyond HLA. Mol Genet Metab. 2004;81(3):187–95.
Gardner JM, Fletcher AL, Anderson MS, Turley SJ. AIRE in the thymus and beyond. Curr Opin Immunol. 2009;21(6):582–9.
Dubois-Lafforgue D, Mogenet L, Thebault K, Jami J, Krief P, Boitard C. Proinsulin 2 knockout NOD mice: a model for genetic variation of insulin gene expression in type 1 diabetes. Diabetes. 2002;51(Suppl 3):S489–93.
Moriyama H, Abiru N, Paronen J, Sikora K, Liu E, Miao D, et al. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. PNAS USA. 2003;100(18):10376–81.
Alleva DG, Maki RA, Putnam AL, Robinson JM, Kipnes MS, Dandona P, et al. Immunomodulation in type 1 diabetes by NBI-6024, an altered peptide ligand of the insulin B(9–23) epitope. Scand J Immunol. 2006;63(1):59–69.
Kash SF, Condie BG, Baekkeskov S. Glutamate decarboxylase and GABA in pancreatic islets: lessons from knock-out mice. Horm Metab Res. 1999;31(5):340–4.
Kubosaki A, Miura J, Notkins AL. IA-2 is not required for the development of diabetes in NOD mice. Diabetologia. 2004;47(1):149–50.
Kubosaki A, Gross S, Miura J, Saeki K, Zhu M, Nakamura S, et al. Targeted disruption of the IA-2beta gene causes glucose intolerance and impairs insulin secretion but does not prevent the development of diabetes in NOD mice. Diabetes. 2004;53(7):1684–91.
Delong T, Baker RL, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, et al. Islet amyloid polypeptide is a target antigen for diabetogenic CD4+ T cells. Diabetes. 2011;60(9):2325–30.
Oeser JK, Parekh VV, Wang Y, Jegadeesh NK, Sarkar SA, Wong R, et al. Deletion of the G6pc2 gene encoding the islet-specific glucose-6-phosphatase catalytic subunit-related protein does not affect the progression or incidence of type 1 diabetes in NOD/ShiLtJ mice. Diabetes. 2011;60(11):2922–7.
Daniel D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur J Immunol. 1995;25(4):1056–62.
Zhang L, Nakayama M, Eisenbarth GS. Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol. 2008;20(1):111–8.
Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. PNAS USA. 2010;107(24):10978–83.
Levisetti MG, Suri A, Petzold SJ, Unanue ER. The insulin-specific T cells of nonobese diabetic mice recognize a weak MHC-binding segment in more than one form. J Immunol. 2007;178(10):6051–7.
Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, et al. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. PNAS USA. 2011;108(40):16729–34.
Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. 2010;11(4):350–4.
Daniel C, Weigmann B, Bronson R. von BH. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med. 2011;208(7):1501–10.
Simone E, Daniel D, Schloot N, Gottlieb P, Babu S, Kawasaki E, et al. T cell receptor restriction of diabetogenic autoimmune NOD T cells. Proc Natl Acad Sci USA. 1997;94(6):2518–21.
Nakayama M, Castoe T, Sosinowski T, He X, Johnson K, Haskins K, et al. Germline TRAV5D-4 T-Cell Receptor Sequence Targets a Primary Insulin Peptide of NOD Mice. Diabetes. 2012;61(4):857–65.
Zhang L, Jasinski JM, Kobayashi M, Davenport B, Johnson K, Davidson H, et al. Analysis of T cell receptor beta chains that combine with dominant conserved TRAV5D-4*04 anti-insulin B:9–23 alpha chains. J Autoimmun. 2009;33(1):42–9.
Liu Z, Cort L, Eberwine R, Herrmann T, Leif JH, Greiner DL, et al. Prevention of type 1 diabetes in the rat with an allele-specific anti-T-cell receptor antibody: Vbeta13 as a therapeutic target and biomarker. Diabetes. 2012;61(5):1160–8.
Zhang L, Stadinski BD, Michels A, Kappler JW, Eisenbarth GS. Immunization with an insulin peptide-MHC complex to prevent type 1 diabetes of NOD mice. Diabetes Metab Res Rev. 2011;27(8):784–9.
Michels AW, Ostrov DA, Zhang L, Nakayama M, Fuse M, McDaniel K, et al. Structure-based selection of small molecules to alter allele-specific MHC class II antigen presentation. J Immunol. 2011;187(11):5921–30.
Prediction of type IA diabetes: the natural history of the prediabetic period, chapter 11. Barbara Davis Center for Diabetes online book. http://barbaradaviscenter.org/. Accessed August 2012.
Acknowledgments
This work was supported by grants from the National Institute of Health (R01 DK 032083, U19AI050864, P30 DK 057516, NO1 AI 15416), the International Autoimmunity Center, the Juvenile Diabetes Research Foundation, the Brehm Coalition, the Helmsley Foundation, and the Children’s Diabetes Foundation.
Conflict of interest
Dr. Eisenbarth is on two university provisional patents for treating autoimmunity with small molecules. There is also a research grant from Novartis in the same area. Part of Dr. Sosinowski’s research is funded by a grant from Novartis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sosinowski, T., Eisenbarth, G.S. Type 1 diabetes: primary antigen/peptide/register/trimolecular complex. Immunol Res 55, 270–276 (2013). https://doi.org/10.1007/s12026-012-8367-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-012-8367-6