
Abstract
Although autoantibodies are the hallmarks of most autoimmune diseases, the mechanisms by which autoreactive B cells are generated and accumulate are still poorly understood. Overexpression of Toll-like receptor 7 (TLR7) that recognizes single-stranded RNAs has been implicated in systemic lupus erythematosus (SLE), although the cellular mechanism by which this receptor drives the disease is unknown. We recently identified a population of CD11c+ age-associated B cells (ABCs) which is driven by TLR7 signaling, secretes autoantibodies and appears in autoimmune-prone mice by the time of onset of autoimmunity. Mice lacking the Mer receptor develop autoantibodies and splenomegaly similar to other mouse models of SLE. Here, we show that Mer−/− mice that lack TLR7 fail to develop anti-chromatin IgG antibodies, perhaps because they also fail to develop ABCs. Moreover, depletion of CD11c+ ABCs from Mer−/− mice leads to rapid reduction in autoantibodies. Together, these data strongly suggest that ABCs and/or their descendants are the primary source of autoantibodies in Mer−/− mice and that TLR7 signaling is crucial for accumulation of ABCs and development of autoantibodies. These data demonstrate for the first time that TLR7, and not TLR9, is responsible for generation of anti-chromatin IgG antibodies in Mer−/− mice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The majority of autoimmune diseases are often identified and characterized by the appearance of antibodies that react with self-antigens. Such autoantibodies often lead to the progression of the disease although in some cases their direct effect on the development of autoimmunity is not clear. In the case of systemic lupus erythematosus (SLE), it is well known that autoantibodies play an important role in the development of kidney pathology, and therefore, autoreactive B cells are one of the major targets in the treatment of SLE disease [1].
There are many animal models of SLE including mice deficient in Mer, a receptor tyrosine kinase, which has many functions including uptake of dead cells. Mer−/− mice are characterized by inefficient clearance of dead cells, splenomegaly, and development of autoantibodies against self-DNA and nuclear proteins and thus behave similarly to many mouse models of SLE [2–4]. In this study, we used Mer−/− mice as a model for lupus-like autoimmunity in which the absence of one well-characterized gene leads to the appearance of autoantibodies.
Recently, we and others identified a population of B cells named age-associated B cells (ABCs), which secretes autoantibodies and accumulates in autoimmune-prone mice, such as Mer−/− mice, at the onset of autoimmunity [5, 6]. We also demonstrated that accumulation of ABCs in aged female C57BL/6 mice depends on intrinsic Toll-like receptor 7 (TLR7) signaling in B cells. TLR7 recognizes single-stranded RNA and potentially can be activated in vivo by self-RNA [7, 8], for example, from necrotic cells that are not cleared by Mer-deficient macrophages. Overexpression of TLR7 has been shown to be sufficient to drive accumulation of autoantibodies and mice with spontaneous translocation of tlr7 gene develop lupus-like disease [9, 10].
Here, we investigated whether TLR7 signaling influences autoimmunity in Mer−/− mice and whether the number of ABCs is dependent on the amount of TLR7 expressed by immune cells in Mer−/− mice. To address these questions we crossed Mer−/− mice to TLR7−/− mice and analyzed mice expressing different amounts of the tlr7 gene. We compared the appearance of ABCs and autoantibodies in these mice over time and demonstrated that both these factors strongly correlate with the number of tlr7 gene copies in the mice. Moreover, mice that had complete ablation of TLR7 not only failed to accumulate ABCs but also did not develop anti-chromatin antibodies throughout the course of the experiment. We also demonstrated that expression of the transcription factor T-bet in B cells correlates with the number of tlr7 gene copies, further suggesting that T-bet might be a lineage-defining factor for ABCs. Additionally, since TLR7 signaling is known to be important for the generation of anti-RNA antibodies, and because anti-chromatin antibodies are thought to be dependent on TLR9 signaling, these data are the first demonstration to our knowledge of the dependence of anti-chromatin antibodies on TLR7 expression.
Research design and methods
Experimental animals
Mer−/− mice on a C57BL/6 genetic background were obtained from Dr. Douglas Graham (University of Colorado, Anschutz Medical Campus). CD11c-DTR/GFP mice were purchased from Jackson Laboratories. Mer−/− CD11c-DTR/GFP mice were obtained by breeding Mer−/− and CD11c-DTR/GFP mice to each other. Mer−/− mice were also bred to TLR7−/− and mice with different number of tlr7 gene copies were maintained. Genotyping for Mer was performed by PCR, and flow cytometry was performed to determine expression of CD11c-DTR/GFP transgene. All manipulations were performed in accordance with the National Jewish Health Institutional Animal Care and Use Committee.
Diphtheria toxin treatment
For depletion of CD11c+ cells, Mer−/− CD11c-DTR/GFP mice were injected intraperitoneally with 4 ng/g body weight Diphtheria toxin (in PBS; Sigma). The efficacy of the depletion was examined using flow cytometry 7 and 15 days after treatment.
Detection of autoantibodies
Concentrations of anti-chromatin IgG antibodies were determined using the protocol of Guth et al. [11]. Briefly, 96-well microplates were coated overnight at 4 °C with mouse chromatin (10 μg/mL), followed by incubation with blocking buffer solution at 37 °C for 2 h. Mouse sera diluted in blocking buffer were added to the trays for 2 h. IgG anti-chromatin antibodies were detected with an alkaline phosphatase (AP)–conjugated goat anti-mouse IgG antibody (Southern Biotechnology Associates, Inc.).
Flow cytometry
Cells were stained under saturating conditions with antibodies to mouse CD3 (clone 145-2C11), B220 (clone RA3-6B2), CD11b (clone M1/70), CD11c (clone N418), CD19 (clone 1D3) and T-bet (clone 4B10) purchased from Ebiosciences or BD Pharmingen, or generated in house. For intracellular, T-bet staining cells were surface-stained, fixed and permeabilized with FoxP3 staining buffer set (eBioscience) and stained with anti-human/mouse T-bet antibodies (clone 4B10). Cells were analyzed by flow cytometry on Cyan (Beckman Coulter) instrument, and data were analyzed using FlowJo software (Treestar).
Results
TLR7 is required for ABCs development in autoimmune-prone Mer−/− mice and for the appearance of anti-chromatin IgG autoantibodies
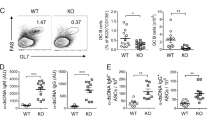
We previously demonstrated that TLR7 is essential for the development of ABCs in aged C57BL/6 female mice and wondered whether this phenomenon persists only in aged wild-type female mice or is it also true for autoimmune-prone mice [5]. Since we recently demonstrated the appearance of ABCs at an early age in Mer−/− mice, we crossed Mer−/− mice to TLR7−/− mice and tested mice with different genotypes for the presence of ABCs. Since tlr7 is encoded on the X chromosome, females have two copies of this gene while males posses only one copy of tlr7. Therefore, the following genotypes were analyzed for the presence of ABCs: Mer−/− TLR7+/+ females, Mer−/− TLR7+/− females, Mer−/− TLR7−/− females, Mer−/− TLR7+ males, Mer−/− TLR7− males. Mice were analyzed when they were 6–8 months old, an age at which Mer−/− mice already possess autoantibodies, for the presence of ABCs in their spleens. As shown in Fig. 1a, ABCs accumulate in Mer−/− mice that possess at least one copy, but not in mice that completely lack, tlr7. Moreover, the percentage of ABCs in these mice directly correlated with the number of copies of tlr7 in the animals. These data are not only in accordance with our previous findings that the accumulation of ABCs requires TLR7 [5], but also suggest a direct correlation between accumulation of ABCs and TLR7 expression.

a TLR7 deficiency prevents accumulation of ABCs in Mer−/− mice. Flow cytometry was performed on splenocytes from Mer-deficient mice with various levels of TLR7 expression. At least 5 mice per group were analyzed. ABCs were identified as CD3−CD19+B220+CD11c+, and the percentage of ABCs among B cells is demonstrated. Bars represent mean (±SEM) of at least 10 mice per group. *p < 0.001 (Student’s two-tailed t test). b TLR7 deficiency results in the absence of autoantibody accumulation. Serum from Mer−/−TLR7+/+ females (solid squares), Mer−/−TLR7+/− females (gradient squares, dashed line), Mer−/−TLR7−/− females (open squares), Mer−/−TLR7+ males (solid circles), Mer−/−TLR7− males (open circles) at different age was analyzed for the presence of anti-chromatin IgG antibodies, and percentage of mice positive for autoantibodies is shown. The sample was counted positive if the titer was 5 times over the titer of control C57BL/6 mice. At least 10 animals per age group were analyzed
Next we tested whether the appearance of autoantibodies in mice with different numbers of copies of the tlr7 gene correlated with the appearance of ABCs. We monitored mice for the appearance of anti-chromatin autoantibodies for 6 months by bleeding mice every month. We determined that 85 % of female mice that possess two copies of tlr7 (TLR7+/+) developed autoantibodies by 4 months of age, while only 50 % of TLR7+/− females and TLR7+ males developed anti-chromatin antibodies by this age (Fig. 1b). Interestingly, none of the mice that completely lacked TLR7 developed autoantibodies throughout the course of study, even by 12 months of age, suggesting that TLR7 is responsible for the appearance of both ABCs and anti-chromatin autoantibodies (Fig. 1b and data not shown). Moreover, we also saw a strong correlation between the percentage of ABCs and the amount of autoantibodies in Mer−/− mice (data not shown). These data demonstrate a very strong correlation between three different factors in Mer−/− mice: TLR7 expression, number of ABCs and development of autoantibodies. We have shown here and previously that accumulation of ABCs depends on TLR7 expression and that ABCs are enriched in autoreactivity, and therefore, the absence of autoantibodies in Mer−/−TLR7−/− mice is very likely due to the absence of ABCs in these mice.
Expression of the transcription factor T-bet in ABCs correlates with TLR7 expression
We have previously shown that RNA levels of the transcription factor, T-bet, are upregulated in ABCs isolated from aged female C57BL/6 mice [5]. Moreover, we have preliminary data suggesting that T-bet expression is required for the development of ABCs (Rubtsova et al., manuscript in preparation). T-bet is not a transcription factor commonly expressed by B cells but rather a characteristic transcription factor for CD4+ Th1 cells and also important for the function of CD8+ T cells and natural killer (NK) cells [12–15]. There were, however, several reports showing that T-bet can be expressed in B cells as well as it has a role in immunoglobulin class switching [16–19]. Since transcription factors are usually the most accurate signature of one or another cell type, we decided to determine whether ABCs from Mer−/− mice also express T-bet. Intracellular staining of splenocytes from Mer−/− mice demonstrated that their ABCs indeed express higher levels of T-bet than conventional follicular (FO) B cells (Fig. 2a). Moreover, analysis of Mer−/− mice with different expression levels of TLR7 revealed that ABCs from females with one or two copies of TLR7 express significantly higher levels of T-bet than ABCs from TLR7+ male mice or than the few ABC-like cells that appear in TLR7−/− mice (Fig. 2b). These data suggest that Mer−/− mice do indeed accumulate ABCs that are identical to the cells identified by us and others previously [5, 6, 20]. Interestingly, the T-bet is also expressed at higher levels in FO B cells from TLR7+/+ mice than in the same population of cells from TLR7−/− mice, although the level (mean fluorescent intensity) of T-bet expression is significantly lower in these cells compared with ABCs (Fig. 2c). Since it has been previously shown that ABCs originate from FO B cells [6], these data suggest that some intrinsic signals in Mer−/− mice initiate the expression of T-bet in FO B cells, promoting their differentiation into ABCs. In support of this hypothesis, we found a strong correlation between the levels of T-bet expression in B cells and the percentages of ABCs in spleens of Mer−/− mice (Fig. 2d).

a ABCs from Mer-deficient mice express higher level of T-bet than FO B cells. Splenocytes from Mer−/− mice were stained with CD19, B220, CD11c, CD3 and T-bet. ABCs (solid black line) were identified as CD3−CD19+B220+CD11c+, and FO B cells (dashed gray line) are CD3−CD19+B220+CD11c−. Mean fluorescence intensity (MFI) of T-bet expression is shown. Staining is a representative of at least five independent experiments. b T-bet expression in ABCs from Mer−/− mice with different expression of TLR7. Average MFI and standard error is shown for at least 3 mice per group. c T-bet expression in FO B cells from Mer−/− mice. Average MFI and standard error is shown for at least 3 mice per each group. *p < 0.005; **p < 0.05 (Student’s two-tailed t test). d Correlation of percentage of ABCs with the expression of T-bet in all B cells. Linear regression analysis was applied, and statistical significance is demonstrated
Depletion of ABCs leads to a decrease in autoantibodies in Mer−/−CD11c-DTR/GFP mice
Next we tested whether depletion of ABCs would affect the level of autoantibodies in Mer−/− mice. Since there is no direct way to deplete ABCs in Mer−/− mice, we crossed these animals to mice expressing GFP and diphtheria toxin receptor (DTR) under the control of the CD11c promoter. In these mice, CD11c expressing cells can be depleted by the injection of diphtheria toxin, and while CD11c is expressed on dendritic cells, it is also expressed by ABCs but not other B cells [5]. Although dendritic cells can play an important role in the development and progression of autoimmunity, their depletion most quickly affects T cell-mediated autoimmunity and not the levels of autoantibodies [21]. In addition, we have shown previously that depletion of ABCs leads to a rapid (by day 7) reduction in the level of autoantibodies [5]. Using Mer−/−CD11c-DTR/GFP mice at the age of 6–8 months which already posses autoantibodies, we demonstrated that injection of diphtheria toxin leads to rapid depletion of ABCs and subsequent reduction in anti-chromatin IgG autoantibodies (Fig. 3a, b). These data suggest that ABCs are the source of autoantibodies in Mer−/− mice. Such rapid reduction in the level of autoantibodies in the blood shortly after diphtheria toxin injection suggests that ABCs, not dendritic cells, are mainly responsible for this effect. These data directly demonstrate that ABCs are responsible for accumulation of autoantibodies although additional experiments, such as generation of bone marrow chimeras, are required to exclude the role of dendritic cells in this process.

a Depletion of ABCs in blood by diphtheria toxin in Mer−/− CD11c-DTR/GFP mice. Mice were bled 0, 7 and 15 days after injection with diphtheria toxin or PBS, and blood was analyzed for the presence of ABCs by flow cytometry. Each treatment group contained 5 mice before treatment. b Titer of anti-chromatin IgG autoantibodies in serum of Mer−/− CD11c-DTR/GFP mice was determined 0, 7 and 15 days after injection of diphtheria toxin. Each treatment group contained 5 mice. *p < 0.01 (Student’s two-tailed t test)
Together these data provide evidence that in Mer−/− mice, ABCs are the main source of autoantibodies and that lack of ABCs, either due to TLR7 deficiency or due to their depletion, leads to significant reduction in autoantibodies.
Discussion
While autoantibodies play an important role in a wide variety of autoimmune diseases, mechanisms by which autoantibody-producing cells are generated and get activated are poorly understood. Multiple mechanisms of tolerance exist to ensure that cells that possess receptors reactive to self-antigens either do not reach the periphery or are silenced and do not participate in the immune response. And yet, in a majority of autoimmune diseases, autoreactive B cells manage to escape tolerance and produce autoantibodies that, in many cases, do significant damage to the body. There are a number of explanations for this phenomenon including failure of negative selection, receptor editing, deficiency in other cells that suppress autoreactive B cells or the nature of the antigen and molecular mimicry [22–24]. The role of TLRs in the activation of autoreactive B cells has also been proposed by several research groups. Such roles include the possibility that certain TLR ligands can bind both antigen receptor and TLRs on or in B cells, thereby delivering two signals at once [25–28]. This idea is particularly relevant in the case of intracellular TLRs (TLR3, 7, 9), as their ligands, nucleic acids in complex with proteins, can be delivered to endosomes via autoantigen-engaging B cell receptors, the immunoglobulin proteins expressed by individual B cells. There is a significant amount of literature supporting the role of TLR7 and TLR9 in autoimmunity, although how these TLRs exactly act is still largely unknown [9, 10, 28–31].
We have previously demonstrated that TLR7 is required for accumulation of a CD11c-expressing B-cell population that accumulates in autoimmune-prone mice and can secrete autoantibodies [5]. Here, we demonstrated that TLR7 ablation is sufficient to prevent accumulation of ABCs, even in autoimmune-prone Mer−/− mice. Moreover, the fact that the percentage of ABCs strongly correlates with the number of copies of the X-linked TLR7 gene expressed in the mice might explain why these cells appear in elderly female and not male C57BL/6 mice. Although there are no direct data suggesting unequal TLR7 expression in male and female mice, it has been demonstrated that female mice produce more type I interferon in response to TLR7 stimulation than male mice [32]. Interestingly, this sex-biased difference is unique to TLR7 and does not persist in case of TLR9 stimulation [32]. Also, in humans, it has been demonstrated that part of the X chromosome that contains TLR7 can exist in a partially Lyonized form [33, 34]. The data presented here support the hypothesis that B cell responsiveness to self-antigens strongly correlates with the number of TLR7 genes encoded in the genome.
Although TLR7 recognizes single-stranded RNAs, here we demonstrated the absence of anti-chromatin IgG antibodies in Mer−/− mice that are deficient in TLR7. To our knowledge, this is the first demonstration of the role of TLR7 in the generation of anti-chromatin autoantibodies. Chromatin-specific antibodies have been previously largely associated with TLR9 and not TLR7 signaling [29–31]. Our data provide evidence that for the production of anti-chromatin autoantibodies, B cells have to receive TLR7 stimulation and, in the absence of such a signal, they fail to produce chromatin-specific antibodies. Since chromatin is a mixture of DNA, RNA and nuclear proteins, it is surprising to see the complete absence of anti-chromatin antibodies in Mer−/−TLR7−/− mice. These data strongly suggest that RNA/protein complexes are more dominant or more abundant than DNA/protein complexes in Mer−/− mice.
Most immune cell populations have specific transcription factors that define their lineage specificity and uniqueness. Previously, we performed gene expression analysis and determined that RNA encoding transcription factor T-bet is expressed in ABCs from wild-type mice at significantly higher amount than in other B-cell populations [5]. Here, we confirmed these data using antibody staining against T-bet in Mer−/− mice and demonstrated a strong correlation between expression of T-bet and TLR7. These results suggest that there might be a direct link between these two proteins. Moreover, our data demonstrate that, although to a lesser extent, T-bet is upregulated in FO B cells in Mer−/− mice. It has been previously shown that ABCs can develop from FO B cells, and our data provide additional support that this is the case. We are currently investigating the role of T-bet in ABC’s development and the exact mechanisms by which T-bet gets turned on in B cells.
These data lead us to propose a model for the development of ABCs in Mer−/− mice (Fig. 4), which fail to efficiently uptake dead cells and therefore possess a source of autoantigens in their blood and secondary lymphoid organs such as the spleen. These autoantigens often represent a complex of DNA or RNA with several nuclear proteins and therefore can bind to specific B cell receptors and get internalized. Alone, this event is not sufficient to activate autoreactive B cells, but since these complexes contain ligands for intracellular TLRs (TLR7 and 9), it is possible that binding of antigen to the B cell receptor allows its delivery to endosomes where intracellular TLRs are localized. Therefore, one complex antigen provides simultaneously two signals: B-cell receptor activation and TLR stimulation. These stimuli together are very efficient in upregulating T-bet and B-cell differentiation into CD11c+ age-associated B cells, which have an activated phenotype and are ready to secrete autoantibodies. Although this model still has some unanswered points, it is strongly supported by our data, and further experiments will elucidate the exact role of ABCs in autoimmunity. For example, according to our model, both TLR7 and TLR9 should facilitate the activation of autoreactive B cells. We have shown previously that only TLR7, and not TLR9, activation leads to accumulation of ABCs [5]. The opposite roles of TLR7 and TLR9 in autoimmunity, with TLR7 being harmful and TLR9 playing a protective role, have been previously described [28, 29, 31]. However, the mechanism explaining these differences has never been found. Similarly, the mechanism explaining why the activation of TLR9 does not lead to the accumulation of ABCs still has to be discovered.

Model showing nature of appearance of autoantibodies in Mer-deficient mice. Inefficient uptake of dead cells leads to the appearance of autoantigens such as DNA, RNA and nucleoproteins in blood, which in turn is recognized by autoreactive B cells through B cell receptor (BCR). After binding, BCR gets internalized and delivers antigen to endosomes where TLR7 is localized. Recognition of RNA and nucleoproteins delivered by BCR initiates TLR7 signaling leading to the upregulation of T-bet and accumulation of ABCs which are the source of autoantibodies in Mer−/− mice
References
Lefkowith JB, Gilkeson GS. Nephritogenic autoantibodies in lupus: current concepts and continuing controversies. Arthr Rheum. 1996;39(6):894–903.
Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196(1):135–40.
Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411(6834):207–11.
Rubtsov AV, Rubtsova K, Kappler JW, Marrack P. Genetic and hormonal factors in female-biased autoimmunity. Autoimmun Rev. 2010;9(7):494–8.
Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c B-cell population is important for the development of autoimmunity. Blood. 2011;118(5):1305–15.
Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. 2011;118(5):1294–304.
Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc Nat Acad Sci USA. 2004;101(15):5598–603.
Diebold SS. Recognition of viral single-stranded RNA by toll-like receptors. Adv Drug Deliv Rev. 2008;60(7):813–23.
Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27(5):801–10.
Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312(5780):1669–72.
Guth A, Detanico T, Smith D, Tung K, Bonorino C, Wysocki L. Spontaneous autoimmunity in mice that carry an IghV partial transgene: a required arginine in VHCDR3. Lupus. 2009;18(4):299–308.
Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, et al. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. 2004;20(4):477–94.
Robbins SH, Tessmer MS, Van Kaer L, Brossay L. Direct effects of T-bet and MHC class I expression, but not STAT1, on peripheral NK cell maturation. Eur J Immunol. 2005;35(3):757–65.
Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med. 2009;206(1):51–9.
Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302(5647):1041–3.
Gerth AJ, Lin L, Peng SL. T-bet regulates T-independent IgG2a class switching. Int Immunol. 2003;15(8):937–44.
Liu N, Ohnishi N, Ni L, Akira S, Bacon KB. CpG directly induces T-bet expression and inhibits IgG1 and IgE switching in B cells. Nat Immunol. 2003;4(7):687–93.
Peng SL, Li J, Lin L, Gerth A. The role of T-bet in B cells. Nat Immunol. 2003;4(11):1041; author reply.
Peng SL, Szabo SJ, Glimcher LH. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Nat Acad Sci USA. 2002;99(8):5545–50.
Rubtsova K, Marrack P, Rubtsov AV. Age-associated B cells: are they the key to understanding why autoimmune diseases are more prevalent in women? Expert Rev Clin Immunol. 2012;8(1):5–7.
Isaksson M, Ardesjo B, Ronnblom L, Kampe O, Lassmann H, Eloranta ML, et al. Plasmacytoid DC promote priming of autoimmune Th17 cells and EAE. Eur J Immunol. 2009;39(10):2925–35.
Halverson R, Torres RM, Pelanda R. Receptor editing is the main mechanism of B cell tolerance toward membrane antigens. Nat Immunol. 2004;5(6):645–50.
Pelanda R, Torres RM. Receptor editing for better or for worse. Curr Opin Immunol. 2006;18(2):184–90.
Ait-Azzouzene D, Skog P, Retter M, Kouskoff V, Hertz M, Lang J, et al. Tolerance-induced receptor selection: scope, sensitivity, locus specificity, and relationship to lymphocyte-positive selection. Immunol Rev. 2004;197:219–30.
Green NM, Marshak-Rothstein A. Toll-like receptor driven B cell activation in the induction of systemic autoimmunity. Semin Immunol. 2011;23(2):106–12.
Crampton SP, Voynova E, Bolland S. Innate pathways to B-cell activation and tolerance. Ann N Y Acad Sci. 2010;1183:58–68.
Avalos AM, Busconi L, Marshak-Rothstein A. Regulation of autoreactive B cell responses to endogenous TLR ligands. Autoimmunity. 2010;43(1):76–83.
Shlomchik MJ. Activating systemic autoimmunity: B’s, T’s, and tolls. Curr Opin Immunol. 2009;21(6):626–33.
Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25(3):417–28.
Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202(9):1171–7.
Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202(2):321–31.
Berghofer B, Frommer T, Haley G, Fink L, Bein G, Hackstein H. TLR7 ligands induce higher IFN-alpha production in females. J Immunol. 2006;177(4):2088–96.
Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434(7031):400–4.
Yen ZC, Meyer IM, Karalic S, Brown CJ. A cross-species comparison of X-chromosome inactivation in Eutheria. Genomics. 2007;90(4):453–63.
Acknowledgments
The authors thank Ms. D. Garcia and Drs. T. Detanico, M. Phillips and M. Munks for assistance. This work was supported in part by USPHS AI-18785, AI-22295, AI-52225 and AI-046374 and Colorado Bioscience Discovery Evaluation Grant Program (#11BGF-20).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Rubtsov, A.V., Rubtsova, K., Kappler, J.W. et al. TLR7 drives accumulation of ABCs and autoantibody production in autoimmune-prone mice. Immunol Res 55, 210–216 (2013). https://doi.org/10.1007/s12026-012-8365-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-012-8365-8