
Abstract
A detailed understanding of the cellular response to human immunodeficiency virus (HIV-1) infection is needed to inform prevention and therapeutic strategies that aim to contain the AIDS pandemic. The cellular immune response plays a critical role in reducing viral load in HIV-1 infection and in the nonhuman primate model of SIV infection. Much of this virus suppressive activity has been ascribed to CD8+T-cell-directed cytolysis of infected CD4+T cells. However, emerging evidence suggests that CD8+T cells can maintain a lowered viral burden through multiple mechanisms. A thorough understanding of the CD8+T-cell functions in HIV-1 infection that correlate with viral control, the populations responsible for these functions, and the elicitation and maintenance of these responses can provide guidance for vaccine design and potentially the development of new classes of antiretroviral therapies. In this review, we discuss the CD8+T-cell correlates of protection in HIV-1 and SIV infection and recent advances in this field.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The CD8+T-cell response to HIV-1 infection
The human immunodeficiency virus (HIV) presents a unique set of challenges to the cellular immune system. The rapidly evolving HIV-1 genome produces an ever-changing milieu of available antigenic epitopes, while targeting cells that play a central role in the cellular immune responses. Thus, the ability of the immune system to respond is impaired. While currently approved therapeutics take advantage of weaknesses in the viral structure and replication cycle, a successful vaccine must rely upon the generation of effective immune responses that are rapidly available and functional at the time of virus challenge.
Engaging the most robust cellular response to limit virus replication for those that do become infected will also have a tremendous impact on the HIV-1 epidemic. Because high plasma viral load correlates with risk of HIV-1 transmission, a reduction in virus load by functional CD8+T cells is likely to decrease the number of new HIV-1 infections at the population level. During acute HIV-1 infection, CD8+T-cell responses correlate with the initial decline in virus replication [1, 2] (reviewed in [3]). Despite this decline in HIV-1 viremia, the majority of HIV-infected individuals are unable to control virus replication below the level of detection without antiretroviral therapy. Studies of immune responses in those few who are able to control HIV-1 replication have found strong associations between the immune response driven by CD8+T cells and viral control. Early CD8+-cell depletion studies in experimentally SIV-infected rhesus macaques demonstrated that CD8+T cells are critical for controlling virus replication in vivo; without these cells, virus replicates wantonly to up to ten times the levels seen in the presence of CD8+T cells [4–6]. Furthermore, it has been well established that those HIV-1 seropositive individuals who maintain high CD4+T-cell counts and are clinically healthy for extended periods of time (long-term nonprogressors) [7–11] and those who control viral replication to low (virus controllers) or undetectable (elite controllers) [1, 12, 13] levels are more likely to have strong, early CD8+T-cell responses than those who progress to disease. Viral control and low setpoint have also been attributed to specific HLA types (e.g., HLA B57, 5801, 27) [14–17], with recent genome-wide association studies ascribing 12% of viral setpoint variability to HLA-related variation [18].
Thus, the CD8+T-cell response to HIV infection has been shown by various means to be critical for the control of HIV-1 and SIV replication once infection has been established. However, CD8+T cells are not a singular entity with a singular function. At any given time, these cells exist along a complicated differentiation pathway and are capable of numerous functions, many of which could affect viral replication. While a great deal of effort has been applied to understanding the character of the CD8+T cells that are responsible for viral control, it is an ongoing and evolving field, particularly in regard to the assays used to measure and characterize CD8+T cells. It is clear from the results of a recent vaccine study [19] that the full anti-HIV functional properties of CD8+T cells must be determined in order to define a correlate for protection. We highlight here some recent advances in the understanding of CD8+T cells and the correlates of viral control in both HIV-1 and SIV infection.
CD8+ T-cell-dependent virus evolution
Key evidence that an immune response is critical for HIV control can be found in the effect that the response has on virus replication and evolution. CTL activity has long been known to arise concurrently with viral decline ([1, 2] (reviewed [20]) (Fig. 1) and to select for escape variants [21–24] (reviewed [3]). These functional virus-specific T-cell responses arise earliest among all of the virus-specific adaptive immune responses that eventually appear following transmission. Recently, these observations have been extended in a study combining mathematical modeling, functional IFNγ ELISpot assays, and intracellular cytokine staining to demonstrate in four acutely infected patients that transmitted/founder viruses can escape from CTL responses as rapidly as 2–4 weeks following screening (Fiebig stage II) [24]. Additional mathematical modeling applied to the observed evolutionary changes in CTL epitopes suggested that the very early responding CD8+T cells in these patients had a causal effect on the control of acute viremia.

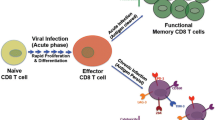
CD8 + T-cell responses impact viral load and setpoint. CD8+T-cell responses arise rapidly following infection. Viral load in plasma (shown in blue) peaks during the acute phase and is typically lowered to a detectable viremic setpoint in the absence of antiretroviral therapy, concurrent with the development of the CD8+T-cells response (shown in red). CD8+T cells contribute toward this viral load reduction through lytic clearance of infected cells and inhibition of virus replication. Successful virus control in the absence of therapy is associated both with strong HLA-dependent CTL activity and increased expression of cytokines, suggesting a role for nonlytic mechanisms as well. (Color figure online)
Subsequently, ultra deep-sequencing of longitudinal samples from three acutely infected patients (two overlapping with the study described above) was applied to an investigation into the early evolution of CD8+T-cell epitopes [25]. The vast majority of sequence diversity within the first month following infection was found within CTL epitopes, suggestive of intense selective pressure exerted by CD8+T cells very early following infection. Likewise, a similar study of eight acutely SIV-infected macaques examining diversity in specific CTL epitopes in Nef, Tat, and Gag [26] found that SIV escape from CTL pressure also coincides with peak CD8+T-lymphocyte response followed by profound, but incomplete, clearance of the transmitted virus species. Balamurali and colleagues demonstrated through real-time PCR analysis of wild-type and CTL escape mutant SHIV-infected macaques that escape mutant–infected cells were cleared by CD8 + T cells at the same rate as cells infected with wild-type virus. These data suggest that CD8 + T-cell-mediated noncytolytic mechanisms may also exert sufficient pressure to drive immune escape [27].
Additional studies of HLA-restricted viral evolution have shown the impact of the CD8+T-cell response on viral fitness and replication capacity. Using replicative fitness assays on Gag variants identified during acute and chronic HIV infection, Brockman and colleagues have shown that CTL escape has a detrimental effect on virus fitness [28]. Furthermore, the replicative defects were more pronounced in escape from HLA-B57, B58, and B13 restricted epitopes and during acute infection. Fitness defects were rescued by compensatory mutation during chronic infection, even in the presence of known protective alleles. This suggests that long-term viral control is determined by CTL responses during the resolution of acute viremia or that additional CD8+T-cell functions are playing a role in controlling virus in these patients during the chronic phase of infection.
Viral targets of CD8+T-cell-mediated inhibition
HIV and SIV encode only nine defined proteins. However, each is recognized to varying degrees as targets for the T-cell response. A large body of evidence suggests that Gag-directed responses are immunodominant in chronic HIV and SIV infection and are associated with low viral loads [29–31]. Recent studies have re-examined the CD8+T-cell-related correlates of control in the context of elite control and CD8+T-cell-mediated antiviral activity. Following peak viremia, elite HIV controllers reduce and maintain plasma viral loads to levels below our ability to detect them (Fig. 1). Recent studies of this unique cohort have used IFNγ ELISpot as well as functional antiviral assays to affirm an important role for Gag-directed responses in viral control [32]. However, a lack of CD8+T-cell-mediated antiviral function in 5 of the 19 elite controllers examined in this study suggests that correlates of control outside of those measured in this study may be responsible for control in these individuals. A second recent study, conducted by Julg and colleagues, examined the role of epitope-specific CD8+T-cell-mediated antiviral activity during the chronic phase of clade C-infected subjects [33]. The breadth of the Gag-specific response, as defined by IFNγ release, was found to associate most strongly with antiviral activity and lower viral loads.
When considering the early effects on virus evolution and fitness, as described above, it becomes clear that CD8+T-cell responses likely change over the course of infection and those responses that occur early following infection may have a dramatic effect on the disease course. Studies examining the earliest CD8+T-cell-mediated responses to SIV infection have described a pattern of changing immunodominant regions immediately following infection and alternative epitopes. Mapping of the presentation kinetics and antiviral activity of CD8+T-cell epitopes in HIV and SIV infection has shown distinct patterns of presentation that, in part, depend upon HLA context [34, 35]. Studies of HIV infection of HLA-B27-positive individuals demonstrated CD8+T-cell-mediated antiviral activity associated with rapid presentation of Gag and Pol epitopes (within 6 h following infection), followed by Vpr epitopes late after infection (at 18 h). However, Gag presentation was still associated with the strongest antiviral response [36]. Following infection, immune responses may be mounted most rapidly to pre-existing targets that do not require de novo synthesis or to targets that are most rapidly generated. Presentation of Vpr- and Rev-directed epitopes in SIV-infected cells has been detected by kinetic intracellular cytokine staining assay (IFNγ and TNFα) at 6 h after infection, with one Rev epitope appearing after only 1 h of infection, suggesting that CD8+T cells recognizing these epitopes could act almost immediately following infection [37]. The dichotomy between the timing of the Vpr response in these two studies may be due to differences in the discreet epitopes being studied, human or animal model, or method used to assess the response. Additionally, the development of very early escape mutations in Nef epitopes suggests that Nef-specific CD8+ T cells can also exert significant selective pressure rapidly after infection [24].
Finally, emerging data have challenged the notion that epitopes are limited to these nine well-characterized proteins by suggesting that CD8+T cells can recognize epitopes derived from alternate reading frames [38, 39]. These cryptic epitopes were found to be immunogenic during both early and chronic HIV infection [39], accounted for up to a quarter of the total CD8+T-cell-mediated anti-SIV response, could be found at high frequency 1–2 weeks following SIV infection [38], and could be generated to high frequencies through vaccination [40]. The clear message from these studies is that infected cells can process and present antigen rapidly following infection and presumably following appropriately targeted vaccination.
Functional correlates of virus inhibition and T-cell differentiation
Two of the most critical questions regarding CD8+T-cell function that we can address in the race to develop an efficacious vaccine are as follows: which CD8+T cells are responsible for virus control and how do they inhibit virus. The studies described above have provided some clarity into the effect that CD8+T cells can have on virus, and the viral targets against which CD8+ cells are most effective. Current research is also directed at understanding which populations along the CD8+-cell differentiation pathway are most effective in inhibiting viral replication, and what functions they use to inhibit.
CD8+T cells differentiate along a pathway from naïve through central memory to terminal effector. It is widely accepted that effector and memory T cells are primed for a rapid secondary response to infection, whereas naïve T cells have not encountered antigen and are not immediately functionally active. Associations have been drawn between CD8+T-cell differentiation state and viral control [41–43]. However, the variable ability of cells within the central memory populations to inhibit viral replication has only recently been evaluated. Julg and colleagues not only addressed the inhibitory capacity of Gag-directed CD8+ cells (as described above) but also demonstrated that those patients who had broad Gag responses and were able to inhibit viral replication better also had higher frequencies of terminal effector CD8+ cells [33]. Yet, no relationship was found between viral inhibition and frequencies of cells within subsets of the central memory compartment. CD8+T cells from high Gag responders were significantly better able to proliferate, suggesting that cells outside of the terminal effectors are likely contributing to the antiviral effect. We have also investigated chronically HIV-infected virus controllers and seronegative vaccinees in an effort to dissect the ability of cells from various differentiation stages to inhibit viral replication by combining phenotypic cell sorts and functional antiviral assays [44]. Naïve cells, not unexpectedly, were unable to inhibit virus replication. However, cells from very early memory stages through terminal effectors were able to potently inhibit virus replication with no significant differences found between memory cells and terminal effectors.
Antiviral function in CD8+T cells is not characterized by a single cellular function, and individual cells may be capable of producing many effector molecules (Fig. 2). Polyfunctionality is often described as the ability of a cell to produce at least 3 measured markers (typically, CD107a, MIP-1β, IFNγ, IL-2, TNFα, and perforin) and has been associated with CD8+T-cell-mediated virus control [9, 45, 46]. However, the correlates of antiviral function are not yet clearly understood. We have demonstrated that CD8+T-cell-mediated antiviral function in virus controllers and vaccinees is associated with Clade B Env and Gag-specific MIP-1β and CD107a expression as monofunctional and dualfunctional populations and that IFNγ correlates with viral inhibition only in the context of co-expression with one or both of these markers [44]. In other studies, epitope-specific evaluation of HLA-B and HLA-C-restricted responses demonstrated a predominance of monofunctional cells, the highest being MIP-1β, followed by IFNγ and CD107a [47]. Higher percentages of polyfunctional cells positively correlated with CD4+T cell counts, but no significant correlations were found between polyfunctionality and viral load. CTL activity is associated with high levels of perforin. In a recent study examining CTL correlates of viral control, Hersperger and colleagues demonstrated clear associations between elite virus control and increased perforin expression in largely terminal effector populations [48]. Furthermore, the level of perforin expressing cells correlated with viral load. IL-2 and TNFα were also expressed to higher levels in elite controllers and virus controllers than in individuals who were viremic and progressed, while MIP-1α and IFNγ expression levels were statistically higher only in virus controllers, but not elite controllers. It is unclear at this time which, if any, of these molecules are entirely responsible for mediating virus inhibition or whether they are acting in concert with co-expressed but currently unmeasured factors.

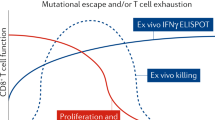
Multifactorial nature of CD8+T-cell-mediated inhibition s of HIV-1 and SIV replication. Illustrated here is a CD8+T cell (red) interacting with HIV-1 or SIV-infected CD4+T cells (blue). CD8+T cells may direct lysis by releasing perforin and granzymes. Additionally, CD8+T cells may inhibit virus replication through secretion of cytokines, chemokines, and other unidentified molecules. (Color figure online)
Mechanisms of CD8+T-cell-mediated antiviral activity
Correlations between perforin expression and viral control is compelling evidence that cytolysis plays an important role in maintaining viral control. However, recent studies from two independent groups studying CD8+T-cell depletion in SIV-infected macaques have come to the singular conclusion that clearance of infected CD4+T cells does not account for the totality of the viral control exerted by CD8+T cells [49, 50]. One of these studies examined the life span of infected CD4+T cells in vivo after depletion of CD8+T cells during the early and late chronic phases by applying measurement of viral load and computer modeling to estimate the rate of decay of productively infected CD4+T cells. [49]. ART was introduced after infection to limit viral production to cells that were infected during the acute phase. While viral load increased significantly following CD8+T-cell depletion, no differences were determined between the decay rate of infected cells in the presence of CD8+T cells or with ART alone. Interestingly, the cytokine plasma levels (MIP-1α, IFNγ, IL-7, TNFα) were also decreased following CD8+T-cell depletion in some but not all of the animals. A second concurrent study also examined CTL clearance of SIV-infected cells following in vivo CD8+T-cell depletion and ART treatment with very similar findings regarding viral load and CD4+T-cell clearance [50]. Taken together, these studies indicate important roles for CD8+T-cell-mediated antiviral functions, beyond cytolytic clearance of infected cells, in controlling virus replication in vivo.
Cytokine release following peptide stimulation, as is seen in IFNγ and TNFα ELISpot assays, and correlations between expression levels of chemokines with known antiviral activities (MIP-1β) and CD8+T-cell-mediated virus inhibition [44] are not evidence of causality. In our study, levels of IFNγ expressing cells did not independently correlate with antiviral capacity, and MIP-1β expression correlated with antiviral activity against X4 viruses, for which there is no known mechanism [44]. Viral inhibition mediated by CD8+T cells is likely mediated by multiple factors with multiple effector mechanisms [51–55]. Measurement of these functions may, therefore, be indicative of cells that are broadly more capable of producing antiviral molecules.
Epigenetic regulation of CD8+T cells could provide a means for regulating the production of multiple inhibitory molecules in CD8+T cells. The rapidity of the memory CD8+T-cell response could be attributed to epigenetic control of transcription factors responsible for perforin, granzyme B, and IFN-γ production through chromatin remodeling [56–58]. The generation of potent antiviral CD8+T-cell responses may depend upon the availability of multiple functions that are differentially expressed throughout cellular maturation. Distinct, epigenetically regulated gene expression patterns have been associated with memory CD8+T-cell differentiation [59]. Chromatin modification by histone deacetylation (HDAC complexes) can play a substantial role in the repression of transcription (reviewed in [60]). We have examined the ability of an HIV-1 suppressive CD8+T-cell line and primary CD8+T cells from an elite controller to regulate histone deacetylation in HIV-1-infected cells [61]. CD8+T cells were able to potently suppress HIV-1 replication. Inhibition of histone deacetylation led to increased viral replication and reversed the CD8+T-cell-mediated suppressive effect in target/effector cell cocultures. However, no changes were observed in the acetylation state of the HIV-1 LTR. Pre-treatment of CD8+T cells with an HDAC inhibitor demonstrated that histone deacetylation within the CD8+-cell chromatin is required for optimal HIV-1 suppression. Conversely, studies of the cytolytic response have shown that HDAC inhibitors augment the cytolytic response of CD8+T lymphocytes [62]. Exposure of CD8+T lymphocytes to a HDAC inhibitor during antigen stimulation increased specific killing and secretion of IFNγ, MIP-1α, and MIP-1β [62]. These data further distinguish the cytolytic and noncytolytic responses in CD8+T lymphocytes and indicate distinct networks of genes that mediate each response. Adding to these observations is a study by Williams and colleagues demonstrating epigenetic regulation of telomerase expression in primary CD8+T cells from HIV-1 controllers [63]. Patterns of hyper- and hypomethylation of the proximal and distal telomerase catalytic subunit promoters differed between elite controllers and progressors. Thus, epigenetic evaluation of CD8+T cells in the context of virus suppression and control has suggested that effective CD8+T cells may be determined by chromatin structure. Further investigations are necessary to dissect the effect of these genetic differences on proliferative capacity and expression of functional molecules.
CD8+T cells can utilize noncytolytic mechanisms to suppress HIV-1 replication through the secretion of soluble factors. We have found that the secretion of MIP-1β, MIP-1α, IP-10, MIG, IL-1α, and interferon gamma correlated most strongly with soluble noncytolytic suppression [64]. Using the ability of histone hyperacetylation to alter the expression of immune-related genes, we identified MIP-1α and IP-10 among the immunemodulatory genes that were downregulated by histone hyperacetylation. This work demonstrated that a multifactorial cytokine profile exists for CD8+T lymphocytes that are capable of mediating noncytolytic suppression of CXCR4-tropic HIV-1 replication. Further characterization of noncytolytic suppressive CD8+T lymphocytes is needed to distinguish between the factors responsible for mediating virus inhibition from those that mark a cytokine signature of noncytolytic suppression.
Mucosal T-cell responses
HIV-1 is predominantly a sexually transmitted disease; thus, a robust mucosal response before HIV-1 infection and virus latency is established could be critical for HIV-1 eradication. Several groups have demonstrated that HIV-1 robustly replicates in gut-associated lymphoid tissue (GALT), during all stages of HIV-1 infection [65] and that cells within these tissues are the initial population of cells infected by HIV-1 [66]. Ferre et al. demonstrated that HIV controllers have polyfunctional HIV-specific T-cell responses in rectal mucosa that could be differentiated from the T-cell responses in the blood [46]. It is unknown whether the polyfunctional nature of the mucosal T cells corresponds to the ability to inhibit HIV replication. Of note, Shacklett et al. [67] found that some controllers had strong mucosal CD8+T-cell responses, as measured by CD107a, IFNγ, MIP-1β, and TNFα, that were discordant from the blood. Furthermore, rectal HIV-1-specific CD8+T cells expressed MIP-1β and CD107a that positively increased with CD4+T-cell counts [68].
In the nonhuman primate model, immunization with an attenuated virus showed protection associated with the presence of CD107-expressing CD8+T cells in the genital tract [69]. Hansen and colleagues [70] demonstrated that vaccination with an RhCMV vector expressing SIV antigens, shown to elicit robust SIV-specific, CD4 + and CD8 + TEM responses at extralymphoid sites (with TNF, IFN-γ, and MIP-1β expression and cytotoxic degranulation), could protect against limiting dose, intrarectal challenge. These finding strongly indicate that T cells, particulary TEM, can be protective against mucosal challenge. Thus, further studies to understand the function of mucosal T cells and how to elicit the most appropriate T-cell response by vaccination are needed. The quality of the CD8+T cell response is clearly important for protection; thus, understanding the cytokine profile and the lytic capability of antigen specific CD8+T cells both systemically and mucosally will be central to our understanding of the type of CD8+T-cell response that needs to be elicited to control HIV-1 replication. Critical to further the design of preventive and therapeutic HIV-1 vaccines that aim to elicit robust mucosal T-cell responses for inhibiting HIV-1 replication will be to determine whether mucosal CD8+T cells can inhibit diverse strains of HIV-1.
Application of CD8+T-cell-mediated correlates of protection to development of therapeutics, vaccines, and efficacy monitoring
CD8+T cells represent an immune response that has been shown to be critical for controlling viral replication. CD8+T cells not only contribute toward the resolution of acute viremia but also exert significant selective pressure on the virus resulting in fitness defects. Antigen presentation has been found to occur swiftly following infection and may account for the rapidity of the T-cell response. It is yet unclear whether the CD8+ T-cell responses that are able to most potently inhibit replication and drive viral fitness defects early in infection are able to be durably maintained or replaced in chronic infection nor, to what extent, the critical epitopes vary among virus controllers with and without known controlling HLA alleles. With additional insight into the dynamics between CD8+T-cell-mediated control and virus evolution, prime boost vaccine strategies may be devised to target changing CD8+T-cell responses in the event that sterilizing immunity through the humoral arm is unsuccessful.
Understanding the functional correlates of CD8+T-cell-mediated immune control may have beneficial implications for the design of both successful therapeutics and efficacious vaccines. New evidence of perforin and MIP-1β associations with viral control and antiviral activity supports roles for both lytic and nonlytic mechanisms in viral control; while CD8+T-cell depletion studies in nonhuman primate models suggest that nonlytic mechanisms may play an even more important role in maintaining viral control than previously thought. We have yet to fully elucidate the relationship between cytokine-/chemokine-driven virus suppression and MHC-dependent elicitation of adaptive responses. A better understanding of this relationship may suggest a means for directing the immune system to produce more efficacious suppressive responses.
While it is not yet known specifically which CD8+T-cell derived molecules mediate potent antiviral effects, correlational studies suggest that the effector molecules are likely to be coexpressed with commonly measured determinants such as MIP-1β, CD107a, and possibly perforin. Epigenetic studies have shown that differences in chromatin structure characterize CD8+T cells from virus controllers and play a role in CD8+T cell-mediated virus suppression, suggesting a mechanism by which suppressive cells may be primed for better proliferation and increased protein expression. Characterizing the molecular mechanisms that CD8+T cells from virus controllers use to control virus replication could lead to new classes of therapeutics. Additionally, the identification of novel correlates of CD8+T cell-mediated control could provide a more accurate means of evaluating vaccine efficacy.
As a therapeutic cure for HIV-1 infection and a sterilizing vaccine currently is elusive, it becomes increasingly important to understand the correlates of immune protection that can protect from disease in the event that infection occurs. While a sterilizing vaccine remains the optimal goal, improving the quality of the immune response following virus breakthrough can have a tremendous impact on the resolution of the global pandemic. Reduction in circulating and compartmentalized virus can improve health of the infected individual: It can also significantly slow the spread of infection by reducing transmission risk. Thus, it remains critical that we continue to evaluate the role that CD8+T cells play in controlling viral replication by establishing clear connections between virus control and CD8+T-cell function and monitoring our ability to incite these beneficial cellular responses.
References
Borrow P, et al. Virus-specific CD8 + cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol. 1994;68(9):6103–10.
Koup RA, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68(7):4650–5.
McMichael AJ, et al. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol. 2010;10(1):11–23.
Jin X, et al. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med. 1999;189(6):991–8.
Schmitz JE, et al. Control of viremia in simian immunodeficiency virus infection by CD8 + lymphocytes. Science. 1999;283(5403):857–60.
Matano T, et al. Administration of an anti-CD8 monoclonal antibody interferes with the clearance of chimeric simian/human immunodeficiency virus during primary infections of rhesus macaques. J Virol. 1998;72(1):164–9.
Klein MR, et al. Kinetics of Gag-specific cytotoxic T lymphocyte responses during the clinical course of HIV-1 infection: a longitudinal analysis of rapid progressors and long-term asymptomatics. J Exp Med. 1995;181(4):1365–72.
Rinaldo C, et al. High levels of anti-human immunodeficiency virus type 1 (HIV-1) memory cytotoxic T-lymphocyte activity and low viral load are associated with lack of disease in HIV-1-infected long-term nonprogressors. J Virol. 1995;69(9):5838–42.
Betts MR, et al. HIV non-progressors preferentially maintain highly functional HIV-specific CD8 + T cells. Blood. 2006;107(12):4781–9.
Cao Y, et al. Virologic and immunologic characterization of long-term survivors of human immunodeficiency virus type 1 infection. N Engl J Med. 1995;332(4):201–8.
Harrer T, et al. Strong cytotoxic T cell and weak neutralizing antibody responses in a subset of persons with stable non-progressing HIV type 1 infection. AIDS Res Hum Retroviruses. 1996;12(7):585–92.
Kloosterboer N, et al. Natural controlled HIV infection: preserved HIV-specific immunity despite undetectable replication competent virus. Virology. 2005;339(1):70–80.
Lambotte O, et al. HIV controllers: a homogeneous group of HIV-1-infected patients with spontaneous control of viral replication. Clin Infect Dis. 2005;41(7):1053–6.
Migueles SA, et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term non-progressors. Proc Natl Acad Sci U S A. 2000;97(6):2709–14.
Flores-Villanueva PO, et al. Control of HIV-1 viremia and protection from AIDS are associated with HLA-Bw4 homozygosity. Proc Natl Acad Sci U S A. 2001;98(9):5140–5.
Leslie A, et al. Additive contribution of HLA class I alleles in the immune control of HIV-1 infection. J Virol. 2010;84(19):9879–88.
Altfeld M, et al. HLA Alleles Associated with Delayed Progression to AIDS Contribute Strongly to the Initial CD8(+) T Cell Response against HIV-1. PLoS Med. 2006;3(10):e403.
Fellay J, et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 2009;5(12):e1000791.
Buchbinder SP, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372(9653):1881–93.
McMichael AJ, et al. The dynamics of the cellular immune response to HIV infection: implications for vaccination. Philos Trans R Soc Lond B Biol Sci. 2000;355(1400):1007–11.
Couillin I, et al. HLA-dependent variations in human immunodeficiency virus Nef protein alter peptide/HLA binding. Eur J Immunol. 1995;25(3):728–32.
Borrow P, et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. 1997;3(2):205–11.
Brumme ZL, et al. Marked epitope- and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J Virol. 2008;82(18):9216–27.
Goonetilleke N, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206(6):1253–72.
Fischer W, et al. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One 2010;5(8).
Love TM, et al. Mathematical modeling of ultradeep sequencing data reveals that acute CD8 + T-lymphocyte responses exert strong selective pressure in simian immunodeficiency virus-infected macaques but still fail to clear founder epitope sequences. J Virol. 2010;84(11):5802–14.
Balamurali M, et al. Does cytolysis by CD8 + T Cells drive immune escape in HIV infection? J Immunol 2010.
Brockman MA, et al. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated in chronic infection. J Virol 2010.
Edwards BH, et al. Magnitude of functional CD8 + T-cell responses to the gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J Virol. 2002;76(5):2298–305.
Kiepiela P, et al. CD8 + T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med. 2007;13(1):46–53.
Riviere Y, et al. Gag-specific cytotoxic responses to HIV type 1 are associated with a decreased risk of progression to AIDS-related complex or AIDS. AIDS Res Hum Retroviruses. 1995;11(8):903–7.
Saez-Cirion A, et al. Heterogeneity in HIV suppression by CD8 T cells from HIV controllers: association with Gag-specific CD8 T cell responses. J Immunol. 2009;182(12):7828–37.
Julg B, et al. Enhanced anti-HIV functional activity associated with Gag-specific CD8 T-cell responses. J Virol. 2010;84(11):5540–9.
Sacha JB, et al. Gag-specific CD8 + T lymphocytes recognize infected cells before AIDS-virus integration and viral protein expression. J Immunol. 2007;178(5):2746–54.
Sacha JB, et al. Pol-specific CD8 + T cells recognize simian immunodeficiency virus-infected cells prior to Nef-mediated major histocompatibility complex class I downregulation. J Virol. 2007;81(21):11703–12.
Payne RP, et al. Efficacious early antiviral activity of HIV Gag- and Pol-specific HLA-B*2705-restricted CD8 + T-cells. J Virol 2010.
Sacha JB, et al. SIV-specific CD8 + T cells recognize Vpr- and Rev-derived epitopes early after infection. J Virol 2010.
Maness NJ, et al. CD8 + T cell recognition of cryptic epitopes is a ubiquitous feature of AIDS virus infection. J Virol 2010.
Bansal A, et al. CD8 T cell response and evolutionary pressure to HIV-1 cryptic epitopes derived from antisense transcription. J Exp Med. 2010;207(1):51–9.
Maness NJ, et al. Robust, vaccine-induced CD8(+) T lymphocyte response against an out-of-frame epitope. J Immunol. 2010;184(1):67–72.
Papagno L, et al. Immune activation and CD8 + T-cell differentiation towards senescence in HIV-1 infection. PLoS Biol. 2004;2(2):E20.
Appay V, Sauce D. Immune activation and inflammation in HIV-1 infection: causes and consequences. J Pathol. 2008;214(2):231–41.
Appay V, et al. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 2008;73(11):975–83.
Freel SA, et al. Phenotypic and functional profile of HIV-inhibitory CD8 T cells elicited by natural infection and heterologous prime/boost vaccination. J Virol. 2010;84(10):4998–5006.
Almeida JR, et al. Antigen sensitivity is a major determinant of CD8 + T-cell polyfunctionality and HIV-suppressive activity. Blood. 2009;113(25):6351–60.
Ferre AL, et al. Mucosal immune responses to HIV-1 in elite controllers: a potential correlate of immune control. Blood. 2009;113(17):3978–89.
Mkhwanazi N, et al. Immunodominant HIV-1-specific HLA-B- and HLA-C-restricted CD8 + T cells do not differ in polyfunctionality. Virology. 2010;405(2):483–91.
Hersperger AR, et al. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog. 2010;6(5):e1000917.
Klatt NR, et al. CD8 + lymphocytes control viral replication in SIVmac239-infected rhesus macaques without decreasing the lifespan of productively infected cells. PLoS Pathog. 2010;6(1):e1000747.
Wong JK, et al. In vivo CD8 + T-cell suppression of siv viremia is not mediated by CTL clearance of productively infected cells. PLoS Pathog. 2010;6(1):e1000748.
Tomaras GD, et al. CD8 + T cell-mediated suppressive activity inhibits HIV-1 after virus entry with kinetics indicating effects on virus gene expression. Proc Natl Acad Sci USA. 2000;97(7):3503–8.
Overman RG, et al. Initiation of human immunodeficiency virus type 1 (HIV-1) transcription is inhibited by non-cytolytic CD8 suppression. Open Virol J. 2007;1:1–7.
Walker CM, et al. CD8 + lymphocytes can control HIV infection in vitro by suppressing virus replication. Science. 1986;234(4783):1563–6.
Cocchi F, et al. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8 + T cells. Science. 1995;270(5243):1811–5.
Mosoian A, et al. Prothymosin-alpha inhibits HIV-1 via Toll-like receptor 4-mediated type I interferon induction. Proc Natl Acad Sci U S A. 2010;107(22):10178–83.
Northrop JK, et al. Epigenetic remodeling of the IL-2 and IFN-gamma loci in memory CD8 T cells is influenced by CD4 T cells. J Immunol. 2006;177(2):1062–9.
Northrop JK, Wells AD, Shen H. Cutting edge: chromatin remodeling as a molecular basis for the enhanced functionality of memory CD8 T cells. J Immunol. 2008;181(2):865–8.
Araki Y, et al. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B). J Immunol. 2008;180(12):8102–8.
Youngblood B, Davis CW, Ahmed R. Making memories that last a lifetime: heritable functions of self-renewing memory CD8 T cells. Int Immunol. 2010;22(10):797–803.
Kadonaga JT. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell. 1998;92(3):307–13.
Saunders KO, et al. Epigenetic regulation of CD8(+) T-lymphocyte mediated suppression of HIV-1 replication. Virology. 2010;405(1):234–42.
Agarwal P, et al. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol. 2009;183(3):1695–704.
Williams K, et al. Epigenetic regulation of telomerase expression in HIV-1-specific CD8 + T cells. AIDS. 2010;24(12):1964–6.
Saunders KO, Ward-Cavines C, Schutte RJ, Freel SA, Glenn Overman R, Thielman NM, et al. Secretion of MIP-1β and MIP-1α by CD8+ T-lymphocytes correlates with HIV-1 inhibition independent of coreceptor usage. Cell. Immunol. 2010. doi:10.1016/j.cellimm.2010.09.011.
Brenchley JM, et al. CD4 + T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200(6):749–59.
Arthos J, et al. HIV-1 envelope protein binds to and signals through integrin alpha4beta7, the gut mucosal homing receptor for peripheral T cells. Nat Immunol. 2008;9(3):301–9.
Shacklett BL, et al. Mucosal T-cell responses to HIV: responding at the front lines. J Intern Med. 2009;265(1):58–66.
Critchfield JW, et al. Magnitude and complexity of rectal mucosa HIV-1-specific CD8 + T-cell responses during chronic infection reflect clinical status. PLoS One. 2008;3(10):e3577.
Genesca M, McChesney MB, Miller CJ. Antiviral CD8 + T cells in the genital tract control viral replication and delay progression to AIDS after vaginal SIV challenge in rhesus macaques immunized with virulence attenuated SHIV 89.6. J Intern Med. 2009;265(1):67–77.
Hansen SG, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med. 2009;15(3):293–9.
Acknowledgments
This work was supported in part by the National Institutes of Health (NIH/NIAID/DAIDS): RO1A5779 and CHAVI AI067854-05.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Freel, S.A., Saunders, K.O. & Tomaras, G.D. CD8+T-cell-mediated control of HIV-1 and SIV infection. Immunol Res 49, 135–146 (2011). https://doi.org/10.1007/s12026-010-8177-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-010-8177-7