
Abstract
The importance of reactive oxygen species-dependent microbial killing by the phagocytic cell NADPH oxidase has been appreciated for some time, although only recently has an appreciation developed for the partnership of lactoperoxidase with related dual oxidases (Duox) within secretions of the airway surface layer. This system produces mild oxidants designed for extracellular killing that are effective against several airway pathogens, including Staphylococcus aureus, Burkholderia cepacia, and Pseudomonas aeruginosa. Establishment of chronic pseudomonas infections involves adaptations to resist oxidant-dependent killing by expression of a redox-active virulence factor, pyocyanin, that competitively inhibits epithelial Duox activity by consuming intracellular NADPH and producing superoxide, thereby inflicting oxidative stress on the host.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Circulating phagocytic blood cells exhibit a remarkable capacity for generating large amounts of reactive oxygen species (ROS) during the engulfment of microbial pathogens, a process referred to as the “respiratory burst” [1]. This activity is attributed to oxygen consumption by the phagocytic NADPH oxidase (phox) complex, an enzyme that donates electrons from NADPH to molecular oxygen to generate superoxide anion, a short-lived precursor of other potent antimicrobial oxidants (hydrogen peroxide (H2O2) and hypochlorous acid) usually produced within the confines of phagosomes. Oxidant-dependent microbial killing by circulating phagocytes has been recognized as an essential component of innate immunity; defects in any one of four genes encoding phagocytic oxidase components result in chronic granulomatous disease (CGD), a hereditary immune deficiency characterized by enhanced susceptibility to low-grade bacterial and fungal pathogens and dysregulated inflammatory responses [2].
More than a decade ago, investigators began to appreciate that deliberate ROS production by the host is not limited to phagocytic cells, and that even low levels of ROS production serve a variety of essential functions, including redox-based cell signaling, vascular regulation, hormone biosynthesis, extracellular matrix cross-linking, oxygen sensing, and alterations in gene expression in response to redox signals [3]. At the same time that these novel roles for ROS were being described, the rapid expansion of genome sequence databases led investigators to realize that the phagocytic NADPH oxidase is but one member of an entire NADPH oxidase (Nox) family. We now know that the Nox family encompasses seven oxidases in man, based on similarities between the catalytic core of the phagocytic system (Nox2 or gp91phox) and other homologues (Nox1, Nox3, Nox4, Nox5, Duox1, and Duox2) [4, 5]. Indeed, many of the proposed functions for ROS in non-phagocytic cells were attributed to these novel Nox family oxidases: Dual oxidases (Duox1 and Duox2) produce hydrogen peroxide needed to support iodide organification during thyroid hormone biosynthesis, Nox1 regulates vascular tone, and Nox3 has developmental or biosynthetic functions in the inner ear that are critical in gravity sensing and balance [6].
Growing evidence is emerging that suggests that several of the non-phagocytic oxidases are also involved in oxidant-based antimicrobial mechanisms (reviewed in [7, 8]). Several oxidases are induced by proinflammatory cytokines, demonstrate responsiveness to pathogen pattern recognition, and appear to accumulate at highest levels on epithelial boundaries of mucosal surfaces. Nox1 and Duox2 are induced by gamma-interferon [9, 10], Duox 1 is induced by Th2 cytokines, IL-4 and IL-13 [10], and Nox4 appears to function downstream of TGF-beta [11, 12]. Nox1 activity is stimulated through either TLR-4 or TLR-5 agonists [13, 14]; its expression is highest in the colon epithelium. Nox4 appears to form complexes with TLR-4 and could participate in downstream responses to microbial recognition [15]. Interestingly, Duox2 is induced in respiratory epithelial cells either by rhinovirus infection or by the viral mimic polyinosine: polycytidylic acid, suggesting that this oxidase serves in antiviral responses [10]. We were the first to propose that Duox isozymes function in supporting the antimicrobial activity of lactoperoxidase (LPO) by demonstrating a close correlation in the expression patterns of LPO and Duox isozymes in exocrine glands and along mucosal surfaces of airways and the gastrointestinal tract [16]. LPO has been recognized as an effective antimicrobial enzyme against both Gram negative and positive bacteria. Its importance in suppressing microbial growth in exocrine secretions such as milk and saliva has been appreciated for decades [17, 18], although the source of hydrogen peroxide supporting its activity had been unclear until recently. A renewed interest in the antimicrobial function of LPO has emerged in the context of airway host defense following detection of LPO and Duox isozymes at high levels in major airways [16, 19, 20].
Extracellular oxidative microbial killing by the Duox-SCN−-LPO system
Duox- and LPO-mediated microbial killing involves generation of oxidative metabolites that are distinct from those produced within phagosomes, in that the major ROS produced for extracellular killing are generally milder, thereby minimizing oxidative damage to host tissues. The phagocytic (Nox2-based) oxidase produces superoxide directly within the interior of phagosomes by transferring electrons from cytosolic NADPH to molecular oxygen. The oxidase components are effectively targeted to newly formed phagosomes by granule fusion and generation of bioactive lipids involved in recruitment of the cytosolic oxidase regulators [21, 22]. Superoxide dismutates rapidly into H2O2, which is then used by myeloperoxidase (MPO) to generate the potent antimicrobial oxidant hypochlorous acid from chloride. In contrast, the Duox isozymes appear to function as dedicated H2O2 generators. Maturation factors are needed to transport Duox to the plasma membrane [23], although it is still unclear whether the Duox enzymes are active within intracellular compartments. No other Duox-supportive co-factors have been described. Calcium mobilizing agonists seem to activate Duox directly to secrete H2O2 from the apical surfaces of airway or thyroid follicular epithelial cells [24, 25]. The Duox isozymes have calcium-binding EF-hands that likely render them directly responsive to elevations in intracellular calcium, as was shown in the case of Nox5 [26]. The Nox-like C-terminal portions should generate superoxide directly from molecular oxygen, although this reactive intermediate is not readily detected in whole cells. The extracellular peroxidase-like ecto-domains of the mammalian dual oxidases lack some of the most conserved heme-binding residues identified in other hemoperoxidases [27]. Thus, the ectodomains may function in the generation of H2O2, rather than its utilization, consistent with the proposed partnership of Duox enzymes with other extracellular hemoperoxidases (i.e., thyroperoxidase and LPO).
The secondary ROS metabolites of the Duox-LPO system are also different from the phagocytic system. Unlike MPO, LPO does not use chloride or generate HOCl, but reacts readily with the pseudohalide thiocyanate to make hypothiocyanite anion: SCN− + H2O2 → OSCN− + H2O. Early studies showed LPO-derived OSCN− is an effective microbicidal or microbistatic oxidant. It is toxic against Escherichia coli [17, 28], Haemophilus influenzae [19], Pseudomonads [28], Staphylococci [29], Streptococci [30–33], viral [34, 35], and fungal [36, 37] pathogens. SCN− is derived from dietary sources and is transported from the blood through the NaI symporter. Concentrations of LPO and SCN− (100–1000 µmol/l) in exocrine secretions are sufficient to favor OSCN- formation [19, 38, 39] while maintaining low H2O2 concentrations in extracellular secretions [40]. Furthermore, it appears that MPO in extracellular secretions uses SCN− as a substrate and that any MPO-derived HOCl in these secretions is readily consumed by SCN− to form the milder oxidant OSCN− [41, 42]. In situ hybridization experiments revealed that each component of Duox-SCN-LPO system is expressed within a distinct location in salivary glands: LPO mRNA is detected in acinar pockets, NaI symporter mRNA is detected in intercalated ducts, and Duox2 mRNA is detected on major terminal duct epithelial cells [16]. Thus, the complete killing system is assembled only in late stages of saliva formation in which the most labile component (H2O2) is produced by Duox2 in terminal ducts. These observations were the first to suggest any host defense-related functions for Duox and the NaI symporter in non-thyroid tissues.
Distinct expression patterns for LPO and Duox were also revealed in major airways, compatible with the notion that Duox and LPO work in partnership as antimicrobial factors in the airway surface layer (ASL) fluid film (Fig. 1a). Duox1 mRNA is detected mainly on the epithelial layer along the lumen of tracheal and bronchial surfaces [16]. LPO accumulates at high levels in ASL secretions, but is synthesized within acini of submucosal glands [16, 19]. We suggested that the enhanced susceptibility of cystic fibrosis (CF) patients to airway infections may reflect a defect in oxidant-based microbial killing when noting that several of the same bacterial species that infect CGD patients are also observed in early stages of CF disease (Burkholderia cepacia, Staphylococcus aureus, Haemophilus influenzae) [16]. Furthermore, we suggested that the cystic fibrosis transmembrane conductance regulator (CFTR), widely recognized as a chloride transporter, could function in supporting the Duox-LPO-based microbicidal system [16], since it also demonstrates efficient SCN− transporter activity [43, 44]. Several groups later obtained evidence supporting the proposed roles of Duox, LPO, and SCN− in microbial killing using human bronchial epithelial cells grown on air–liquid interface (ALI) culture models [45–47]. Normal primary human bronchial epithelial (NHBE) cells can grow as a differentiated polarized cell layer on matrix-coated permeable membranes exposed to air, mimicking the airway environment. Duox1 appears in the late phases (2.5–3 weeks) of differentiation of these ALI cultures, coinciding with ciliogenesis [47]. These cultures are capable of killing several bacterial species that infect the airways of CF patients (Pseudomonas aeruginosa, Burkholderia cepacia, Staphylococcus aureus, Haemophilus influenzae) in a Duox-, SCN−-, and LPO-dependent manner [45–47]. Two groups have provided evidence indicating that the defect in CFTR of CF airway cells is sufficient in limiting SCN− transport to the extent of compromising Duox- and LPO-dependent microbial killing on ALI cultures [45, 46]. Recently, two other SCN− transporters have been described in NHBE cells that are cytokine inducible and may serve as important SCN− transporters under inflammatory conditions [48, 49]. More work is needed to explore whether SCN− transport through CFTR and its role as a LPO substrate are most critical in preventing the colonization of airways by P. aeruginosa and other oxidant-sensitive pathogens.

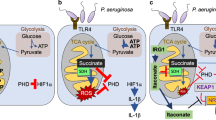
The oxidative antimicrobial Duox/SCN-/LPO system of major airways and counteroffensive mechanisms of Pseudomonas aeruginosa imposed by the redox active virulence factor pyocyanin. a Lactopreoxidase is produced in the submucosal glands of the airways and accumulates in the airway surface layer, where it converts thiocyanate into microbicidal hypothiocanite using Duox-derived hydrogen peroxide. b The redox-active Pseudomonas pigment, pyocyanin, enters airway cells, inhibits Duox activity by competing for its substrate (NADPH) and by inhibiting Duox expression. The airway peroxidases, lactoperoxidase (LPO) and myeloperoxidase (MPO), can detoxify pyocyanin using Duox-derived hydrogen peroxide. (Adapted from refs. [7] and [47])
Redox-based counter-offenses of Pseudomonas aeruginosa
Pseudomonas aeruginosa is unique as an adaptable, opportunistic airway pathogen that infects individuals with weakened immune systems (CF, burn, and other immunosuppressed patients, but not CGD), who usually develop more serious complications following the establishment of chronic infections [50]. Early isolates of Pseudomonas aeruginosa are in general non-mucoid, mobile, and greatly sensitive to antibiotics. Later in the course of the infection, the bacteria undergo adaptive changes through quorum sensing mechanisms leading to a mucoid, alginate-producing phenotype that establishes chronic infection by forming biofilms [50]. The bacterium has a well-defined life cycle: the swimming, planktonic forms attach to new surfaces and form microcolonies that then develop into macrocolonies and biofilms, which are more resistant to killing.
Since the airway Duox-LPO-SCN-system is able to kill P. aeruginosa in vitro, we explored whether bacterial culture density or phase of growth could influence its own chances for survival against human airway defenses. In a screen using Duox-expressing NCI-H292 airway epithelial cells, we found that supernatants of overgrown P. aeruginosa cultures strongly inhibit Duox activation [47]. Furthermore, co-incubation of the bacteria from long-term cultures with NCI-H292 cells for several hours resulted in complete loss of Duox function [47]. The bacterium harbors a variety of virulence determinants that at different stages of the infection could contribute to enhanced survival in human airways, including LPS, flagellum, pilus, alginate, type III secretion system, extracellular proteases, exotoxin-A, phospholipases, rhamnolipid, and phenazines [51]. Among the virulence factors secreted in late phases of bacterial growth, phenazines caught our attention as potential effectors of Duox, since they are secreted, redox active, small molecular weight tricyclic compounds that can easily cross biomembranes [52]. Pyocyanin, a blue-green phenazine virulence factor, is produced by most clinical isolates of Pseudomonas aeruginosa infecting CF patients [53]. Phenazines have been considered for a long time as secondary metabolites, although several recent studies provide new information about their functions: their production is precisely regulated by quorum sensing, they are important virulence factors toxic to both prokaryotic and eukaryotic cells, and they can modify a variety of cell functions, including gene expression and protein synthesis in bacteria and higher organisms [54]. Pyocyanin is cytotoxic against other bacteria, fungi, worms, flies, and mammals [55]. Most of the toxic effects of pyocyanin have been attributed to its ability to inflict intracellular oxidative stress (Table 1 ). As it enters airway epithelial cells, pyocyanin oxidizes intracellular pools of NADPH, NADH, and GSH directly by accepting electrons [56, 57]. The reduced pyocyanin donates this electron to molecular oxygen under aerobic conditions, thereby forming superoxide anion, which is then converted into H2O2 within cells.
In comparing the effects of different Pseudomonas aeruginosa strains, we found that inhibition of Duox activity correlated well with their ability to secrete pyocyanin [47]. The strong inhibition of Duox exhibited by the wild-type strain PA14 was abolished when its phenazine-deficient mutant was used [47]. These observations were confirmed in experiments using purified pyocyanin obtained from the supernatant of long-term cultures of P. aeruginosa. The toxin inhibited Duox activation in both NCI-H292 and NHBE cells, while producing intracellular superoxide and exposing the cell interior to oxidative stress [47]. We proposed that competition between Duox and pyocyanin for a common substrate, NADPH, could account for these observations as a possible mechanism (Fig. 1b).
Besides these immediate effects of pyocyanin, long-term exposure to the toxin inhibited both differentiation- and cytokine-induced up-regulation of Duox protein in airway cells [47]. These suppressive effects of pyocyanin were prevented by addition of antioxidants (NAC, GSH) to the cell culture medium, again suggesting that its inhibitory effects on Duox expression are related to oxidative stress [47]. We then tested whether the toxin interferes with killing of Pseudomonas aeruginosa by mature differentiated NHBE cells. Addition of the toxin to primary ALI airway cells blocked the killing of the bacterium, verifying the crucial role of pyocyanin toxicity against the Duox-based antimicrobial system, even after short-term pyocyanin exposure [47]. These effects of pyocyanin on the airway dual oxidases can be generalized to include other members of the Nox NADPH oxidase family, notably the Nox2-based oxidase of circulating phagocytic cells. Previous work demonstrated inhibitory effects of pyocyanin on superoxide release by neutrophils as it depletes intracellular stores of NADPH (Table 1 ). We confirmed these effects in a Nox2-reconstituted cell model, showing that pyocyanin inhibits extracellular superoxide release by the oxidase while producing intracellular superoxide in a dose-dependent manner [47].
Duox- and peroxidase-mediated detoxification of pyocyanin—another redox-based defensive mechanism
Recent studies have indicated that pyocyanin is also subject to irreversible oxidative metabolism, as it was shown to be inactivated by treatments with H2O2 and peroxidase mimics, hemin or microperoxidase 11 (a proteolytic peptide of cytochrome c covalently bound to heme) [58]. The oxidized pyocyanin could not induce IL-8 release from A549 cells nor was it reduced by NADH. Therefore, we investigated whether the two hemoperoxidases prevalent in airway secretions (LPO and MPO) were also capable of a similar pyocyanin inactivating mechanism. Both peroxidases consumed pyocynin in a H2O2-dependent manner, producing derivatives that were less capable of producing superoxide in treated airway cells [47]. Thus, it appears that the Duox-peroxidase airway system can also function in detoxification of pyocyanin, suggesting physiological mechanisms for eliminating this virulence factor.
Conclusions
In summary, our findings extend a growing body of observations related to a complex redox-based interplay between airway pathogens and the airway innate immune system. It appears that the airway epithelium is capable of significant reactive oxygen species release by Duox that in partnership with secreted hemoperoxidases can kill several known airway pathogens. Pseudomonas aeruginosa, an adaptable opportunistic pathogen, produces a redox active toxin pyocyanin with counteroffensive capabilities for overcoming the Duox-based killing system, while imposing oxidative stress on the host. Finally, the effects of LPO and MPO in detoxification of pyocyanin suggest another novel role for Duox-derived H2O2 in the elimination of the toxin by the peroxidases, which may be of therapeutic interest.
The inhibitory effect of pyocyanin on the novel Duox-based airway host defense mechanism represents one more example of how the pro-oxidant-related cytotoxicity of pyocyanin suppresses a variety of innate and adaptive host immune functions (Table 1). In addition to supporting the antimicrobial and detoxifying activities of LPO, Duox1 was suggested to participate in other functions of airway cells including acid secretion [59], mucin expression [60], and wound healing [61]. Therefore, pyocyanin could affect these airway epithelial cell activities, which all relate to host defense or barrier formation against pathogens. The ability of pyocyanin to trigger IL-8 release from epithelial cells could contribute to the significant recruitment of neutrophils to infected lungs [62, 63], another remarkable feature of advanced CF disease with chronic pseudomonas infection. Despite their recruitment in large numbers, their antimicrobial functions are effectively incapacitated by at least two other ROS-related pyocyanin effects: direct inhibition of the phagocytic oxidase and induction of apoptosis leading to release of neutrophil granule components that can damage lung tissues [64, 65]. This process is further exacerbated by pyocyanin-mediated inhibition of the uptake of apoptotic neutrophils by macrophages, which also involves pyocyanin-induced oxidative stress [66]. Thus, the effects of pyocyanin on the airway epithelium are part of a complex cascade of redox-related events that can tip the balance of power between host and microbe, leading to compromised host immunity and enhanced lung inflammation.
It is clear that oxidative stress is a significant component of advanced CF disease accompanied by chronic P. aeruginosa infection, although the extent to which pyocyanin contributes to this burden needs further clarification. Future work should define critical cellular targets subject to oxidation and explore redox-based changes in host gene expression patterns that compromise host immunity and promote inflammation to delineate the precise mechanisms by which pyocyanin-mediated oxidative stress affects the host. Such studies would provide a better understanding of the pathogenesis of P. aeruginosa infection and may suggest novel effective redox-based therapies.
References
Leto TL. The respiratory burst oxidase. In: Gallin JI, Snyderman R, editors. Inflammation. Basic principles and clinical correlates. Philadelphia: Lippincott Williams and Wilkins; 1999. p. 769–86.
Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore). 2000;79:170–200.
Finkel T. Reactive oxygen species and signal transduction. IUBMB Life. 2001;52:3–6.
Geiszt M, Leto TL. The Nox family of NAD(P)H oxidases: host defense and beyond. J Biol Chem. 2004;279:51715–18.
Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–9.
Nauseef WM. Biological roles for the NOX family NADPH oxidases. J Biol Chem. 2008;283:16961–5.
Rada B, Leto TL. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib Microbiol. 2008;15:164–87.
Leto TL, Geiszt M. Role of Nox family NADPH oxidases in host defense. Antioxid Redox Signal. 2006;8:1549–61.
Geiszt M, Lekstrom K, Brenner S, Hewitt SM, Dana R, Malech HL, et al. NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91phox in the regulated production of superoxide by phagocytes. J Immunol. 2003;171:299–306.
Harper RW, Xu C, Eiserich JP, Chen Y, Kao CY, Thai P, et al. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 2005;579:4911–17.
Sturrock A, Huecksteadt TP, Norman K, Sanders K, Murphy TM, Chitano P, et al. Nox4 mediates TGF-beta1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1543–55.
Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, et al. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L661–73.
Kawahara T, Kuwano Y, Teshima-Kondo S, Takeya R, Sumimoto H, Kishi K, et al. Role of nicotinamide adenine dinucleotide phosphate oxidase 1 in oxidative burst response to Toll-like receptor 5 signaling in large intestinal epithelial cells. J Immunol. 2004;172:3051–8.
Kawahara T, Kohjima M, Kuwano Y, Mino H, Teshima-Kondo S, Takeya R, et al. Helicobacter pylori lipopolysaccharide activates Rac1 and transcription of NADPH oxidase Nox1 and its organizer NOXO1 in guinea pig gastric mucosal cells. Am J Physiol Cell Physiol. 2005;288:C450–7.
Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol. 2004;173:3589–93.
Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003;17:1502–4.
Reiter B, Marshall VM, Bjorck L, Rosen CG. Nonspecific bactericidal activity of the lactoperoxidases-thiocyanate-hydrogen peroxide system of milk against Escherichia coli and some gram-negative pathogens. Infect Immun. 1976;13:800–7.
Pruitt KM. The salivary peroxidase system: thermodynamic, kinetic and antibacterial properties. J Oral Pathol. 1987;16:417–20.
Wijkstrom-Frei C, El-Chemaly S, Ali-Rachedi R, Gerson C, Cobas MA, Forteza R, et al. Lactoperoxidase and human airway host defense. Am J Respir Cell Mol Biol. 2003;29:206–12.
Ratner AJ, Prince A. Lactoperoxidase. New recognition of an “old” enzyme in airway defenses. Am J Respir Cell Mol Biol. 2000;22:642–4.
Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122:277–91.
Ueyama T, Kusakabe T, Karasawa S, Kawasaki T, Shimizu A, Son J, et al. Sequential binding of cytosolic Phox complex to phagosomes through regulated adaptor proteins: evaluation using the novel monomeric Kusabira-Green System and live imaging of phagocytosis. J Immunol. 2008;181:629–40.
Grasberger H, Refetoff S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem. 2006;281:18269–72.
Forteza R, Salathe M, Miot F, Conner GE. Regulated hydrogen peroxide production by Duox in human airway epithelial cells. Am J Respir Cell Mol Biol. 2005;32:462–9.
Ameziane-El Hassani R, Morand S, Boucher JL, Frapart YM, Apostolou D, Agnandji D, et al. Duox2 has intrinsic Ca2+-dependent H2O2-generating activity. J Biol Chem. 2005;280:30046–54.
Banfi B, Tirone F, Durussel I, Knisz J, Moskwa P, Molnar GZ, et al. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5). J Biol Chem. 2004;279:18583–91.
Edens WA, Sharling L, Cheng G, Shapira R, Kinkade JM, Lee T, et al. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J Cell Biol. 2001;154:879–91.
Bjorck L, Rosen C, Marshall V, Reiter B. Antibacterial activity of the lactoperoxidase system in milk against pseudomonads and other gram-negative bacteria. Appl Microbiol. 1975;30:199–204.
Johansen C, Falholt P, Gram L. Enzymatic removal and disinfection of bacterial biofilms. Appl Environ Microbiol. 1997;63:3724–8.
Oram JD, Reiter B. The inhibition of streptococci by lactoperoxidase, thiocyanate and hydrogen peroxide. The oxidation of thiocyanate and the nature of the inhibitory compound. Biochem J. 1966;100:382–8.
Oram JD, Reiter B. The inhibition of streptococci by lactoperoxidase, thiocyanate and hydrogen peroxide. The effect of the inhibitory system on susceptible and resistant strains of group N streptococci. Biochem J. 1966;100:373–81.
Thomas EL, Milligan TW, Joyner RE, Jefferson MM. Antibacterial activity of hydrogen peroxide and the lactoperoxidase-hydrogen peroxide-thiocyanate system against oral streptococci. Infect Immun. 1994;62:529–35.
Thomas EL, Pera KA, Smith KW, Chwang AK. Inhibition of Streptococcus mutans by the lactoperoxidase antimicrobial system. Infect Immun. 1983;39:767–78.
Courtois P, van Beers D, de Foor M, Mandelbaum IM, Pourtois M. Abolition of herpes simplex cytopathic effect after treatment with peroxidase generated hypothiocyanite. J Biol Buccale. 1990;18:71–4.
Pourtois M, Binet C, Van Tieghem N, Courtois P, Vandenabbeele A, Thiry L. Inhibition of HIV infectivity by lactoperoxidase-produced hypothiocyanite. J Biol Buccale. 1990;18:251–3.
Lenander-Lumikari M. Inhibition of Candida albicans by the peroxidase/SCN−/H2O2 system. Oral Microbiol Immunol. 1992;7:315–20.
Popper L, Knorr D. Inactivation of yeast and filamentous fungi by the lactoperoxidase-hydrogen peroxide-thiocyanate-system. Nahrung. 1997;41:29–33.
Paul BD, Smith ML. Cyanide and thiocyanate in human saliva by gas chromatography-mass spectrometry. J Anal Toxicol. 2006;30:511–15.
Jalil RA. Concentrations of thiocyanate and hypothiocyanite in the saliva of young adults. J Nihon Univ Sch Dent. 1994;36:254–60.
Ferreira IM, Hazari MS, Gutierrez C, Zamel N, Chapman KR. Exhaled nitric oxide and hydrogen peroxide in patients with chronic obstructive pulmonary disease: effects of inhaled beclomethasone. Am J Respir Crit Care Med. 2001;164:1012–15.
van Dalen CJ, Whitehouse MW, Winterbourn CC, Kettle AJ. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem J. 1997;327(Pt 2):487–92.
Ashby MT, Carlson AC, Scott MJ. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J Am Chem Soc. 2004;126:15976–7.
Linsdell P, Hanrahan JW. Adenosine triphosphate-dependent asymmetry of anion permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. J Gen Physiol. 1998;111:601–14.
Illek B, Tam AW, Fischer H, Machen TE. Anion selectivity of apical membrane conductance of Calu 3 human airway epithelium. Pflugers Arch. 1999;437:812–22.
Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB Jr, Nauseef WM, et al. A novel host defense system of airways is defective in cystic fibrosis. Am J Respir Crit Care Med. 2007;175:174–83.
Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M. The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett. 2007;581:271–8.
Rada B, Lekstrom K, Damian S, Dupuy C, Leto TL. The pseudomonas toxin pyocyanin inhibits the dual oxidase-based antimicrobial system as it imposes oxidative stress on airway epithelial cells. J Immunol. 2008;181:4883–93.
Pedemonte N, Caci E, Sondo E, Caputo A, Rhoden K, Pfeffer U, et al. Thiocyanate transport in resting and IL-4-stimulated human bronchial epithelial cells: role of pendrin and anion channels. J Immunol. 2007;178:5144–53.
Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008; PMID: 18772398.
Campodonico VL, Gadjeva M, Paradis-Bleau C, Uluer A, Pier GB. Airway epithelial control of Pseudomonas aeruginosa infection in cystic fibrosis. Trends Mol Med. 2008;14:120–33.
Lau GW, Hassett DJ, Britigan BE. Modulation of lung epithelial functions by Pseudomonas aeruginosa. Trends Microbiol. 2005;13:389–97.
Price-Whelan A, Dietrich LE, Newman DK. Rethinking ‘secondary’ metabolism: physiological roles for phenazine antibiotics. Nat Chem Biol. 2006;2:71–8.
Wilson R, Sykes DA, Watson D, Rutman A, Taylor GW, Cole PJ. Measurement of Pseudomonas aeruginosa phenazine pigments in sputum and assessment of their contribution to sputum sol toxicity for respiratory epithelium. Infect Immun. 1988;56:2515–17.
Dietrich LE, Teal TK, Price-Whelan A, Newman DK. Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science. 2008;321:1203–6.
Lau GW, Hassett DJ, Ran H, Kong F. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol Med. 2004;10:599–606.
O’Malley YQ, Reszka KJ, Spitz DR, Denning GM, Britigan BE. Pseudomonas aeruginosa pyocyanin directly oxidizes glutathione and decreases its levels in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L94–103.
Muller PK, Krohn K, Muhlradt PF. Effects of pyocyanine, a phenazine dye from Pseudomonas aeruginosa, on oxidative burst and bacterial killing in human neutrophils. Infect Immun. 1989;57:2591–6.
Reszka KJ, O’Malley Y, McCormick ML, Denning GM, Britigan BE. Oxidation of pyocyanin, a cytotoxic product from Pseudomonas aeruginosa, by microperoxidase 11 and hydrogen peroxide. Free Radic Biol Med. 2004;36:1448–59.
Schwarzer C, Machen TE, Illek B, Fischer H. NADPH oxidase-dependent acid production in airway epithelial cells. J Biol Chem. 2004;279:36454–61.
Shao MX, Nadel JA. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc Natl Acad Sci USA. 2005;102:767–72.
Wesley UV, Bove PF, Hristova M, McCarthy S, van der Vliet A. Airway epithelial cell migration and wound repair by ATP-mediated activation of dual oxidase 1. J Biol Chem. 2007;282:3213–20.
Look DC, Stoll LL, Romig SA, Humlicek A, Britigan BE, Denning GM. Pyocyanin and its precursor phenazine-1-carboxylic acid increase IL-8 and intercellular adhesion molecule-1 expression in human airway epithelial cells by oxidant-dependent mechanisms. J Immunol. 2005;175:4017–23.
Denning GM, Wollenweber LA, Railsback MA, Cox CD, Stoll LL, Britigan BE. Pseudomonas pyocyanin increases interleukin-8 expression by human airway epithelial cells. Infect Immun. 1998;66:5777–84.
Usher LR, Lawson RA, Geary I, Taylor CJ, Bingle CD, Taylor GW, et al. Induction of neutrophil apoptosis by the Pseudomonas aeruginosa exotoxin pyocyanin: a potential mechanism of persistent infection. J Immunol. 2002;168:1861–8.
Allen L, Dockrell DH, Pattery T, Lee DG, Cornelis P, Hellewell PG, et al. Pyocyanin production by Pseudomonas aeruginosa induces neutrophil apoptosis and impairs neutrophil-mediated host defenses in vivo. J Immunol. 2005;174:3643–9.
Bianchi SM, Prince LR, McPhillips K, Allen L, Marriott HM, Taylor GW, et al. Impairment of apoptotic cell engulfment by pyocyanin, a toxic metabolite of Pseudomonas aeruginosa. Am J Respir Crit Care Med. 2008;177:35–43.
Kanthakumar K, Cundell DR, Johnson M, Wills PJ, Taylor GW, Cole PJ, et al. Effect of salmeterol on human nasal epithelial cell ciliary beating: inhibition of the ciliotoxin, pyocyanin. Br J Pharmacol. 1994;112:493–8.
Munro NC, Barker A, Rutman A, Taylor G, Watson D, McDonald-Gibson WJ, et al. Effect of pyocyanin and 1-hydroxyphenazine on in vivo tracheal mucus velocity. J Appl Physiol. 1989;67:316–23.
Dormehl I, Ras G, Taylor G, Hugo N. Effect of Pseudomonas aeruginosa-derived pyocyanin and 1-hydroxyphenazine on pulmonary mucociliary clearance monitored scintigraphically in the baboon model. Int J Rad Appl Instrum B. 1991;18:455–9.
Ran H, Hassett DJ, Lau GW. Human targets of Pseudomonas aeruginosa pyocyanin. Proc Natl Acad Sci USA. 2003;100:14315–20.
Kong F, Young L, Chen Y, Ran H, Meyers M, Joseph P, et al. Pseudomonas aeruginosa pyocyanin inactivates lung epithelial vacuolar ATPase-dependent cystic fibrosis transmembrane conductance regulator expression and localization. Cell Microbiol. 2006;8:1121–33.
Schwarzer C, Fu Z, Fischer H, Machen TE. Redox-independent activation of NF-kB by P. aeruginosa pyocyanin in a CF airway epithelial cell line. J Biol Chem. 2008;283:27144–53.
Schwarzer C, Fischer H, Kim EJ, Barber KJ, Mills AD, Kurth MJ, et al. Oxidative stress caused by pyocyanin impairs CFTR Cl(-) transport in human bronchial epithelial cells. Free Radic Biol Med. 2008; PMID: 18845244.
O’Malley YQ, Abdalla MY, McCormick ML, Reszka KJ, Denning GM, Britigan BE. Subcellular localization of Pseudomonas pyocyanin cytotoxicity in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;284:L420–30.
Muller M. Premature cellular senescence induced by pyocyanin, a redox-active Pseudomonas aeruginosa toxin. Free Radic Biol Med. 2006;41:1670–7.
Denning GM, Railsback MA, Rasmussen GT, Cox CD, Britigan BE. Pseudomonas pyocyanine alters calcium signaling in human airway epithelial cells. Am J Physiol. 1998;274:L893–900.
O’Malley YQ, Reszka KJ, Rasmussen GT, Abdalla MY, Denning GM, Britigan BE. The Pseudomonas secretory product pyocyanin inhibits catalase activity in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1077–86.
Britigan BE, Railsback MA, Cox CD. The Pseudomonas aeruginosa secretory product pyocyanin inactivates alpha1 protease inhibitor: implications for the pathogenesis of cystic fibrosis lung disease. Infect Immun. 1999;67:1207–12.
Shellito J, Nelson S, Sorensen RU. Effect of pyocyanine, a pigment of Pseudomonas aeruginosa, on production of reactive nitrogen intermediates by murine alveolar macrophages. Infect Immun. 1992;60:3913–5.
Lauredo IT, Sabater JR, Ahmed A, Botvinnikova Y, Abraham WM. Mechanism of pyocyanin- and 1-hydroxyphenazine-induced lung neutrophilia in sheep airways. J Appl Physiol. 1998;85:2298–304.
Ulmer AJ, Pryjma J, Tarnok Z, Ernst M, Flad HD. Inhibitory and stimulatory effects of Pseudomonas aeruginosa pyocyanine on human T and B lymphocytes and human monocytes. Infect Immun. 1990;58:808–15.
Miller KM, Dearborn DG, Sorensen RU. In vitro effect of synthetic pyocyanine on neutrophil superoxide production. Infect Immun. 1987;55:559–63.
Ras GJ, Anderson R, Taylor GW, Savage JE, Van Niekerk E, Wilson R, et al. Proinflammatory interactions of pyocyanin and 1-hydroxyphenazine with human neutrophils in vitro. J Infect Dis. 1990;162:178–85.
Muller M, Sorrell TC. Modulation of neutrophil superoxide response and intracellular diacylglyceride levels by the bacterial pigment pyocyanin. Infect Immun. 1997;65:2483–7.
Muller M, Sorrell TC. Production of leukotriene B4 and 5-hydroxyeicosatetraenoic acid by human neutrophils is inhibited by Pseudomonas aeruginosa phenazine derivatives. Infect Immun. 1991;59:3316–8.
Muller M, Sorrell TC. Leukotriene B4 omega-oxidation by human polymorphonuclear leukocytes is inhibited by pyocyanin, a phenazine derivative produced by Pseudomonas aeruginosa. Infect Immun. 1992;60:2536–40.
Nutman J, Berger M, Chase PA, Dearborn DG, Miller KM, Waller RL, et al. Studies on the mechanism of T cell inhibition by the Pseudomonas aeruginosa phenazine pigment pyocyanine. J Immunol. 1987;138:3481–7.
Nutman J, Chase PA, Dearborn DG, Berger M, Sorensen RU. Suppression of lymphocyte proliferation by Pseudomonas aeruginosa phenazine pigments. Isr J Med Sci. 1988;24:228–32.
Cheluvappa R, Jamieson HA, Hilmer SN, Muller M, Le Couteur DG. The effect of Pseudomonas aeruginosa virulence factor, pyocyanin, on the liver sinusoidal endothelial cell. J Gastroenterol Hepatol. 2007;22:1350–1.
Britigan BE, Roeder TL, Rasmussen GT, Shasby DM, McCormick ML, Cox CD. Interaction of the Pseudomonas aeruginosa secretory products pyocyanin and pyochelin generates hydroxyl radical and causes synergistic damage to endothelial cells. Implications for Pseudomonas-associated tissue injury. J Clin Invest. 1992;90:2187–96.
Kamath JM, Britigan BE, Cox CD, Shasby DM: Pyocyanin from Pseudomonas aeruginosa inhibits prostacyclin release from endothelial cells. Infect Immun. 1995;63:4921–3.
Warren JB, Loi R, Rendell NB, Taylor GW. Nitric oxide is inactivated by the bacterial pigment pyocyanin. Biochem J. 1990;266:921–3.
Muller M. Pyocyanin induces oxidative stress in human endothelial cells and modulates the glutathione redox cycle. Free Radic Biol Med. 2002;33:1527–33.
Muller M, Sztelma K, Sorrell TC. Inhibition of platelet eicosanoid metabolism by the bacterial phenazine derivative pyocyanin. Ann N Y Acad Sci. 1994;744:320–2.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rada, B., Leto, T.L. Redox warfare between airway epithelial cells and Pseudomonas: dual oxidase versus pyocyanin. Immunol Res 43, 198–209 (2009). https://doi.org/10.1007/s12026-008-8071-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-008-8071-8