
Abstract
The fourth edition of the World Health Organization (WHO) classification of endocrine tumours contains substantial new findings for the adrenal tumours. The tumours are presented in two chapters labelled as “Tumours of the adrenal cortex” and “Tumours of the adrenal medulla and extra-adrenal paraganglia.” Tumours of the adrenal cortex are classified as cortical carcinoma, cortical adenoma, sex cord stromal tumours, adenomatoid tumour, mesenchymal and stromal tumours (myelolipoma and schwannoma), haematological tumours, and secondary tumours. Amongst them, schwannoma and haematological tumours are newly documented. The major updates in adrenal cortical lesions are noted in the genetics of the cortical carcinoma and cortical adenoma based on the data from The Cancer Genome Atlas (TCGA). Also, a system for differentiation of oncocytoma from oncocytic cortical carcinoma is adopted. Tumours of the adrenal medulla and extra-adrenal paraganglia comprise pheochromocytoma, paraganglioma (head and neck paraganglioma and sympathetic paraganglioma), neuroblastic tumours (neuroblastoma, nodular ganglioneuroblastoma, intermixed ganglioneuroblastoma, and ganglioneuroma), composite pheochromocytoma, and composite paraganglioma. In this group, neuroblastic tumours are newly included in the classification. The clinical features, histology, associated pathologies, genetics, and predictive factors of pheochromocytoma and paraganglioma are the main changes introduced in this chapter of WHO classification of endocrine tumours. The term “metastatic pheochromocytoma/paraganglioma” is used to replace “malignant pheochromocytoma/paraganglioma.” Also, composite pheochromocytoma and composite paraganglioma are now documented in separate sections instead of one. Overall, the new classification incorporated new data on pathology, clinical behaviour, and genetics of the adrenal tumours that are important for current management of patients with these tumours.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
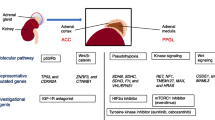
The fourth edition of the World Health Organization (WHO) classification of endocrine tumours published in 2017 contains substantial modification in the topics of adrenal tumours when compared with the third edition that was published in 2004 [1, 2]. These modifications are mainly based on new knowledge of the genetics as well as the clinical behaviour of these tumours. In the 2017 classification, the information on adrenal tumours was discussed in two chapters instead of one chapter in the 2004 classification. The first chapter (Chap. 4) is labelled as “Tumours of the adrenal cortex” which comprises tumours that arise from or affecting predominately the adrenal cortex. The second chapter (Chap. 5) is named as “Tumours of the adrenal medulla and extra-adrenal paraganglia.” Table 1 summarizes the categories of tumours in the updated WHO classification of this group of tumours. The key changes noted in this edition of WHO classification are the incorporation of the molecular genetic findings in adrenal cortical carcinoma and pheochromocytoma/paraganglioma as well as the inclusion of a few entities in both adrenal cortical and medullary tumours. The following sections will discuss the classification, provide updates, and highlight the changes noted in new WHO classification of the adrenal tumours.
TUMOURS of the Adrenal Cortex
Cortical Carcinoma
Amongst the primary adrenal tumours, cortical carcinoma is uncommon. It is less common than cortical adenoma and pheochromocytoma [3]. Nevertheless, great progress has been made in the understanding of the cancer as a result of genomic studies which mainly is from The Cancer Genome Atlas (TCGA) [4,5,6,7,8]. The current WHO classification has updated the histological features as well as the potential application of the new knowledge of the genomic landscape noted in adrenal cortical carcinoma.
One of the characteristics of the patients having adrenal cortical carcinoma is the bimodal age distribution. The tumour is common in the first and fifth decades [9]. There is a predilection for females for adrenal cortical carcinoma. Also, approximately half of the patients with adrenal cortical carcinomas are asymptomatic or presents with symptoms related to mechanical effects of tumour growth. The other half of patients with adrenal cortical carcinoma present with signs and symptoms of steroid hormone excess (aldosterone-producing, cortisol-producing, and sex hormone-producing). Cortisol-producing adrenal cortical carcinoma is the most common amongst the functioning tumours.
The European Network for the Study of Adrenal Tumours (ENSAT) is recommended for use in staging of adrenal cortical carcinoma. In this classification, adrenal cortical carcinomas are classified on criteria based on size and extent of the tumours. The organ confined stages are stage I and stage II [10]. Stage I tumours are less than 50 mm in diameter whereas stage II tumours are larger than 50 mm in diameter. On the other hand, stage III tumours are represented by involvement of surrounding tissue, regional lymph nodes, or regional veins whereas stage IV tumours are characterized by distant metastasis. In fact, many adrenal cortical carcinomas could reach massive size and weight. For instance, adrenal cortical carcinoma with size over 25 cm and 2 kg has been reported [11]. Areas of necrosis and haemorrhages are frequently noted in adrenal cortical carcinoma.
The WHO defines adrenal cortical carcinoma as a malignant epithelial tumour of adrenal cortical cells. Histologically, the classical features of adrenal cortical carcinoma comprise tumour cells with adrenal cortical differentiation and necrosis. The tumour often has a thick capsule. The distinction from adrenal cortical adenoma is often straightforward. There are many diagnostic algorithms proposed in the previous WHO edition to differentiate cortical carcinoma from adenoma. Weiss score, although imperfect, is adopted to by current WHO classification as a primary determinant of malignancy [12]. In pediatric patients (less than 15 years old), Weiss criteria may overdiagnose cortical tumour as adrenal cortical carcinoma. Using Weiss criteria, malignancy could be diagnosed based on at least three of the following features—high nuclear grade, high mitotic rate (>5 mitoses per 50 high-power field), atypical mitotic figures, <25% clear cells, diffuse architecture, tumour necrosis, venous invasion, sinusoidal invasion, and capsular invasion (Fig. 1). Reticular staining could be to identify the nested pattern of cells. Disruption of the reticulin network could be assessed by reticulin stain whereas interruption of basal lamina could be reviewed by using immunohistochemical antibodies to laminin or collagen type IV. The diffuse architecture (one of the criteria in Weiss score) of carcinoma could be highlighted by loss of this regular network.

Adrenocortical carcinoma: showing marked necrosis and tumour cells with prominent nuclear pleomorphism invasion (haematoxylin × 20)
Apart from the conventional adrenocortical carcinoma, the new WHO classification also recognized a few histological variants. They are in decreasing order of frequency—oncocytic, myxoid, and sarcomatoid carcinomas. Adrenal myxoid carcinomas and sarcomatoid carcinomas are rare. Myxoid carcinoma has abundant extracellular mucin in the stroma. Using Weiss score, a patient with this subtype of adrenal cortical carcinoma may be underdiagnosed. Sarcomatoid carcinoma reveals loss of cortical differentiation. The carcinoma could have typical cortical carcinoma area (biphasic). Sarcomatoid carcinoma needs to be differentiated from retroperitoneal sarcoma involving the adrenal gland especially when that lacks a typical cortical carcinoma area (monophasic) [1].
Adrenal oncocytic carcinoma is characterized by tumour cells having abundant eosinophilic cytoplasm. Weiss score cannot be applied in the differentiation of oncocytic adenoma from oncocytic carcinoma. This is because three of the criteria in the Weiss score—high nuclear grade, <25% clear cells, and diffuse architecture are characteristics of adrenal oncocytic tumours (Fig. 2). Thus, Lin-Weiss-Bisceglia system is recommended in the updated WHO classification for differentiation of oncocytic adenoma from oncocytic carcinoma [12]. In this system, the presence of any one of the three major criteria—high mitotic rate (>5 mitoses per 50 high-power field), atypical mitotic figures and venous invasion—indicates malignancy. These criteria are actually amongst the nine features noted in Weiss score. In addition, the presence of one to four minor criteria in adrenal cortical neoplasm (necrosis, sinusoidal invasion, capsular invasion, and large size/weight (size >10 cm and/or weight >200 g)) indicates that it is of uncertain malignant potential (Table 2).

Oncocytic adenoma: showing eosinophilic cytoplasm, diffuse architecture, and high nuclear grade (haematoxylin × 4)
Immunohistochemical markers such as SF1 (steroidogenic factor-1), inhibin alpha, melan-A, and calretinin could be used to identify the adrenal cortical carcinomas. SF-1 is an orphan nuclear receptor that is expressed in the adrenal gland, gonads, spleen, ventromedial hypothalamus, and pituitary gonadotropic cells and has a role in steroidogenesis and development. It has high sensitivity and specificity in identifying the cortical origin of the adrenal mass. The expression of SF-1 also correlated with poorer clinical outcome in patients with adrenal cortical carcinoma. However, its expression is limited by pre-analytic factor. On the other hand, synaptophysin is commonly expressed in adrenal cortical carcinoma. The expression of synaptophysin in cortical carcinoma should be interpreted with caution so as not to confuse the tumour with pheochromocytoma. Also, adrenal cortical carcinoma can be positive for cytokeratin.
In addition to the accepted diagnostic algorithms, immunostaining for Ki-67 may potentially be used to differentiate adrenocortical adenoma from cortical carcinoma. Cortical adenomas show a Ki-67 proliferation index of <5% and carcinomas of >5%. Also, insulin-like growth factor 2 (IGF2) could use to support the diagnosis of cortical carcinoma. It is a growth factor with structural similarity to insulin and mainly responsible for foetal growth. IGF2 activates the MAPK and PI3K pathways in cancer. The protein is expressed in a much high proportion of cortical carcinomas when compared to lower proportion of expression in cortical adenomas. In addition, the presence of IGF2 may explain the tumour-associated hypoglycaemia in some patients with adrenocortical carcinoma.
TCGA analysis of adrenal cortical carcinoma showed (1) high prevalence of IGF2 overexpression; (2) high frequency of p53 mutations, which are dominant in paediatric cases and associated with aggressive diseases; (3) frequent and diverse Wnt pathway defects (CTNNB1 point mutations and ZNRF3 deletions); (4) copy number alterations and whole genome doubling as important mechanisms of disease progression; (5) telomere and telomerase reactivation; and (6) relative lack of targetable hotspot mutations [4,5,6,7,8]. Many of the molecular changes could aid in the differentiation between good and poor prognosis in patients with cortical carcinoma. Some of the molecular changes may also aid to differentiate cortical carcinoma from cortical adenoma. From the therapeutic standpoint, drugs with combined inhibition of IGF2 and Wnt pathways may be investigated for the use in the treatment of metastatic or recurrent adrenocortical carcinoma.
The 5-year survival rate of patients with adrenal cortical carcinoma is less than 50% [13]. Prognostic factors in patients with adrenal cortical carcinoma include advanced clinical staging as well as GRAS parameters (grade of the carcinoma, resection status, patient age (≥50 years) and symptoms related to tumour or to hormone excess) [13]. Also, rupture of a tumour capsule may be an important factor in the prognosis of patients with adrenal cortical carcinoma. The prognostic roles of Ki-67 index are validated by dividing the adrenal cortical carcinoma into three groups based on percentages of Ki-67-positive cancer cells. These groups are <10, 10–19, and >20%, respectively. The proliferative index correlates with multi-platform molecular profiling (includes methylation, copy number, mRNA, whole genome doubling, and gene mutations) from the data of TCGA [14, 15].
Cortical Adenoma
Adrenal cortical adenoma is the most common primary adrenal tumour as well as the most common adrenal incidentaloma [3]. The distinction between hyperplastic nodule and adenoma of adrenal gland is difficult and arbitrary. Clinically, cortical adenoma could be non-functioning and functioning (aldosterone-producing, cortisol-producing, and sex hormone-producing). It is often yellow in colour (Fig. 3).

Cortical adenoma: with bright yellow cut surface
Adrenal cortical adenoma typically consists of lipid-rich cells with abundant intracytoplasmic lipid resembling zona fasciculata and lipid-poor compact cells resembling zona reticularis in different proportions (Fig. 4). Features like spironolactone bodies, lipomatous, or myelolipomatous metaplasia may be seen. In cortisol-producing cortical adenoma, the non-neoplastic adrenal cortex often becomes atrophic, with marked thinning of the zona fasciculata and zona reticularis. The glomerulosa is spared and can appear hyperplasia.

Cortical adenoma: abundant lipid-rich tumour cells with some lipid-poor compact cells
In the current WHO classification, two variants of cortical adenoma were mentioned, namely black adenoma and oncocytoma. Black adenoma is a cortical adenoma that is dark in colour due to accumulation of lipofuscin [16] (Fig. 5). Adrenal cortical oncocytoma is a cortical tumour with large cells having dense eosinophilic cytoplasm, isolated nuclear pleomorphism, and prominent nucleoli [17]. It is often non-functioning.

Black adenoma: cortical adenoma with dark colour on cut surface as a result of lipofuscin deposition. It is easier to appreciate on macroscopic examination than microscopic examination
Immunohistochemical profiles of adrenal cortical adenomas are defined in the current WHO classification. As in adrenal cortical carcinoma, cortical adenoma is a positive maker of cortical origin, namely SF-1, inhibin, and melan-A. They are useful for differentiating the cortical adenoma from metastatic tumours. Also, as in cortical carcinoma, synaptophysin staining is positive and non-specific. In the current WHO classification, CYP11B2 is reported to be useful in identifying aldosterone-secreting adenoma.
In the current WHO classification, the findings of next generation sequencing studies in adrenal cortical adenomas are presented. In aldosterone-secreting adenoma, mutations in KCNJ5, ATP1A1, ATP2B3, and CACNA1D that interfere with ion transport across the plasma membrane are documented [18]. In cortisol-secreting adenoma, mutation of PRKACA is reported [19]. Mutation of PRKAR1A is noted in cortisol-secreting adenoma in the setting of Carney complex [20]. In addition, non-functioning adenoma could have CTNNB1 mutation resulting in nuclear localization of beta-catenin [21].
The above-mentioned molecular changes noted since the last WHO classification help in the understanding of pathogenesis of different types of cortical adenoma. Also, it is clear that adenoma and carcinoma of adrenal cortex have different molecular profiles. However, diagnostically, we still rely on clinical biochemistry and absence of malignant features to make the diagnosis of adrenal cortical adenoma.
Sex Cord Stromal Tumours
Sex cord stromal tumour of the adrenal gland is rare. There were three granulosa cell tumours and three Leydig cell tumours reported in the literature [22,23,24,25,26,27]. All of them were reported in postmenopausal women. Patients with Leydig cell tumour presented with virilisation whereas those with granulosa cell tumours had uterine bleeding or refractory hypertension. The histology and immunochemical profiles are similar to their counterparts in the ovary.
Adenomatoid Tumour
Adrenal adenomatoid tumour is a benign tumour of mesothelial derivation. The tumour has a significant male predilection. Adrenal adenomatoid tumours are mostly asymptomatic but could produce hormone-related symptoms [28, 29]. They show similar morphology as in adenomatoid tumours in the other portion of urogenital tract except that stromal smooth muscle proliferation is often less common. Composite tumours (with adenoma or myelolipoma) rarely occur [30, 31].
Mesenchymal and Stromal Tumours
In the current WHO classification, when compared to the third edition, only myelolipoma and schwannoma are discussed as well as in more details under the category of mesenchymal and stromal tumours.
Adrenal myelolipoma is a benign cortical tumour composed of adipose tissue and bone marrow elements. It is the second most common adrenal cortical tumour [3]. The current WHO classification has updated the clinical and pathological details of the tumour. Calcification or osseous metaplasia is sometimes noted in adrenal myelolipoma [32, 33]. Differential diagnoses of the adrenal tumour include other less common adrenal lipomatous lesions such as lipoma, teratoma, and angiomyolipoma [34,35,36,37,38]. Myelolipoma has been reported to be associated with many diseases as it was commonly being detected incidentally during workup for other diseases. The tumour was often detected in patients with chronic diseases such as cancers, diabetes mellitus, and haematological disorders [39]. In these conditions, endogenous adrenocorticotropic hormone (ACTH) could be elevated which could be contributed to the pathogenesis of myelolipoma. The tumour is more common in females and located twice more common on the right adrenal gland [40]. In the English literature, approximately 40 cases of bilateral myelolipomas were noted [41]. Biochemically, the majority of the adrenal myelolipomas are non-functioning. Endocrine dysfunction was noted in less than 10% of adrenal myelolipomas [34, 41]. All adrenal myelolipomas are benign.
The management for large, functioning, and complicated cases are by surgery.
Adrenal schwannoma is rare with less than 60 cases reported in the English literature [42,43,44]. It is an entity not documented in third edition of WHO classification of endocrine tumours. The tumour is more common in women. It has similar histology to schwannoma that occurs in other location. Adrenal schwannoma needs to be differentiated from retroperitoneal schwannoma as the latter can undergo malignant transformation by clinical and morphological assessment. Adrenal schwannoma should also be differentiated from other rarer spindle cell lesions in the adrenal such as leiomyoma and solitary fibrous tumour. These spindle cell lesions are rare and can easily be differentiated from schwannoma by morphological examination and immunohistochemistry. Management of adrenal schwannoma is surgical excision as there is no way to differentiate the tumour from other adrenal tumours and many adrenal schwannomas are large in size. No malignant progression of the tumour has been reported. In addition, in contrast to schwannoma noted in other locations, the adrenal schwannoma has not been reported to be associated with neurofibromatosis.
Haematological Tumours
Primary haematological tumours are mostly lymphomas and rarely plasmacytoma. They were not discussed in the last edition of WHO classification of endocrine tumours. Less than 200 cases of primary adrenal lymphoma were reported in the literature [45]. Majority of the patients with primary adrenal lymphoma are male adults presented with B symptoms as well as adrenal insufficiency. Involvement of the adrenal glands presents with effacement of both cortex and medulla. Bilateral involvement of adrenal glands as well as involvement of the adjacent para-aortic lymph nodes could be seen. To be classified as primary adrenal lymphoma, the lymphoma in the adrenal gland must be more prominent than in the adjacent lymph nodes. Histologically, the most common lymphoma is diffuse large B cell lymphoma which accounted for slightly less than 80% of cases [45]. Many of these cases have Bcl-6 gene rearrangement [46]. The second most common adrenal lymphoma noted is peripheral T cell lymphoma.
Secondary Tumours
Secondary tumours of the adrenal cortex could be from direct infiltration by adjacent cancer or more commonly by haematogenous metastases. Adrenal metastases are common in patients with advanced cancer. Imaging approaches such as CT scan, MRI, PET, endoscopic ultrasound, and fine needle aspiration were adopted for the study of secondary tumours. Involvement of both adrenal glands is noted in half of the cases [47] (Fig. 6). Macroscopically, the colour and texture of metastatic lesions are different from adrenal cortical adenoma. Carcinoma, primarily adenocarcinoma, comprises more than 90% of the secondary tumours. The common primary sites of the metastatic carcinoma depend mainly on the prevalence of the different cancers in the population. It may be difficult to distinguish secondary tumours from primary cortical tumours. Also, identifying the primary site of the lesion may be challenging at the time of adrenal biopsy. Immunohistochemistry is of great help in these settings. The prognosis of the patients with secondary tumours in adrenal gland is generally poor. Nevertheless, improved patients’ survival in some cases could occur after surgical resection of adrenal metastases [47, 48]. Patients with resection of adrenal metastases had better response to adjuvant therapies.

Bilateral adrenal metastases from carcinoma of caecum detected at autopsy
Tumours of the Adrenal Medulla and Extra-adrenal Paraganglia
By definition, pheochromocytomas arise from adrenal medulla whereas paragangliomas arise from extra-adrenal paraganglia. In this chapter of the WHO classification, the advances in knowledge from this group of tumours attract the most attention from pathologists, scientists, and clinicians worldwide.
Firstly, there is exponential increase in genetic discoveries in this group of tumours as a result of advances in next generation sequencing technology in the recent years [49, 50]. At least 30% of these tumours are now known to be hereditary. Thus, this group of tumours is the most strongly hereditary amongst all human tumours. There are now more than 20 sporadic or hereditary susceptibility genes related to the pathogenesis of pheochromocytoma and paraganglioma [50]. Genetic mutations of many of the pheochromocytoma and paraganglioma could be grouped into two major clusters. Cluster 1 mutations are involved with the pseudohypoxic pathway. Also, they resulted in a marked increase in vascularization and in the expression of vascular endothelial growth factor (VEGF) and its receptors. In addition, some members of the group have impaired DNA demethylation. Cluster 2 mutations are associated with abnormal activation of kinase signalling pathways. Examples of the kinase pathways involved are PI3Kinase/AKT, RAS/RAF/ERK, and mTOR pathways. Overall, known genetic mutations account for the pathogenesis of approximately 60% of pheochromocytomas and paragangliomas [50]. Therapies targeting these pathways may have roles in the treatment of metastasizing as well as recurrent pheochromocytoma and paraganglioma. Drugs targeting angiogenesis and demethylation agents could be used for cluster 1 tumours whereas agents targeting mTOR pathway could be used for cluster 2 tumours in future clinical trials.
Amongst the cluster I and 2 gene mutations, the relative common mutations in hereditary pheochromocytoma and paraganglioma noted include mutations in VHL (von Hippel-Lindau), SDHB (succinate dehydrogenase B), SDHD (succinate dehydrogenase D), RET (rearranged during transfection), and NF1 (neurofibromatosis 1). The approximate frequencies of these mutations in the tumours are 9, 6–8, 5–7, 5, and 2% respectively [1]. Mutations of VHL, RET, and NF1 are predominately in pheochromocytoma and rare in paraganglioma whereas mutations of SDHB and SDHD are common in paraganglioma but uncommon in pheochromocytoma. SDHB mutation is predominately noted in sympathetic paraganglioma but uncommon in head and neck paraganglioma. On the other hand, SDHD mutation has similar distribution between sympathetic and head and neck paraganglioma [1].
Amongst these genetic changes, mutations in genes encoding succinate dehydrogenase (SDH) subunits, collectively known as SDH gene family (SDHB, SDHD, SDHC, SDHA, SDHAF2), in combination have been found to be the most common genetic cause of hereditary pheochromocytoma and paraganglioma.
Mutations of any of the SDH gene family will lead to loss of SDHB protein which could be detected by immunohistochemistry [51]. Thus, the detection of SDHB protein is an important marker to perform in this group of tumours. Of interest, NF1 accounted for only approximately 2% of the hereditary pheochromocytoma and paraganglioma but it is the most common somatic mutation in this group of tumours [52].
The discovery of the genes responsible for pheochromocytoma and paraganglioma provides the genetic base that they are linked to other tumours in some clinical syndromes. The classical syndromes comprise—neurofibromatosis, multiple neuroendocrine neoplasia (MEN) (II and III) syndromes, and von Hippel-Lindau syndrome [51]. Also, mutations in SDH genes contribute to the understanding of hereditary paraganglioma—pheochromocytoma syndromes, Carney’s triad, and Carney-Stratakis syndrome [51]. Other than known association of paraganglioma with gastrointestinal stromal tumour (GIST), lesions newly known to be associated with the genetic mutations in pheochromocytoma/paraganglioma include succinate dehydrogenase-deficient renal cell carcinoma, pituitary tumour, and pancreatic neuroendocrine tumour [51, 53]. On the other hand, there are some hereditary pheochromocytomas or paragangliomas (with TMEM127 or KIF1B mutations) that are not syndromic [51, 54]. Also, there is transmission of some hereditary pheochromocytoma or paraganglioma subject to inheritance that involves a parent-of-origin effect in which the disease usually manifests only following paternal transmission (SDHD, SDHAF2, and MAX) [55].
Lastly, the biological behaviour of this group of tumours could not be identified based on the histological features. There are two systems mentioned in the new WHO classification for reference. In 2002, PASS (pheochromocytoma of the adrenal gland scaled score) was developed by Thompson after review of 100 cases of pheochromocytoma [56]. PASS is established on a set of 12 histological features. The drawbacks of using PASS are that the score is based purely on histological features and could only apply for pheochromocytoma [56]. The prognostic values of PASS have been tested by different research groups and usefulness of PASS has been questioned [57, 58]. In 2005, Kimura and colleagues from Japan proposed another system for assessment of malignant potential of this group of tumours based on 146 pheochromocytoma and paraganglioma [59]. The system was refined in 2014 based on a total of 163 tumours, including 40 metastatic tumours, collected by the Pheochromocytoma Study Group in Japan (PHEO-J) [60]. The system is called grading system for pheochromocytoma and paraganglioma (GAPP). GAPP omits several PASS parameters. The system was based on four histological features (histological pattern, cellularity, comedo-type necrosis, capsular/vascular invasion), proliferative index (Ki-67), and hormones secreted by the pheochromocytoma/paraganglioma. GAPP takes into account of biochemical properties of paraganglioma or pheochromocytoma. Tumours with norepinephrine secretion are given higher score (more aggressive) than non-functioning tumours or tumour secreting epinephrine. In the GAPP follow-up study, SDHB immunohistochemistry was added as a parameter [61]. Both systems are not universally adopted in the current WHO classification of endocrine tumours as the findings are still preliminary and without international validations. They were documented at current in the WHO classification for information. It is worth noting that some other parameters like the size of the tumour, microRNAs, and genomic results may also predict the behaviour of this group of tumour [62,63,64].
Pheochromocytoma
The terms “malignant pheochromocytoma” and “benign pheochromocytoma” from the 2004 WHO classification of endocrine tumours are abandoned in the current WHO classification of endocrine tumours. The two sections in the 2004’s WHO classification of endocrine tumours (“Benign pheochromocytoma” and “Malignant pheochromocytoma”) were combined into a single section—“Pheochromocytoma” in the 2017 WHO classification of endocrine tumours. This is because there is no histological system that is currently endorsed for the biological aggressiveness of this group of tumours. Thus, all pheochromocytomas could have metastatic potential. By the same token, the term “malignant” is not used but replaced with “metastatic” in this group of tumours. This also eliminates the confusion in trying to differentiate between the locally invasive and distant metastatic tumours. This change in terminology also apply to the paraganglioma.
Pheochromocytoma is an intra-adrenal sympathetic paraganglioma. Most pheochromocytomas occur in the fourth to fifth decades of life and with roughly equal sex distribution [65]. It is typically encapsulated tumour that is pinkish-grey to tan on cut sections (Fig. 7). Cystic changes and haemorrhages could be noted.

Pheochromocytoma: the colour is usually light brown on cut sections
The different catecholamines are discriminators of different hereditary forms of pheochromocytoma [66,67,68,69,70,71]. Pheochromocytomas associated with MEN II or neurofibromatosis type 1 typically produce epinephrine. Also, von Hippel-Lindau syndrome-associated pheochromocytomas have isolated increases in normetanephrine and norepinephrine. In addition, dopamine metabolite (3-methoxytyramine) points to the presence of SDHB, SDHD, or SDHC mutations and is linked to potentially metastatic tumours.
The current WHO classification labels medullary nodule less than 1 cm (previously arbitrarily classified as hyperplastic nodule based on its small size) as small pheochromocytoma based on the molecular findings. The main tumour cells have granular cytoplasm in a vascular stroma (Fig. 8). They are identified by neuroendocrine markers. Sustentacular cells around the main tumour cells could be highlighted by S-100. Nuclear pseudoinclusions, nuclear pleomorphism, and intracytoplasmic globules are often noted [65]. Lipofuscin, neuromelanin, or melanin pigments are sometimes found [72]. Less common features such as amyloid deposits, spindle cell, clear cell, and oncocytic changes could be seen [65, 73]. Approximately 5 to 10% of pheochromocytomas metastasize [74].

Pheochromocytoma: granular cytoplasm in a vascular stroma (haematoxylin × 4)
Paraganglioma
Paragangliomas are extra-adrenal non-epithelial tumours originating from neural crest-derived paraganglion cells situated in the region of autonomic nervous system ganglia and accompanying nerves. The histology is similar to pheochromocytoma. As a group, they had higher frequency of metastases when compared to pheochromocytoma [74].
Instead of having four sections of paragangliomas from different sites in the 2004 version, the current WHO classification of endocrine tumours presents the information in one section for paraganglioma. Paraganglioma is divided into two groups: one from the parasympathetic system and one from the sympathetic system based on the clinical and biological behaviour. Paraganglioma from parasympathetic paraganglia is primary located in the head and neck and less frequently located in the thorax and pelvis. Thus, paraganglioma is described under two subtypes in the section. They are (1) head and neck paraganglioma and (2) sympathetic paraganglioma.
Head and Neck Paraganglioma
Head and neck paragangliomas are generally non-functioning [75, 76]. They are named according to the anatomical sites of origin as carotid body paraganglioma, jugulotympanic paraganglioma (middle ear), vagal paraganglioma, and laryngeal paraganglioma (Fig. 9). These four entities are described in details in separate sections in a chapter in the 2017 World Health Organization (WHO) classification of head and neck tumours by the same group of authors [77]. Head and neck paraganglioma is the most common paraganglioma which accounts for approximately 20% of all paragangliomas [78]. Amongst them, carotid body paraganglioma is the most common and accounts for over half of the head and neck paraganglioma [75, 76]. As a whole, less than 5% of head and neck paragangliomas metastasize. The lower risk of metastasis was noted in carotid body paraganglioma [78].

Middle ear paraganglioma: it is highly vascular and may be mistaken for a haemangioma (haematoxylin × 4)
Hereditary cases of head and neck paraganglioma could be multiple and occur in association with sympathetic paraganglioma. The germline mutation is slightly less than 20% in patients who appear as having apparently sporadic tumours and much higher in patients with family history [79]. The mutation most commonly noted in one of the succinate dehydrogenase gene (SDHx) and could be screened by immunohistochemical staining for SDHB protein. Paragangliomas associated with SDHB mutations have a high risk of metastasis. Thus, even in the absence of family history, genetic testing could be recommended for at least the most common genes in all patients, depending on local resources.
Sympathetic Paraganglioma
Approximately 85% of sympathetic paragangliomas arise below the diaphragm. Sympathetic paraganglioma could be seen in retroperitoneum around the adrenal/renal area, around the organ of Zuckerkandl, or in the urinary bladder [75, 80]. The other sympathetic paragangliomas are noted in the thorax, heart, and other locations [81,82,83]. Sympathetic paragangliomas are more likely to be functioning when compared to head and neck paragangliomas [84]. Patients with sympathetic paragangliomas usually have elevated norepinephrine only or norepinephrine and dopamine. The patients do not have elevated epinephrine as the enzyme required for the formation of epinephrine is only found in adrenal medulla [85]. Paediatric onset paragangliomas are almost always hereditary [86]. Pseudorosette pattern may be more common in SDHB mutation-associated tumours [61]. Sympathetic paragangliomas had high risk of metastases and those with SDHB mutation had even higher risk of metastases (could up to 50%) [87].
Neuroblastic Tumour of the Adrenal Gland
Neuroblastic tumours of adrenal gland are now included in Chap. 5 of the WHO classification of endocrine tumours. Neuroblastic tumours arise from the sympathoadrenal lineage of the neural crest during development. The primary site of this group of tumour is often abdominal (around 70%) and many of them are located in the adrenal gland [88,89,90]. The other sites include abdominal ganglia, thoracic ganglia, pelvic ganglia, cervical sympathetic ganglia, and paratesticular region. They are divided into four categories based on the morphology, clinical features, and behaviour by the International Neuroblastoma Pathology Classification (INPC). These four categories are neuroblastoma, nodular ganglioneuroblastoma, intermixed ganglioneuroblastoma, and ganglioneuroma.
Neuroblastoma is a cellular neuroblastic tumour without prominent Schwannian stroma. The tumour occurs slightly more common in boys [88, 89]. It occurs in patients from prenatal to 4 years. Also, the tumour is often clinically aggressive and could present with metastases in many different organs such as lymph node, liver, and bones. Neuroblastoma is further subtyped into undifferentiated (no clearly identifiable neurophil formation), poorly differentiated (with neurophil, <5% differentiating neuroblasts and often Homer Wright rosette), and differentiating (usually with abundant neurophil and >5% differentiating neuroblasts). The differentiating neuroblasts are characterized by synchronous differentiation of the nucleus (enlarged, eccentrically located, with vesicular chromatin, and prominent nucleoli) and of the cytoplasm (eosinophilic or amphophilic with diameter at least twice that of the nucleus). Immunohistochemical stains are needed to differentiate the undifferentiated subtype from other small round cell tumours. Neuronal makers like synaptophysin, chromogranin, and CD56 as well as specific neural crest markers such as tyrosine hydroxylase and PHOX2b are useful for identifying this group of tumours [91, 92].
Ganglioneuroma is a benign neuroblastic tumour. It occurs in patients of older age groups than those with neuroblastoma or ganglioneuroblastoma. Adrenal ganglioneuroma is usually in the 4th decade [93]. Macroscopically, the tumour is lobulated and grey-white (Fig. 10). The tumour is composed of Schwannian stroma with individually distributed neuronal elements. It is subdivided into maturing and mature subtypes. Maturing subtype contains maturing and mature ganglion cells whereas mature subtype contains exclusively mature ganglion cells surrounded by satellite cells. The ganglion cells are positive for S-100.

a Ganglioneuroma: grey-white lobulated mass in adrenal gland. b Ganglioneuroma: microscopic examination showing the ganglion cells in fibrous stroma (haematoxylin × 20)
Ganglioneuroblastoma comprised of mixture of neuroblasts and ganglion cells in varying proportions. It occurs in intermediate position between neuroblastoma and ganglioneuroma in terms of morphology, clinical presentation, and prognosis. In the INPC, it is divided into two types. The first type is nodular ganglioneuroblastoma. The tumour is identified by a grossly visible neuroblastic nodular component (stroma-poor) which is usually haemorrhagic and/or necrotic coexisting with an intermixed ganglioneuroblastoma (stroma-rich) or ganglioneuroma (stroma-dominant) component. The other type is intermixed ganglioneuroblastoma which is composed of intermixing of neuroblastic cells and ganglion cells. By definition, more than 50% of tumour tissue shows a ganglioneuroma appearance.
Two systems, the International Neuroblastoma Staging System (INSS) and the International Neuroblastoma Risk Group (INRG) staging system are widely recognized to predict the prognosis of patients with neuroblastoma [94,95,96].
INSS based on (1) molecular factors, (2) patient age (the younger the patient, the worse the prognosis), (3) INPC (International Neuroblastoma Pathology Classification) histology grouping, and (4) other clinical parameters (e.g. surgical resection details, symptomatic or not). The molecular factors include MYCN status and DNA index (ploidy). The MYCN oncogene is amplified in approximately 20 to 25% of neuroblastoma. The amplification could be detected by FISH or by immunohistochemistry. The INPC histology grouping divided the patients with neuroblastic tumours into two groups—favourable histology group and unfavourable histology group based on age of the patients, histological types as mentioned earlier, and mitosis-karyorrhexis index (MKI, defined as number of cells undergoing mitosis or karyorrhexis per 5000 cells).
Similarly, INRG based on (1) patient age, (2) histological category, (3) grade of tumour differentiation, and (4) molecular factors. The molecular factors are MYCN status, ploidy, and presence of unbalanced 11q aberration (higher risk with presence of the aberration).
Composite Pheochromocytoma
Composite pheochromocytoma is a tumour consisting of pheochromocytoma combined with a developmentally related neurogenic tumour such as ganglioneuroma, ganglioneuroblastoma, neuroblastoma, or peripheral nerve sheath tumour. Majority of the cases had ganglioneuroma as the neurogenic component. Many cases were reported as case reports in the literature. There are only four relatively larger series [97,98,99,100]. Lam and Lo in 1999 reported the first series with four patients and reviewed the features of 31 cases in the literature [97]. To date, slightly more than 70 cases were reported. Composite pheochromocytomas were seen in older patients and were bigger than conventional pheochromocytomas [97]. There is no definite gender predilection. The syndrome of watery diarrhoea, hypokalaemia, and achlorhydria caused by ectopic secretion of vasoactive intestinal peptide (VIP) is most often detected in composite pheochromocytoma than conventional pheochromocytoma [101,102,103]. The tumour could be associated with hereditary syndromes as well having metastases similar to conventional pheochromocytoma. Some of the composite tumours had distinctive components whereas some have mixed cells. Immunohistochemical stains could help to identify the presence of composite tumour.
Composite Paraganglioma
Composite paraganglioma is much rarer than composite pheochromocytoma. In the updated WHO classification, they were described separately from composite pheochromocytoma. Composite paragangliomas occurred in patients of wide age range (15 months to 81 years) [104, 105]. Slightly more than 20 cases have been reported. Majority of the cases were noted in the retroperitoneum and urinary bladder [104,105,106,107,108,109,110,111,112]. Cases have been described in cauda equina and posterior mediastinum [113, 114]. No case has been reported in the head and neck region. Some of the cases were symptomatic with catecholamine-related signs [104]. Majority of the reported cases consisted of paraganglioma and ganglioneuroma [104, 106,107,108,109,110,111,112,113,114]. Also, there are a few examples of composite paraganglioma and neuroblastoma [105, 115, 116]. One pigmented composite paraganglioma was noted in the urinary bladder [111]. Armstrong and colleagues has reported SDHB deletion in one retroperitoneum composite paraganglioma (with neuroblastoma) in a 13-year-old girl [115]. Nearly all the cases are non-metastasizing. Nevertheless, Fritzsche and colleagues had reported a composite paraganglioma and neuroblastoma having bone and liver metastasis and died 10 months after diagnosis [116].
References
Lloyd RV, Osamura RY, Kloppel G, Rosai J. WHO classification of tumours: pathology and genetics of tumours of endocrine organs. 4th ed. Lyon: IARC; 2017.
DeLellis RA, Lloyd RV, Heitz PU, Eng C. WHO classification of tumours: pathology and genetics of tumours of endocrine organs. 3rd ed. Lyon: IARC; 2004.
Lam KY. Adrenal tumors in Chinese. Virchows Arch A 421:13–16, 1992.
Pinto EM, Chen X, Easton J, Finkelstein D, Liu Z, Pounds S, Rodriguez-Galindo C, Lund TC, Mardis ER, Wilson RK, Boggs K, Yergeau D, Cheng J, Mulder HL, Manne J, Jenkins J, Mastellaro MJ, Figueiredo BC, Dyer MA, Pappo A, Zhang J, Downing JR, Ribeiro RC, Zambetti GP. Genomic landscape of paediatric adrenocortical tumours. Nat Commun 6: 6302, 2015.
Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G, Kim S, Assie G, Morozova O, Akbani R, Shih J, Hoadley KA, Choueiri TK, Waldmann J, Mete O, Robertson AG, Wu HT, Raphael BJ, Shao L, Meyerson M, Demeure MJ, Beuschlein F, Gill AJ, Sidhu SB, Almeida MQ, Fragoso MC, Cope LM, Kebebew E, Habra MA, Whitsett TG, Bussey KJ, Rainey WE, Asa SL, Bertherat J, Fassnacht M, Wheeler DA; Cancer Genome Atlas Research Network., Hammer GD, Giordano TJ, Verhaak RG. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 29: 723–736, 2016.
Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G, Kim S, Assie G, Morozova O, Akbani R, Shih J, Hoadley KA, Choueiri TK, Waldmann J, Mete O, Robertson AG, Wu HT, Raphael BJ, Shao L, Meyerson M, Demeure MJ, Beuschlein F, Gill AJ, Sidhu SB, Almeida MQ, Fragoso MC, Cope LM, Kebebew E, Habra MA, Whitsett TG, Bussey KJ, Rainey WE, Asa SL, Bertherat J, Fassnacht M, Wheeler DA; Cancer Genome Atlas Research Network., Hammer GD, Giordano TJ, Verhaak RG. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell. 30: 363, 2016.
Assié G, Letouzé E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, René-Corail F, Elarouci N, Sbiera S, Kroiss M, Allolio B, Waldmann J, Quinkler M, Mannelli M, Mantero F, Papathomas T, De Krijger R, Tabarin A, Kerlan V, Baudin E, Tissier F, Dousset B, Groussin L, Amar L, Clauser E, Bertagna X, Ragazzon B, Beuschlein F, Libé R, de Reyniès A, Bertherat J. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 46: 607–612, 2014.
Assié G, Jouinot A, Bertherat J. The “omics” of adrenocortical tumours for personalized medicine. Nat Rev Endocrinol 10: 215–228, 2014.
Dworakowska D, Drabarek A, Wenzel I, Babińska A, Świątkowska-Stodulska R, Sworczak K. Adrenocortical cancer (ACC)—literature overview and own experience. Endokrynol Pol 65: 492–502, 2014
Lughezzani G, Sun M, Perrotte P, Jeldres C, Alasker A, Isbarn H, Budäus L, Shariat SF, Guazzoni G, Montorsi F, Karakiewicz PI. The European network for the study of adrenal tumors staging system is prognostically superior to the international union against cancer-staging system: a North American validation. Eur J Cancer 46: 713–719, 2010.
Straka M, Soumarova R, Bulejcik J, Banik M, Pura M, Skrovina M. Giant adrenocortical carcinoma with 27-month disease-free survival by surgical resection alone: a case report. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 158: 474–478, 2014.
Papotti M, Libè R, Duregon E, Volante M, Bertherat J, Tissier F. The Weiss score and beyond—histopathology for adrenocortical carcinoma. Horm Cancer. 2: 333–340, 2011.
Libé R, Borget I, Ronchi CL, Zaggia B, Kroiss M, Kerkhofs T, Bertherat J, Volante M, Quinkler M, Chabre O, Bala M, Tabarin A, Beuschlein F, Vezzosi D, Deutschbein T, Borson-Chazot F, Hermsen I, Stell A, Fottner C, Leboulleux S, Hahner S, Mannelli M, Berruti A, Haak H, Terzolo M, Fassnacht M, Baudin E ENSAT network. Prognostic factors in stage III–IV adrenocortical carcinomas (ACC): a European Network for the Study of Adrenal Tumor (ENSAT) study. Ann Oncol 26: 2119–2125, 2015.
Beuschlein F, Weigel J, Saeger W, Kroiss M, Wild V, Daffara F, Libé R, Ardito A, Al Ghuzlan A, Quinkler M, Oßwald A, Ronchi CL, de Krijger R, Feelders RA, Waldmann J, Willenberg HS, Deutschbein T, Stell A, Reincke M, Papotti M, Baudin E, Tissier F, Haak HR, Loli P, Terzolo M, Allolio B, Müller HH, Fassnacht M. Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab 100: 841–849, 2015.
Duregon E, Molinaro L, Volante M, Ventura L, Righi L, Bolla S, Terzolo M, Sapino A, Papotti MG. Comparative diagnostic and prognostic performances of the hematoxylin-eosin and phospho-histone H3 mitotic count and Ki-67 index in adrenocortical carcinoma. Mod Pathol 27: 1246–1254, 2014.
Lam KY, Wat MS. Adrenal cortical black adenoma: report of two cases and review of the literature. J Urol Pathol 4: 183–190, 1996.
Mearini L, Del Sordo R, Costantini E, Nunzi E, Porena M. Adrenal oncocytic neoplasm: a systematic review. Urol Int 91: 125–133, 2013.
Nakamura Y, Yamazaki Y, Tezuka Y, Satoh F, Sasano H. Expression of CYP11B2 in aldosterone-producing adrenocortical adenoma: regulatory mechanisms and clinical significance. Tohoku J Exp Med 240:183–190, 2016.
Thiel A, Reis AC, Haase M, Goh G, Schott M, Willenberg HS, Scholl UI. PRKACA mutations in cortisol-producing adenomas and adrenal hyperplasia: a single-center study of 60 cases. Eur J Endocrinol 172: 677–685, 2015.
Azevedo MF, Stratakis CA. The transcriptome that mediates increased cyclic adenosine monophosphate signaling in PRKAR1A defects and other settings. Endocr Pract. 17S3: 2–7, 2011.
Bonnet S, Gaujoux S, Launay P, Baudry C, Chokri I, Ragazzon B, Libé R, René-Corail F, Audebourg A, Vacher-Lavenu MC, Groussin L, Bertagna X, Dousset B, Bertherat J, Tissier F. Wnt/β-catenin pathway activation in adrenocortical adenomas is frequently due to somatic CTNNB1-activating mutations, which are associated with larger and nonsecreting tumors: a study in cortisol-secreting and -nonsecreting tumors. J Clin Endocrinol Metab 96: E419-E426, 2011.
Orselli RC, Bassler TJ. Theca granuloma cell tumor arising in adrenal. Cancer 31: 474–477, 1973
Trost BN, Koenig MP, Zimmermann A, Zachmann M, Müller J. Virilization of a post-menopausal woman by a testosterone-secreting Leydig cell type adrenal adenoma. Acta Endocrinol (Copenh). 98: 274–282, 1981
Vasiloff J, Chideckel EW, Boyd CB, Foshag LJ. Testosterone-secreting adrenal adenoma containing crystalloids characteristic of Leydig cells. Am J Med 79: 772–776, 1985.
Pollock WJ, McConnell CF, Hilton C, Lavine RL. Virilizing Leydig cell adenoma of adrenal gland. Am J Surg Pathol. 10: 816–822, 1986.
Hameed A, Coleman RL. Fine-needle aspiration cytology of primary granulosa cell tumor of the adrenal gland: a case report. Diagn Cytopathol. 22: 107–109, 2000.
Cheng JY, Gill AJ, Kumar SK. Granulosa cell tumour of the adrenal. Pathology 47: 487–489, 2015.
Simpson PR. Adenomatoid tumor of the adrenal gland. Arch Pathol Lab Med 114: 725–727, 1990.
El-Daly H, Rao P, Palazzo F, Gudi M. A rare entity of an unusual site: adenomatoid tumour of the adrenal gland: a case report and review of the literature. Patholog Res Int. 2010: 702472, 2010.
Taskin OC, Gucer H, Mete O. An unusual adrenal cortical nodule: composite adrenal cortical adenoma and adenomatoid tumor. Endocr Pathol 26: 370–373, 2015.
Timonera ER, Paiva ME, Lopes JM, Eloy C, van der Kwast T, Asa SL. Composite adenomatoid tumor and myelolipoma of adrenal gland: report of 2 cases. Arch Pathol Lab Med. 132: 265–267, 2008.
Kenney PJ, Wagner BJ, Rao P, Heffess CS. Myelolipoma: CT and pathologic features. Radiology 208:87–95, 1998
Mitsui Y, Yasumoto H, Hiraki M, Arichi N, Ishikawa N, Harada Y, Maruyama R, Shiina H. Coordination of bone morphogenetic protein 2 (BMP2) and aberrant canonical Wnt/β-catenin signaling for heterotopic bone formation in adrenal myelolipoma: a case report. Can Urol Assoc J 8: E104–E107, 2014.
Lam A. Lipomatous tumours in adrenal gland: WHO updates and clinical implications Endocr Relat Cancer 24: R65-R79, 2017
Khong PL, Lam KY, Ooi GC, Liu MJ, Metreweli C. Mature teratomas of the adrenal gland: imaging features. Abdom Imagining 2002; 27: 347–350.
Lam KY, Lo CY. Adrenal lipomatous tumours: a 30 year clinicopathological experience at a single institution. J Clin Pathol 54: 707–712, 2001.
Lam KY, Lo CY. Teratoma in the region of adrenal gland: a unique entity masquerading as lipomatous adrenal tumor. Surgery 126: 90–94, 1999.
Lam KY, Chan ACL, Ng IOL. Giant adrenal lipoma: a report of two cases and review of the literature. Scand J Urol Nephrol 31:89–90, 1997.
Au WY, Tam PC, Ma SK, Lam KY. Giant myelolipoma in a patient with thalassemia intermedia. Am J Hematol 65: 265–266, 2000.
Zhao J, Sun F, Jing X, Zhou W, Huang X, Wang H, Zhu Y, Yuan F, Shen Z. The diagnosis and treatment of primary adrenal lipomatous tumours in Chinese patients: a 31-year follow-up study. Can Urol Assoc J 8: E132-E136, 2014.
Shenoy VG, Thota A, Shankar R, Desai MG. Adrenal myelolipoma: controversies in its management. Indian J Urol 31: 94–101, 2015.
Li SQ, Zhang YS, Shi J, Li HZ. Clinical features and retroperitoneal laparoscopic resection of adrenal schwannoma in 19 patients. Endocr Pract 21: 323–329, 2015.
Mohiuddin Y, Gilliland MG. Adrenal schwannoma: a rare type of adrenal incidentaloma. Arch Pathol Lab Med 137: 1009–1014, 2013.
Xiao C, Xu B, Ye H, Yang Q, Wang L, Sun YH. Experience with adrenal schwannoma in a Chinese population of six patients. J Endocrinol Invest 34: 417–421, 2011.
Rashidi A, Fisher SI. Primary adrenal lymphoma: a systematic review. Ann Hematol 92: 1583–1593, 2013
Mozos A, Ye H, Chuang WY, Chu JS, Huang WT, Chen HK, Hsu YH, Bacon CM, Du MQ, Campo E, Chuang SS. Most primary adrenal lymphomas are diffuse large B-cell lymphomas with non-germinal center B-cell phenotype, BCL6 gene rearrangement and poor prognosis. Mod Pathol 22: 1210–1217, 2009.
Lam KY, Lo CY. Metastatic tumours of the adrenal glands: a 30-year experience in a teaching hospital. Clin Endocrinol 56: 95–101, 2002.
Gryn A, Peyronnet B, Manunta A, Beauval JB, Bounasr E, Nouhaud FX, Rioux-Leclercq N, Caron P, Thoulouzan M, Verhoest G, Soulie M, Bensalah K, Huyghe E. Patient selection for laparoscopic excision of adrenal metastases: a multicenter cohort study. Int J Surg 24: 75–80, 2015.
Pillai S, Gopalan V, Lo CY, Liew V, Smith RA, Lam AK. Silent genetic alterations identified by targeted next-generation sequencing in pheochromocytoma/paraganglioma: a clinicopathological correlations. Exp Mol Pathol. 102: 41–46, 2017.
Pillai S, Gopalan V, Smith RA, Lam AK. Updates on the genetics and the clinical impacts on phaeochromocytoma and paraganglioma in the new era. Crit Rev Oncol Hematol 100: 190–208, 2016.
Lam AK. Update on paragangliomas and pheochromocytomas. Turk Patoloji Derg 31S1: 105–112, 2015.
Burnichon N, Buffet A, Parfait B, Letouzé E, Laurendeau I, Loriot C, Pasmant E, Abermil N, Valeyrie-Allanore L, Bertherat J, Amar L, Vidaud D, Favier J, Gimenez-Roqueplo AP. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum Mol Genet 21: 5397–5405, 2012.
Niemeijer ND, Papathomas TG, Korpershoek E, de Krijger RR, Oudijk L, Morreau H, Bayley JP, Hes FJ, Jansen JC, Dinjens WN, Corssmit EP. Succinate dehydrogenase (SDH)-deficient pancreatic neuroendocrine tumor expands the SDH-related tumor spectrum. J Clin Endocrinol Metab. 2015; 100: E1386–E1393.
Toledo SP, Lourenço DM Jr, Sekiya T, Lucon AM, Baena ME, Castro CC, Bortolotto LA, Zerbini MC, Siqueira SA, Toledo RA, Dahia PL. Penetrance and clinical features of pheochromocytoma in a six-generation family carrying a germline TMEM127 mutation. J Clin Endocrinol Metab 100: E308-E318, 2015.
Hoekstra AS, Devilee P, Bayley JP. Models of parent-of-origin tumorigenesis in hereditary paraganglioma. Semin Cell Dev Biol 43:117–124, 2015.
Thompson LD. Pheochromocytoma of the adrenal gland scaled score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 26: 551–566, 2002.
Agarwal A, Mehrotra PK, Jain M, Gupta SK, Mishra A, Chand G, Agarwal G, Verma AK, Mishra SK, Singh U. Size of the tumor and pheochromocytoma of the adrenal gland scaled score (PASS): can they predict malignancy? World J Surg 34:3022–3028, 2010.
Mlika M, Kourda N, Zorgati MM, Bahri S, Ben Ammar S, Zermani R. Prognostic value of pheochromocytoma of the adrenal gland scaled score (PASS score) tests to separate benign from malignant neoplasms. Tunis Med 91: 209–215, 2013.
Kimura N, Watanabe T, Noshiro T, Shizawa S and Miura Y. Histological grading of adrenal and extra-adrenal pheochromocytomas and relationship to prognosis: a clinicopathological analysis of 116 adrenal pheochromocytomas and 30 extra-adrenal sympathetic paragangliomas including 38 malignant tumors. Endocr Pathol 16: 23–32, 2005.
Kimura N, Takayanagi R, Takizawa N, Itagaki E, Katabami T, Kakoi N, Rakugi H, Ikeda Y, Tanabe A, Nigawara T, Ito S, Kimura I, Naruse M; Phaeochromocytoma Study Group in Japan. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer 21:405–414, 2014.
Kimura N, Takekoshi K, Horii A, Morimoto R, Imai T, Oki Y, Saito T, Midorikawa S, Arao T, Sugisawa C, Yamada M, Otuka Y, Kurihara I, Sugano K, Nakane M, Fukuuchi A, Kitamoto T, Saito J, Nishikawa T, Naruse M. Clinicopathological study of SDHB mutation-related pheochromocytoma and sympathetic paraganglioma. Endocr Relat Cancer. 21: L13–L16, 2014.
Parenti G, Zampetti B, Rapizzi E, Ercolino T, Giachè V, Mannelli M. Updated and new perspectives on diagnosis, prognosis, and therapy of malignant pheochromocytoma/paraganglioma. J Oncol 2012: 872713, 2012.
Castro-Vega LJ, Letouzé E, Burnichon N, Buffet A, Disderot PH, Khalifa E, Loriot C, Elarouci N, Morin A, Menara M, Lepoutre-Lussey C, Badoual C, Sibony M, Dousset B, Libé R, Zinzindohoue F, Plouin PF, Bertherat J, Amar L, de Reyniès A, Favier J, Gimenez-Roqueplo AP. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun. 6: 6044, 2015.
Patterson E, Webb R, Weisbrod A, Bian B, He M, Zhang L, Holloway AK, KrishnaR, Nilubol N, Pacak K, Kebebew E. The microRNA expression changes associated with malignancy and SDHB mutation in pheochromocytoma. Endocr Relat Cancer 19: 157–166, 2012.
Lam KY, Chan ACL, Wong WM, Lam KSL. A review of clinicopathologic features of pheochromocytomas in Hong Kong Chinese. Eur J Surg Oncol 19: 421–427, 1993.
Eisenhofer G, Pacak K, Huynh TT, Qin N, Bratslavsky G, Linehan WM, Mannelli M, Friberg P, Grebe SK, Timmers HJ, Bornstein SR, Lenders JW. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr Relat Cancer 18: 97–111, 2010.
Eisenhofer G, Peitzsch M. Laboratory evaluation of pheochromocytoma and paraganglioma. Clin Chem 60:1486–1499, 2014.
Eisenhofer G, Goldstein DS, Kopin IJ, Crout JR. Pheochromocytoma: rediscovery as a catecholamine-metabolizing tumor. Endocr Pathol 14:193–212, 2003.
Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev 56: 331–349, 2004.
Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, Mannelli M, Linehan WM, Adams K, Timmers HJ, Pacak K. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer 48: 1739–1749, 2012.
Eisenhofer G, Lenders JW, Timmers H, Mannelli M, Grebe SK, Hofbauer LC, Bornstein SR, Tiebel O, Adams K, Bratslavsky G, Linehan WM, Pacak K. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem 57: 411–420, 2011.
Kakkar A, Kaur K, Kumar T, Cherian LB, Kaushal R, Sharma MC, Dhar A, Seth A, Jain D. Pigmented pheochromocytoma: an unusual variant of a common tumor. Endocr Pathol. 27: 42–45, 2016.
Kasem K, Lam AK. Adrenal oncocytic phaeochromocytoma with putative adverse histologic features: a unique case report and review of the literature. Endocr Pathol 25: 416–421, 2014.
Kim KY, Kim JH, Hong AR, Seong MW, Lee KE, Kim SJ, Kim SW, Shin CS, Kim SY. Disentangling of malignancy from benign pheochromocytomas/paragangliomas. PLoS One 11: e0168413, 2016.
Lam KY, Lo CY, Wat NMS, Luk JM, Lam KSL. The clinicopathological features and importance of p53, Rb and mdm2 expression in phaeochromocytomas and paragangliomas. J Clin Pathol 54:443–448, 2001.
Lam KY, Chan ACL. Paragangliomas: a comparative clinical, histologic and immunohistochemical study. Int J Surg Pathol 1:111–116, 1993.
Chan JKC, Kimura N, Capella C, Gill A, Komminoth P, Lam AKY, Tischler AS, Williams MD. Paraganglion tumours. In: WHO classification of head and neck tumours. El-Nigger AK, Chan JKC, Grandis JF, Takata T, Slootweg PJ (eds), Chapter 10. 4th ed. Lyon: IARC; 2017 pp275–284.
Sajid MS, Hamilton G, Baker DM; Joint Vascular Research Group. A multicenter review of carotid body tumour management. Eur J Vasc Endovasc Surg 34:127–130, 2007.
Piccini V, Rapizzi E, Bacca A, Di Trapani G, Pulli R, Giachè V, Zampetti B, Lucci-Cordisco E, Canu L, Corsini E, Faggiano A, Deiana L, Carrara D, Tantardini V, Mariotti S, Ambrosio MR, Zatelli MC, Parenti G, Colao A, Pratesi C, Bernini G, Ercolino T, Mannelli M. Head and neck paragangliomas: genetic spectrum and clinical variability in 79 consecutive patients. Endocr Relat Cancer 19:149–155, 2012.
Lam KY, Chan ACL. Paraganglioma of urinary bladder: an immunohistochemical study and report of an unusual association with intestinal carcinoid. Aust NZ J Surg 63: 740–745, 1993.
Garg A, Mishra D, Bansal M, Maharia HR, Goyal V. Right atrial paraganglioma: an extremely rare primary cardiac neoplasm mimicking myxoma. J Cardiovasc Ultrasound 24: 334–336, 2016.
Michałowska I, Ćwikła J, Prejbisz A, Kwiatek P, Szperl M, Michalski W, Wyrwicz L, Kuśmierczyk M, Januszewicz A, Maciejczyk A, Roszczynko M, Pęczkowska M. Mediastinal paragangliomas related to SDHx gene mutations. Kardiochir Torakochirurgia Pol 13:276–282, 2016.
Soomro NH, Zahid AB, Zafar AA. Non-functional paraganglioma of the mediastinum. J Pak Med Assoc 66: 609–611, 2016.
Blanchet EM, Martucci V, Pacak K. Pheochromocytoma and paraganglioma: current functional and future molecular imaging. Front Oncol. 2012 1: 58, 2012.
Blumenfeld J, Cohen N, Anwar M, Teitelman G, Laragh JH, Ruggiero DA. Hypertension and a tumor of the glomus jugulare region. Evidence for epinephrine biosynthesis. Am J Hypertens 6: 382–387, 1993.
Waguespack SG, Rich T, Grubbs E, Ying AK, Perrier ND, Ayala-Ramirez M, Jimenez C. A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 95: 2023–2037, 2010.
Assadipour Y, Sadowski SM, Alimchandani M, Quezado M, Steinberg SM, Nilubol N, Patel D, Prodanov T, Pacak K, Kebebew E. SDHB mutation status and tumor size but not tumor grade are important predictors of clinical outcome in pheochromocytoma and abdominal paraganglioma. Surgery 161: 230–239, 2017.
Mehdiabadi GB, Arab E, Rafsanjani KA, Ansari S, Moinzadeh AM. Neuroblastoma in Iran: an experience of 32 years at a referral children’s hospital. Asian Pac J Cancer Prev 14: 2739–2742, 2013.
Cotterill SJ, Pearson AD, Pritchard J, Foot AB, Roald B, Kohler JA, Imeson J. Clinical prognostic factors in 1277 patients with neuroblastoma: results of the European Neuroblastoma Study Group “Survey” 1982–1992. Eur J Cancer 36: 901–908, 2000.
Spinelli C, Rossi L, Barbetta A, Ugolini C, Strambi S. Incidental ganglioneuromas: a presentation of 14 surgical cases and literature review. J Endocrinol Invest 38: 547–554, 2015.
Bielle F, Fréneaux P, Jeanne-Pasquier C, Maran-Gonzalez A, Rousseau A, Lamant L, Paris R, Pierron G, Nicolas AV, Sastre-Garau X, Delattre O, Bourdeaut F, Peuchmaur M. PHOX2B immunolabeling: a novel tool for the diagnosis of undifferentiated neuroblastomas among childhood small round blue-cell tumors. Am J Surg Pathol 36: 1141–1149, 2012.
Hata JL, Correa H, Krishnan C, Esbenshade AJ, Black JO, Chung DH, Mobley BC. Diagnostic utility of PHOX2B in primary and treated neuroblastoma and in neuroblastoma metastatic to the bone marrow. Arch Pathol Lab Med 139: 543–546, 2015.
Shawa H, Elsayes KM, Javadi S, Morani A, Williams MD, Lee JE, Waguespack SG, Busaidy NL, Vassilopoulou-Sellin R, Jimenez C, Habra MA. Adrenal ganglioneuroma: features and outcomes of 27 cases at a referral cancer centre. Clin Endocrinol (Oxf) 80: 342–347, 2014.
Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, Favrot M, Hedborg F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11:1466–1477, 1993
Brodeur GM, Seeger RC, Barrett A, Berthold F, Castleberry RP, D'Angio G, De Bernardi B, Evans AE, Favrot M, Freeman AI, et al. International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol 6:1874–1881, 1988.
Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, Kaneko M, London WB, Matthay KK, Nuchtern JG, von Schweinitz D, Simon T, Cohn SL, Pearson AD; INRG Task Force. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27: 298–303, 2009.
Lam KY, Lo CY. Composite pheochromocytoma-ganglioneuroma of the adrenal gland: an uncommon entity with distinctive clinicopathologic features. Endocr Pathol 1999;10: 343–352.
Comstock JM, Willmore-Payne C, Holden JA, Coffin CM. Composite pheochromocytoma: a clinicopathologic and molecular comparison with ordinary pheochromocytoma and neuroblastoma. Am J Clin Pathol 132:69–73, 2009.
Shawa H, Elsayes KM, Javadi S, Sircar K, Jimenez C, Habra MA. Clinical and radiologic features of pheochromocytoma/ganglioneuroma composite tumors: a case series with comparative analysis. Endocr Pract 20: 864–869, 2014.
Shida Y, Igawa T, Abe K, Hakariya T, Takehara K, Onita T, Sakai H. Composite pheochromocytoma of the adrenal gland: a case series. BMC Res Notes 8: 257, 2015.
Loehry CA, Kingham JG, Whorwell PJ. Watery diarrhoea and hypokalaemia associated with a phaeochromocytoma. Postgrad Med J 51:416–419, 1975.
Jiang J, Zhang L, Wu Z, Ai Z, Hou Y, Lu Z, Gao X. A rare case of watery diarrhea, hypokalemia and achlorhydria syndrome caused by pheochromocytoma. BMC Cancer 14: 553, 2014.
Tischler AS, Dayal Y, Balogh K, Cohen RB, Connolly JL, Tallberg K. The distribution of immunoreactive chromogranins, S-100 protein, and vasoactive intestinal peptide in compound tumors of the adrenal medulla. Hum Pathol 18: 909–917, 1987.
Lam KY, Loong F, Shek TWH, Chu SM. Composite paraganglioma-ganglioneuroma of the urinary bladder: a cliniopathologic, immunohistochemical, and ultrastructural study of a case and review of the literature. Endocr Pathol 1998;9: 363–373.
Monclair T, Ruud E, Holmstrøm H, Aagenæs I, Asplin M, Beiske K. Extra-adrenal composite phaeochromocytoma/neuroblastoma in a 15-month-old child. J Ped Surg Case Reports 3: 348e350, 2015.
Ohtsuki Y, Watanabe R, Okada Y, Matsuka Y, Lee GH, Furihata M. Composite paraganglioma and ganglioneuroma in the retroperitoneum: a case report. Med Mol Morphol 45: 168–172, 2012.
Ito H, Kurokawa T, Yokoyama O. Composite paraganglioma with ganglioneuroma in the retroperitoneal space. Int J Urol 17: 385–386, 2010.
Hirasaki S, Kanzaki H, Okuda M, Suzuki S, Fukuhara T, Hanaoka T. Composite paraganglioma-ganglioneuroma in the retroperitoneum. World J Surg Oncol 7: 81, 2009.
Chen CH, Boag AH, Beiko DT, Siemens DR, Froese A, Isotalo PA. Composite paraganglioma-ganglioneuroma of the urinary bladder: a rare neoplasm causing hemodynamic crisis at tumour resection. Can Urol Assoc J 3: E45–E48, 2009.
Usuda H, Emura I. Composite paraganglioma-ganglioneuroma of the urinary bladder. Pathol Int 55: 596–601, 2005.
Dundr P, Dudorkinová D, Povýsil C, Pesl M, Babjuk M, Dvorácek J, Zelinka T. Pigmented composite paraganglioma-ganglioneuroma of the urinary bladder. Pathol Res Pract 199:765–769, 2003.
Hu J, Wu J, Cai L, Jiang L, Lang Z, Qu G, Liu H, Yao W, Yu G. Retroperitoneal composite pheochromocytoma-ganglioneuroma: a case report and review of literature. Diagn Pathol 8: 63, 2013.
Shankar GM, Chen L, Kim AH, Ross GL, Folkerth RD, Friedlander RM. Composite ganglioneuroma-paraganglioma of the filum terminale. J Neurosurg Spine 12: 709–713, 2010.
de Montpréville VT, Mussot S, Gharbi N, Dartevelle P, Dulmet E. Paraganglioma with ganglioneuromatous component located in the posterior mediastinum. Ann Diagn Pathol. 9: 110–114, 2005.
Armstrong R, Greenhalgh KL, Rattenberry E, Judd B, Shukla R, Losty PD, Maher ER. Succinate dehydrogenase subunit B (SDHB) gene deletion associated with a composite paraganglioma/neuroblastoma. J Med Genet 46: 215–216, 2009.
Fritzsche FR, Bode PK, Koch S, Frauenfelder T. Radiological and pathological findings of a metastatic composite paraganglioma with neuroblastoma in a man: a case report. J Med Case Rep 4: 374, 2010.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
None.
Conflict of Interest
The author declares that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Lam, A.Ky. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocr Pathol 28, 213–227 (2017). https://doi.org/10.1007/s12022-017-9484-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12022-017-9484-5